
Tumor Lymphatic Interactions Induce CXCR2-CXCL5 Axis and Alter Cellular Metabolism and Lymphangiogenic Pathways to Promote Cholangiocarcinoma

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal Models
2.3. Cell Culture and Treatments
2.4. RNA Isolation and Real Time Quantitative Reverse Transcriptase PCR (qPCR)
2.5. Cell Lysis and Western Blot
2.6. Boyden Chamber Assay
2.7. Cytokine-Chemokine Array Analysis
2.8. Scratch Wound Assay
2.9. Immunofluorescence (IF) and Immunohistochemistry (IHC)
2.10. Tube Formation Assay
2.11. ELISA for CXCL5
2.12. Seahorse Assay
2.13. Lactate and Glucose-Glo™ Assay
2.14. MitoSOX Assay
2.15. Statistical Analysis
3. Results
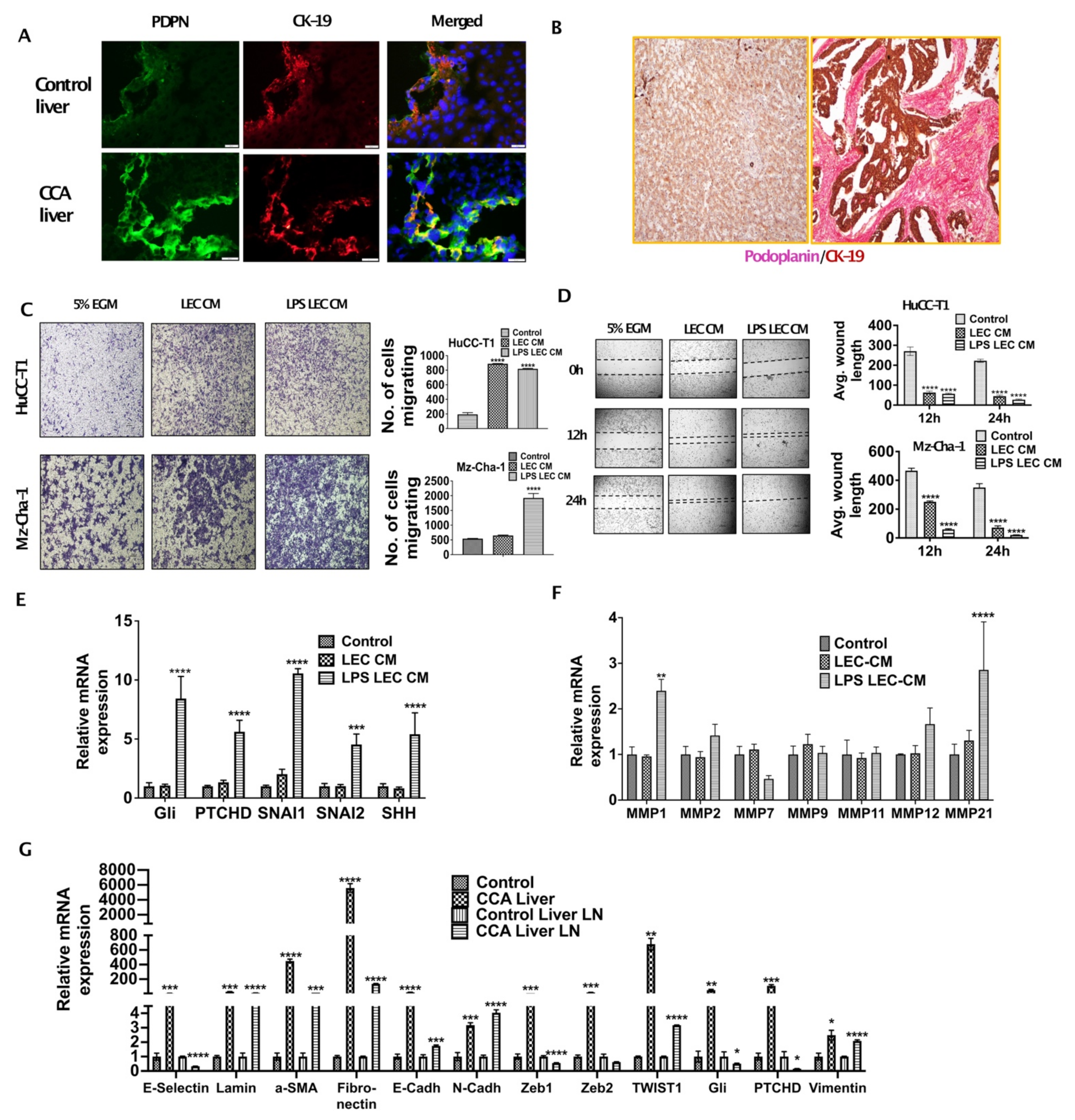
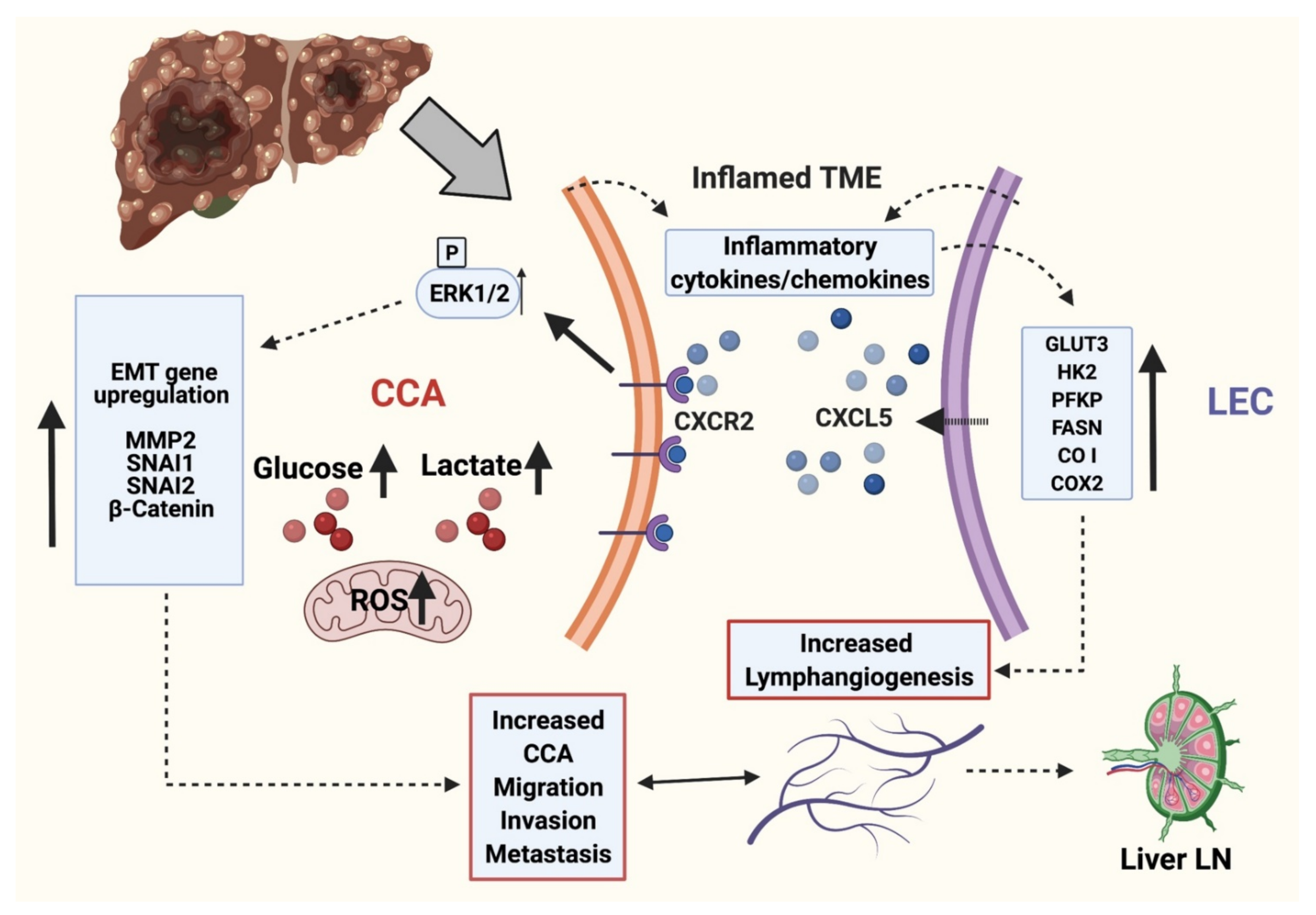
3.1. Inflamed LECs Enhance Tumor Migration and Activation of Critical EMT Regulators in CCA
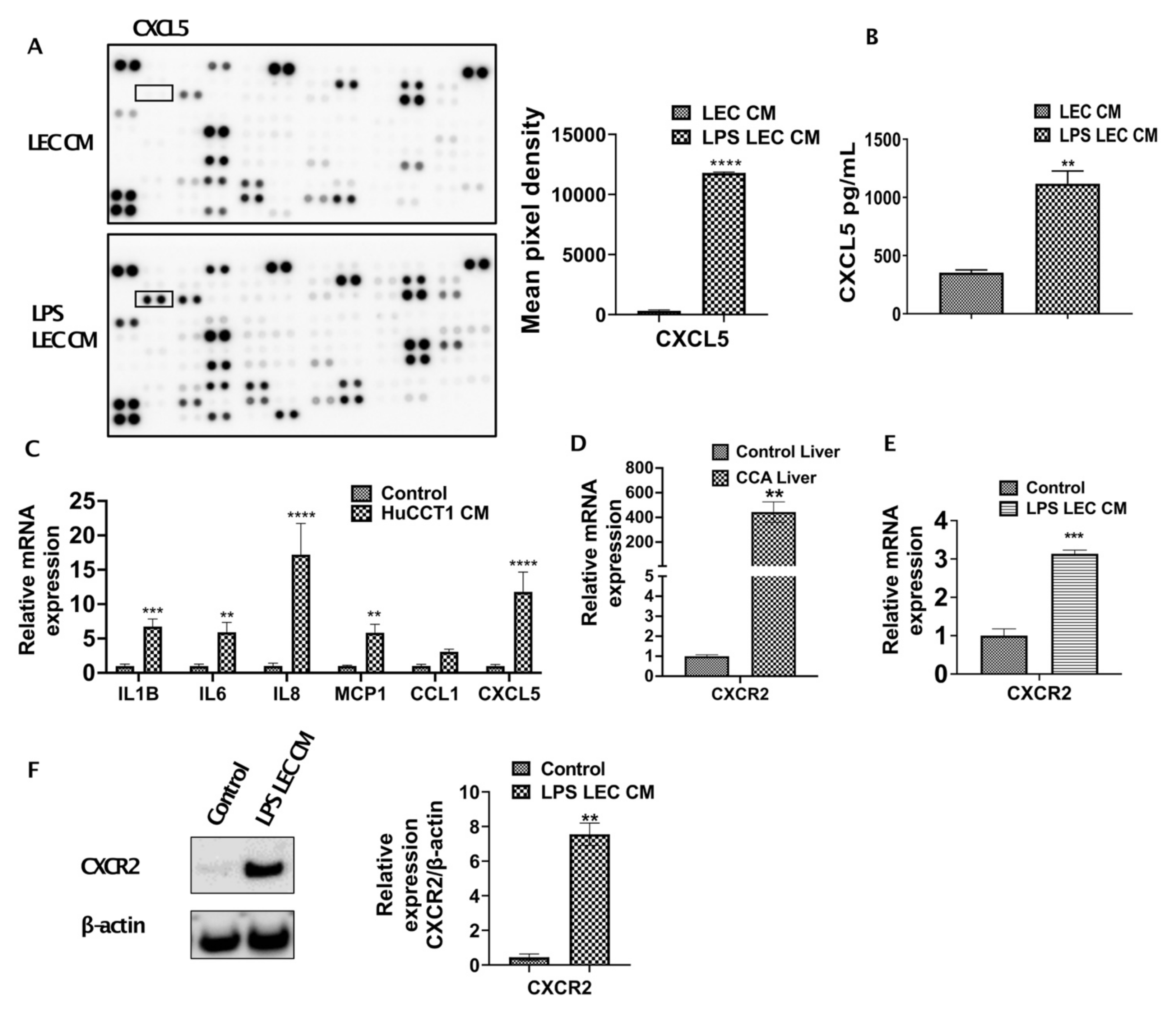
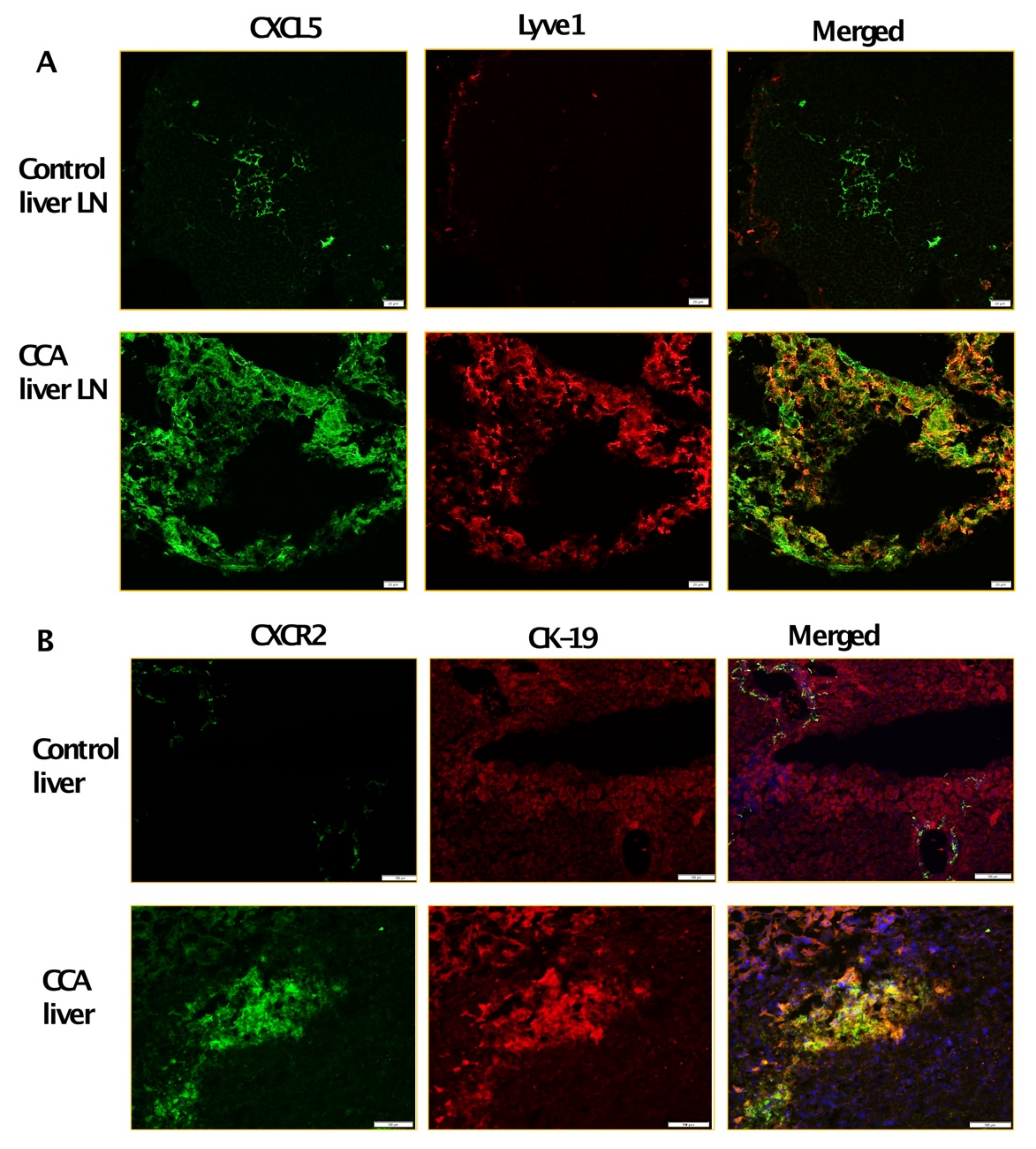
3.2. Inflamed LECs Express High Levels of CXCL5 and Induce the Upregulation of CXCR2 Expression in CCA Cells
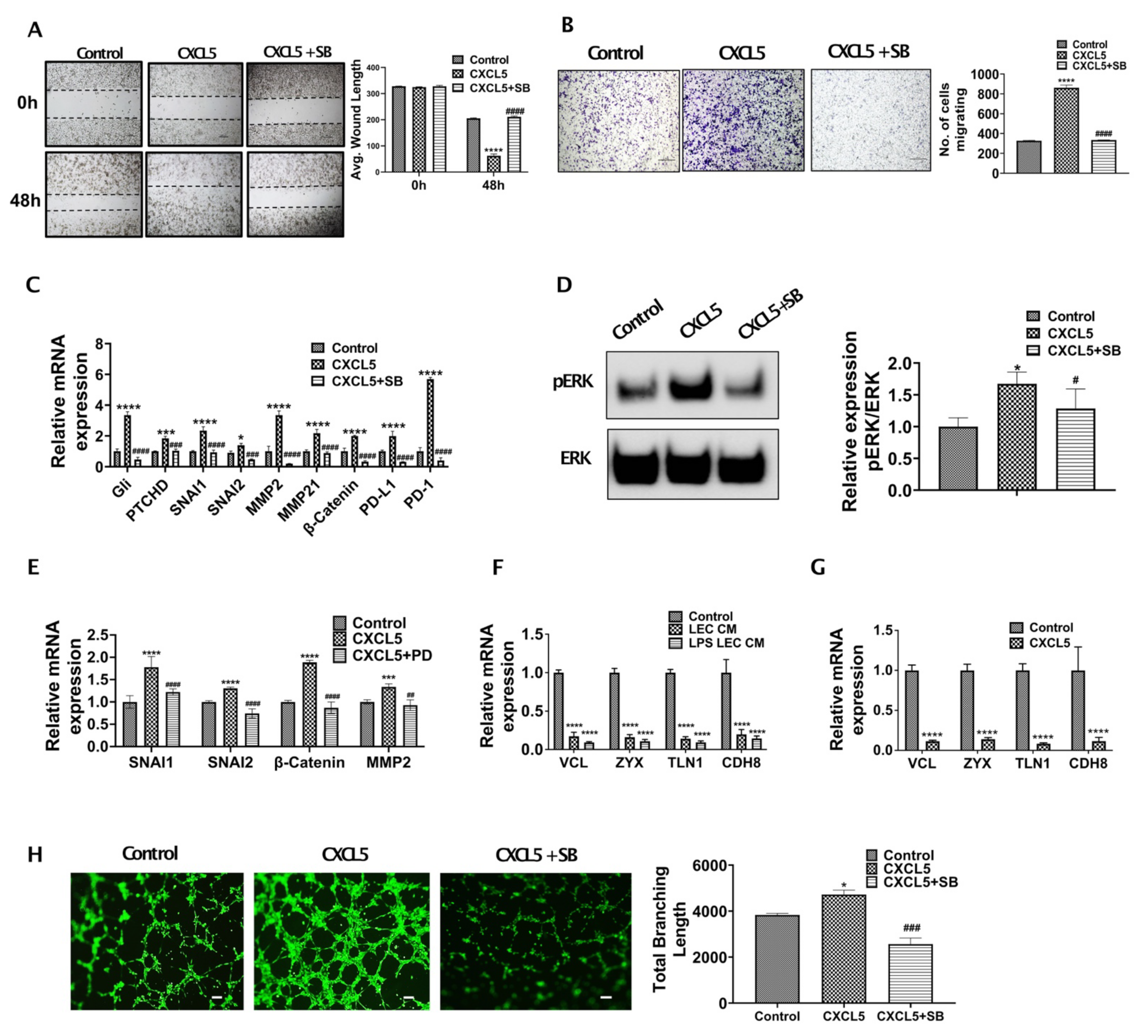
3.3. CXCL5 Increases Migration and Tumor Promoting Pathways by Inducing Critical EMT Determinants and Focal Adhesion Molecules in CCA and Also Enhance Lymphangiogenesis
3.4. CXCR2-CXCL5 Signaling Enhances Lymphatic Tube Formation
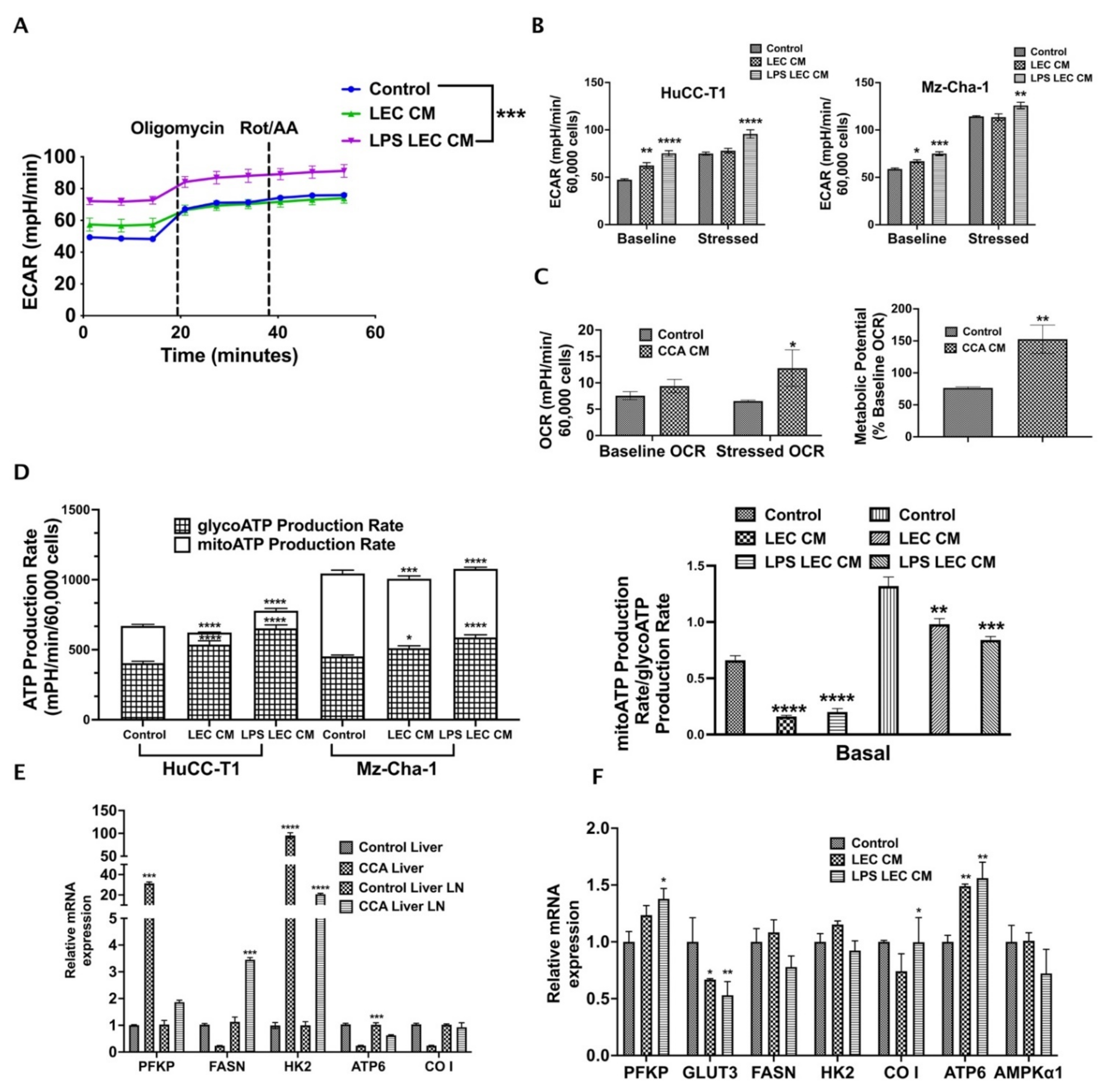
3.5. Crosstalk with Inflamed LECs Alter Cellular Bioenergetics and Critical Metabolic Gene Expression in CCA Cells
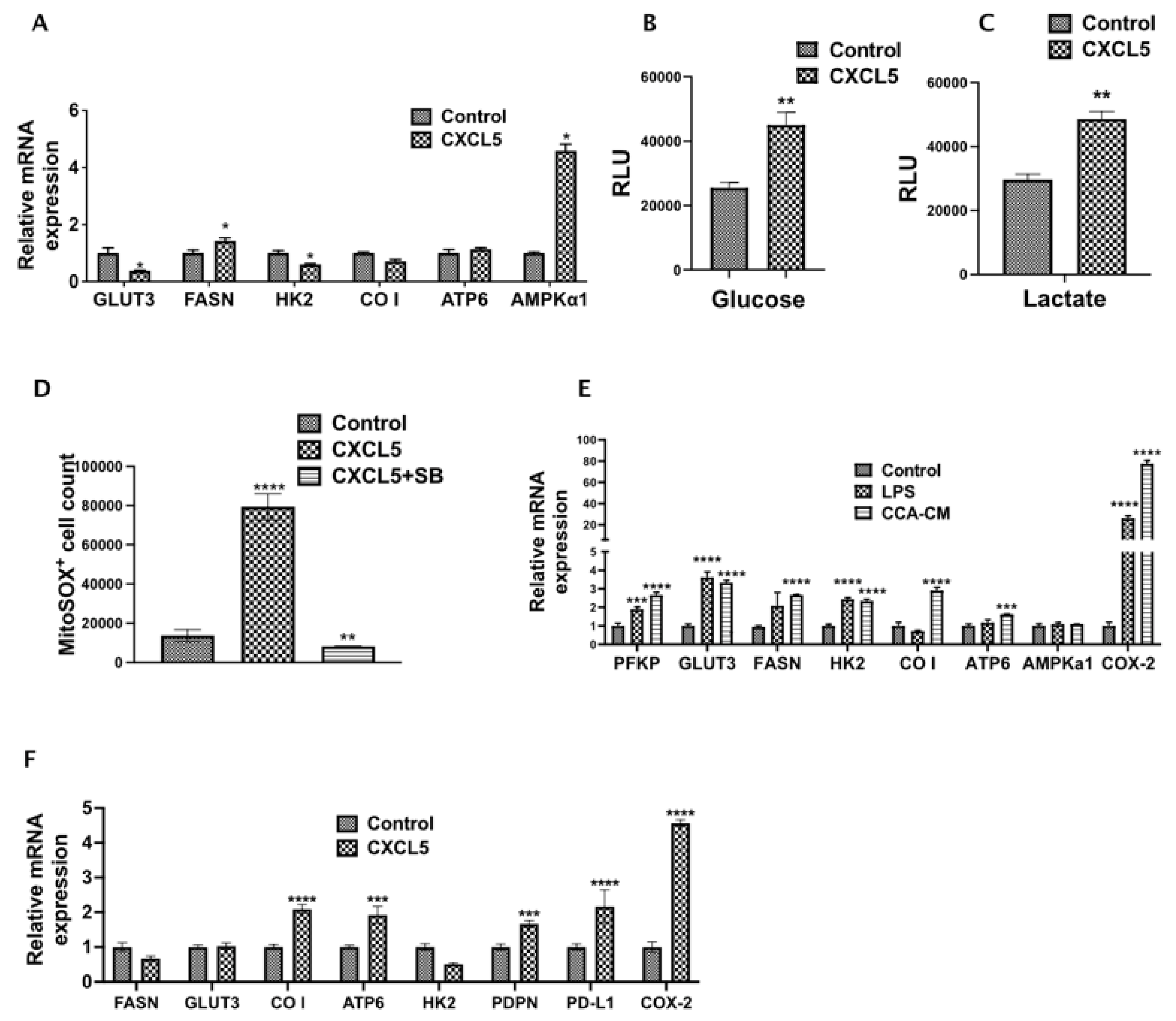
3.6. CXCL5 Directly Contributes to the Altered CCA Metabolism and Increases Lactate Production, Glucose Uptake and Induces Mitochondrial Reactive Oxygen Species (mitoROS)
3.7. CCA-LEC Crosstalk Induce Metabolic Gene Expression in LECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Baiocchi, L.; Sato, K.; Ekser, B.; Kennedy, L.; Francis, H.; Ceci, L.; Lenci, I.; Alvaro, D.; Franchitto, A.; Onori, P.; et al. Cholangiocarcinoma: Bridging the translational gap from preclinical to clinical development and implications for future therapy. Expert Opin. Investig. Drugs 2021, 30, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Glaser, S.; Chakraborty, S. Inflammation and Progression of Cholangiocarcinoma: Role of Angiogenic and Lymphangiogenic Mechanisms. Front. Med. 2019, 6, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, M.; Jeong, S.; Wang, X.; Tong, Y.; Cao, J.; Sun, H.Y.; Xia, L.; Xu, N.; Xi, Z.F.; Zhang, J.J.; et al. Tumor-associated lymphangiogenesis predicts unfavorable prognosis of intrahepatic cholangiocarcinoma. BMC Cancer 2019, 19, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabris, L.; Alvaro, D. The prognosis of perihilar cholangiocarcinoma after radical treatments. Hepatology 2012, 56, 800–802. [Google Scholar] [CrossRef]
- Alitalo, A.; Detmar, M. Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene 2012, 31, 4499–4508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Skobe, M. Lymphatic vessel activation in cancer. Ann. N. Y. Acad. Sci. 2008, 1131, 235–241. [Google Scholar] [CrossRef]
- Lee, E.; Pandey, N.B.; Popel, A.S. Lymphatic endothelial cells support tumor growth in breast cancer. Sci. Rep. 2014, 4, 5853. [Google Scholar] [CrossRef] [Green Version]
- Stacker, S.A.; Williams, S.P.; Karnezis, T.; Shayan, R.; Fox, S.B.; Achen, M.G. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 2014, 14, 159–172. [Google Scholar] [CrossRef]
- Cadamuro, M.; Morton, S.D.; Strazzabosco, M.; Fabris, L. Unveiling the role of tumor reactive stroma in cholangiocarcinoma: An opportunity for new therapeutic strategies. Transl. Gastrointest. Cancer 2013, 2, 130–144. [Google Scholar] [CrossRef]
- Aishima, S.; Nishihara, Y.; Iguchi, T.; Taguchi, K.; Taketomi, A.; Maehara, Y.; Tsuneyoshi, M. Lymphatic spread is related to VEGF-C expression and D2-40-positive myofibroblasts in intrahepatic cholangiocarcinoma. Mod. Pathol. 2008, 21, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Cadamuro, M.; Brivio, S.; Mertens, J.; Vismara, M.; Moncsek, A.; Milani, C.; Fingas, C.; Cristina Malerba, M.; Nardo, G.; Dall’Olmo, L.; et al. Platelet-derived growth factor-D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J. Hepatol. 2019, 70, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Koh, Y.J.; Kim, K.E.; Koh, B.I.; Nam, D.H.; Alitalo, K.; Kim, I.; Koh, G.Y. CXCR4 signaling regulates metastasis of chemoresistant melanoma cells by a lymphatic metastatic niche. Cancer Res. 2010, 70, 10411–10421. [Google Scholar] [CrossRef] [Green Version]
- Kumaravel, S.; Singh, S.; Roy, S.; Venkatasamy, L.; White, T.K.; Sinha, S.; Glaser, S.S.; Safe, S.H.; Chakraborty, S. CXCL11-CXCR3 Axis Mediates Tumor Lymphatic Cross Talk and Inflammation-Induced Tumor, Promoting Pathways in Head and Neck Cancers. Am. J. Pathol. 2020, 190, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Zhang, J.; Shi, Y.; Li, W.; Shi, H.; Ji, R.; Mao, F.; Qian, H.; Xu, W.; Zhang, X. CXCL5 promotes gastric cancer metastasis by inducing epithelial-mesenchymal transition and activating neutrophils. Oncogenesis 2020, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, H.; Sun, M.; Deng, X.; Wu, X.; Ma, Y.; Li, M.; Shuoa, S.M.; You, Q.; Miao, L. CXCL5/CXCR2 axis in tumor microenvironment as potential diagnostic biomarker and therapeutic target. Cancer Commun. 2020, 40, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Ma, X.L.; Wei, Y.Q.; Wei, X.W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim. Biophys. Acta. Rev. Cancer 2019, 1871, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.L.; Dai, Z.; Zhou, Z.J.; Chen, Q.; Wang, Z.; Xiao, Y.S.; Hu, Z.Q.; Huang, X.Y.; Yang, G.H.; Shi, Y.H.; et al. CXCL5 contributes to tumor metastasis and recurrence of intrahepatic cholangiocarcinoma by recruiting infiltrative intratumoral neutrophils. Carcinogenesis 2014, 35, 597–605. [Google Scholar] [CrossRef]
- Kang, H.; Kim, H.; Lee, S.; Youn, H.; Youn, B. Role of Metabolic Reprogramming in Epithelial (-) Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2019, 20, 2042. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.; Fischbach, S.R.; Bronk, S.F.; Hirsova, P.; Krishnan, A.; Dhanasekaran, R.; Smadbeck, J.B.; Smoot, R.L.; Vasmatzis, G.; Gores, G.J. YAP-associated chromosomal instability and cholangiocarcinoma in mice. Oncotarget 2018, 9, 5892–5905. [Google Scholar] [CrossRef] [Green Version]
- Knuth, A.; Gabbert, H.; Dippold, W.; Klein, O.; Sachsse, W.; Bitter-Suermann, D.; Prellwitz, W.; Meyer zum Buschenfelde, K.H. Biliary adenocarcinoma. Characterisation of three new human tumor cell lines. J. Hepatol. 1985, 1, 579–596. [Google Scholar] [CrossRef]
- Meng, F.; DeMorrow, S.; Venter, J.; Frampton, G.; Han, Y.; Francis, H.; Standeford, H.; Avila, S.; McDaniel, K.; McMillin, M.; et al. Overexpression of membrane metalloendopeptidase inhibits substance P stimulation of cholangiocarcinoma growth. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G759–G768. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Leng, J.; Han, C.; Demetris, A.J. The cyclooxygenase-2 inhibitor celecoxib blocks phosphorylation of Akt and induces apoptosis in human cholangiocarcinoma cells. Mol. Cancer Ther. 2004, 3, 299–307. [Google Scholar] [PubMed]
- Safe, S.; Kasiappan, R. Natural Products as Mechanism-based Anticancer Agents: Sp Transcription Factors as Targets. Phytother. Res. 2016, 30, 1723–1732. [Google Scholar] [CrossRef]
- Singh, S.; Kumaravel, S.; Dhole, S.; Roy, S.; Pavan, V.; Chakraborty, S. Neuropeptide Substance P Enhances Inflammation-Mediated Tumor Signaling Pathways and Migration and Proliferation of Head and Neck Cancers. Indian J. Surg. Oncol. 2021, 12, 93–102. [Google Scholar] [CrossRef]
- Chakraborty, S.; Zawieja, D.C.; Davis, M.J.; Muthuchamy, M. MicroRNA signature of inflamed lymphatic endothelium and role of miR-9 in lymphangiogenesis and inflammation. Am. J. Physiol. Cell Physiol. 2015, 309, C680–C692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan, C.I.; Tsai, M.H.; Chiu, C.F.; Huang, Y.P.; Liu, C.J.; Chang, N.W. Fenofibrate Suppresses Oral Tumorigenesis via Reprogramming Metabolic Processes: Potential Drug Repurposing for Oral Cancer. Int. J. Biol. Sci. 2016, 12, 786–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Chakraborty, S.; Meininger, C.J.; Muthuchamy, M. Insulin resistance disrupts cell integrity, mitochondrial function, and inflammatory signaling in lymphatic endothelium. Microcirculation 2018, 25, e12492. [Google Scholar] [CrossRef]
- Pathak, T.; Gueguinou, M.; Walter, V.; Delierneux, C.; Johnson, M.T.; Zhang, X.; Xin, P.; Yoast, R.E.; Emrich, S.M.; Yochum, G.S.; et al. Dichotomous role of the human mitochondrial Na+/Ca2+/Li+ exchanger NCLX in colorectal cancer growth and metastasis. eLife 2020, 9, e59686. [Google Scholar] [CrossRef] [PubMed]
- Barnoud, T.; Parris, J.L.D.; Murphy, M.E. Tumor cells containing the African-Centric S47 variant of TP53 show increased Warburg metabolism. Oncotarget 2019, 10, 1217–1223. [Google Scholar] [CrossRef] [Green Version]
- Ji, R.C. Lymphatic endothelial cells, tumor lymphangiogenesis and metastasis: New insights into intratumoral and peritumoral lymphatics. Cancer Metastasis. Rev. 2006, 25, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Gores, G.J. Molecular pathogenesis of cholangiocarcinoma. Dig. Dis. 2014, 32, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnsworth, R.H.; Karnezis, T.; Maciburko, S.J.; Mueller, S.N.; Stacker, S.A. The Interplay Between Lymphatic Vessels and Chemokines. Front. Immunol. 2019, 10, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trussoni, C.E.; Tabibian, J.H.; Splinter, P.L.; O’Hara, S.P. Lipopolysaccharide (LPS)-Induced Biliary Epithelial Cell NRas Activation Requires Epidermal Growth Factor Receptor (EGFR). PLoS ONE 2015, 10, e0125793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawieja, S.D.; Wang, W.; Chakraborty, S.; Zawieja, D.C.; Muthuchamy, M. Macrophage alterations within the mesenteric lymphatic tissue are associated with impairment of lymphatic pump in metabolic syndrome. Microcirculation 2016, 23, 558–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labib, P.L.; Goodchild, G.; Pereira, S.P. Molecular Pathogenesis of Cholangiocarcinoma. BMC Cancer 2019, 19, 185. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Chakraborty, S.; Zawieja, S.D.; Wang, W.; Lee, Y.; Wang, Y.J.; von der Weid, P.Y.; Zawieja, D.C.; Muthuchamy, M. Lipopolysaccharide modulates neutrophil recruitment and macrophage polarization on lymphatic vessels and impairs lymphatic function in rat mesentery. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H2042–H2057. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Liu, B.; Liu, Y.; Yu, X.; Cheng, G. Dual effects of active ERK in cancer: A potential target for enhancing radiosensitivity. Oncol. Lett. 2020, 20, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Deschenes-Simard, X.; Kottakis, F.; Meloche, S.; Ferbeyre, G. ERKs in cancer: Friends or foes? Cancer Res. 2014, 74, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Zhai, B.; Sun, W.; Hu, F.; Cheng, H.; Xu, J. Activation of phosphatidylinositol 3-kinase/AKT/snail signaling pathway contributes to epithelial-mesenchymal transition-induced multi-drug resistance to sorafenib in hepatocellular carcinoma cells. PLoS ONE 2017, 12, e0185088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallet, A.; Therond, P.P. Temporal modulation of the Hedgehog morphogen gradient by a patched-dependent targeting to lysosomal compartment. Dev. Biol. 2005, 277, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.H.; Wang, L.; Zhang, J.B.; Liu, Y.; Li, X.Q.; Guo, L.; Zhang, B.; Zhu, W.W.; Ye, Q.H. MST4 promotes hepatocellular carcinoma epithelial-mesenchymal transition and metastasis via activation of the p-ERK pathway. Int. J. Oncol. 2014, 45, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Zhang, R.; Singh, S.; Poddar, M.; Xu, E.; Oertel, M.; Chen, X.; Ganesh, S.; Abrams, M.; Monga, S.P. Targeting beta-catenin in hepatocellular cancers induced by coexpression of mutant beta-catenin and K-Ras in mice. Hepatology 2017, 65, 1581–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Zhu, S.; Cao, J.; Zhang, L.; Li, W.; Liu, K.; Zhong, J.; Shang, C.; Chen, Y. Inhibition of MMP-2 Expression Enhances the Antitumor Effect of Sorafenib in Hepatocellular Carcinoma by Suppressing the PI3K/AKT/mTOR Pathway. Oncol. Res. 2017, 25, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Aparna, M.; Rao, L.; Kunhikatta, V.; Radhakrishnan, R. The role of MMP-2 and MMP-9 as prognostic markers in the early stages of tongue squamous cell carcinoma. J. Oral. Pathol. Med. 2015, 44, 345–352. [Google Scholar] [CrossRef]
- Di Mitri, D.; Mirenda, M.; Vasilevska, J.; Calcinotto, A.; Delaleu, N.; Revandkar, A.; Gil, V.; Boysen, G.; Losa, M.; Mosole, S.; et al. Re-education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer. Cell Rep. 2019, 28, 2156–2168.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra67. [Google Scholar] [CrossRef]
- Katoh, H.; Wang, D.; Daikoku, T.; Sun, H.; Dey, S.K.; Dubois, R.N. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013, 24, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Richards, D.M.; Hettinger, J.; Feuerer, M. Monocytes and macrophages in cancer: Development and functions. Cancer Microenviron. 2013, 6, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, J.; Liu, Y.; Dai, N.; Hoffmann, C.; Hudock, K.M.; Zhang, P.; Guttentag, S.H.; Kolls, J.K.; Oliver, P.M.; Bushman, F.D.; et al. Cxcr2 and Cxcl5 regulate the IL-17/G-CSF axis and neutrophil homeostasis in mice. J. Clin. Investig. 2012, 122, 974–986. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subimerb, C.; Pinlaor, S.; Lulitanond, V.; Khuntikeo, N.; Okada, S.; McGrath, M.S.; Wongkham, S. Circulating CD14+ CD16+ monocyte levels predict tissue invasive character of cholangiocarcinoma. Clin. Exp. Immunol. 2010, 161, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; King, J.; Moro, A.; Sugi, M.D.; Dawson, D.W.; Kaplan, J.; Li, G.; Lu, X.; Strieter, R.M.; Burdick, M.; et al. Overexpression of CXCL5 is associated with poor survival in patients with pancreatic cancer. Am. J. Pathol. 2011, 178, 1340–1349. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Chung, J.; Burger, M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L1094–L1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [Green Version]
- San-Millan, I.; Julian, C.G.; Matarazzo, C.; Martinez, J.; Brooks, G.A. Is Lactate an Oncometabolite? Evidence Supporting a Role for Lactate in the Regulation of Transcriptional Activity of Cancer-Related Genes in MCF7 Breast Cancer Cells. Front. Oncol. 2019, 9, 1536. [Google Scholar] [CrossRef]
- Polet, F.; Feron, O. Endothelial cell metabolism and tumour angiogenesis: Glucose and glutamine as essential fuels and lactate as the driving force. J. Intern. Med. 2013, 273, 156–165. [Google Scholar] [CrossRef]
- Dhup, S.; Dadhich, R.K.; Porporato, P.E.; Sonveaux, P. Multiple biological activities of lactic acid in cancer: Influences on tumor growth, angiogenesis and metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Vegran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [Green Version]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Baumann, F.; Leukel, P.; Doerfelt, A.; Beier, C.P.; Dettmer, K.; Oefner, P.J.; Kastenberger, M.; Kreutz, M.; Nickl-Jockschat, T.; Bogdahn, U.; et al. Lactate promotes glioma migration by TGF-beta2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol. 2009, 11, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Li, X.B.; Gu, J.D.; Zhou, Q.H. Review of aerobic glycolysis and its key enzymes—New targets for lung cancer therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef]
- Moon, J.S.; Kim, H.E.; Koh, E.; Park, S.H.; Jin, W.J.; Park, B.W.; Park, S.W.; Kim, K.S. Kruppel-like factor 4 (KLF4) activates the transcription of the gene for the platelet isoform of phosphofructokinase (PFKP) in breast cancer. J. Biol. Chem. 2011, 286, 23808–23816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastos, D.C.; Paupert, J.; Maillard, C.; Seguin, F.; Carvalho, M.A.; Agostini, M.; Coletta, R.D.; Noel, A.; Graner, E. Effects of fatty acid synthase inhibitors on lymphatic vessels: An in vitro and in vivo study in a melanoma model. Lab. Investig. 2017, 97, 194–206. [Google Scholar] [CrossRef]
- DeWaal, D.; Nogueira, V.; Terry, A.R.; Patra, K.C.; Jeon, S.M.; Guzman, G.; Au, J.; Long, C.P.; Antoniewicz, M.R.; Hay, N. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018, 9, 446. [Google Scholar] [CrossRef]
- Ko, Y.H.; Verhoeven, H.A.; Lee, M.J.; Corbin, D.J.; Vogl, T.J.; Pedersen, P.L. A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: From bench side to bedside. J. Bioenerg. Biomembr. 2012, 44, 163–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwagi, S.; Hosono, K.; Suzuki, T.; Takeda, A.; Uchinuma, E.; Majima, M. Role of COX-2 in lymphangiogenesis and restoration of lymphatic flow in secondary lymphedema. Lab. Investig. 2011, 91, 1314–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, X.; Majumder, M.; Girish, G.V.; Mohindra, V.; Maruyama, T.; Lala, P.K. Targeting COX-2 and EP4 to control tumor growth, angiogenesis, lymphangiogenesis and metastasis to the lungs and lymph nodes in a breast cancer model. Lab. Investig. 2012, 92, 1115–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Yang, Y.; Xiao, J.; Lv, Y.; Liu, Y.; Yang, H.; Zhao, L. COX-2-mediated regulation of VEGF-C in association with lymphangiogenesis and lymph node metastasis in lung cancer. Anat. Rec. 2010, 293, 1838–1846. [Google Scholar] [CrossRef]
- Baracca, A.; Sgarbi, G.; Mattiazzi, M.; Casalena, G.; Pagnotta, E.; Valentino, M.L.; Moggio, M.; Lenaz, G.; Carelli, V.; Solaini, G. Biochemical phenotypes associated with the mitochondrial ATP6 gene mutations at nt8993. Biochim. Biophys. Acta 2007, 1767, 913–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaffarpour, M.; Mahdian, R.; Fereidooni, F.; Kamalidehghan, B.; Moazami, N.; Houshmand, M. The mitochondrial ATPase6 gene is more susceptible to mutation than the ATPase8 gene in breast cancer patients. Cancer Cell Int. 2014, 14, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Primers | Forward Sequence | Reverse Sequence |
| Gli | AGGGCTGCAGTAAAGCCTTCA | CTTGACATCTTTTCGCAGCG |
| PTCHD | TGAGACTGACCACGGCCTG | ACCCTCAGTTGGAGCTGCTTG |
| SNAI1 | CAGACCCACTCAGATGTCAA | CATAGTTAGTCACACCTCGT |
| SNAI2 | ACATTAGAACTCACACGGGGA | GTGTGCTACACAGCAGCCAGA |
| SHH | CCCAATTACAACCCCGACATC | TCACCCGCAGTTTCACTCCT |
| MMP1 | ACAGCCCAGTACTTATTCCCTTTG | GGGCTTGAAGCTGCTTACGA |
| MMP2 | GCTGGCTGCCTTAGAACCTTTC | GAACCATCACTATGTGGGCTGAGA |
| MMP7 | GGGACATTCCTCTGATCCTAATGC | GAATTACTTCTCTTTCCATATCGTTTCTGAATGC |
| MMP9 | GCACGACGTCTTCCAGTACC | GCACTGCAGGATGTCATAGGT |
| MMP11 | CAACATACCTCAATCCTGTCCC | CAATGGCTTTGGAGGATAGC |
| MMP12 | TTGAATATGACTTCCTACTCCAACG | GTGGTACACTGAGGACATAGCAAAT |
| MMP21 | AACAATAGGACACGCTATGG | CATCTCTTTTCCATGTCCAG |
| IL1β | AATCTGTACCTGTCCTGCGTGTT | TGGGTAATTTTTGGGATCTACACTCT |
| IL6 | GTAGCCGCCCCACACAGA | CATGTCTCCTTTCTCAGGGCTG |
| IL8 | GTGCAGTTTTGCCAAGGAGT | TTATGAATTCTCAGCCCTCTTCAAAAACTTCTC |
| MCP1 | ACTCTCGCCTCCAGCATGAA | TTGATTGCATCTGGCTGAGC |
| CCL1 | GGAAGATGTGGACAGCAAGAGC | TGTATGGCTGTTAGTTTCGG |
| CXCL5 | TGGACGGTGGAAACAAGG | TGGACGGTGGAAACAAGG |
| CXCR2 | CACCGATGTCTACCTGCTGA | CACAGGGTTGAGCCAAAAGT |
| β-Catenin | AAAATGGCAGTGCGTTTAG | TTTGAAGGCAGTCTGTCGTA |
| PD-L1 | ACAGCTGAATTGGTCATCCCA | CACATCCATCATTCTCCCTTTTC |
| PD1 | CGTGGCCTATCCACTCCTCA | ATCCCTTGTCCCAGCCACTC |
| VCL | TCAGATGAGGTGACTCGGTTGG | GGGTGCTTATGGTTGGGATTCG |
| ZYX | GCAGAATGTGGCTGTCAACGAAC | TGAAGCAGGCGATGTGGAACAG |
| TLN1 | TTGGAGATGCCAGCAAGCGACT | CCAGTTCTGTGGCTGCCTGATT |
| CDH8 | CTACTGAAATTAGGAACCACAGTCAGAT | CTAACAGTTTGAATGACTTGGCCG |
| PFKP | CGGAAGTTCCTGGAGCACCTCTC | AAGTACACCTTGGCCCCCACGTA |
| GLUT3 | ACTTTGACGGACAAGGGAAATG | ACCAGTGACAGCCAACAGG |
| FASN | CGCGTGGCCGGCTACTCCTAC | CGGCTGCCACACGCTCCTCT |
| HK2 | GAGCCACCACTCACCCTACT | CCAGGCATTCGGCAATGTG |
| CO I | CTCTTGCGGTACTCATTGAAG | GAGCTGCTGTTCGGTGTC |
| ATP6 | GAAGCGCCACCCTAGCAATA | GCTTGGATTAAGGCGACAGC |
| AMPKα1 | TGCGTGTACGAAGGAAGAATCC | TGTGACTTCCAGGTCTTGGAGTT |
| COX2 | GAATGGGGTGATGAGCAGTT | CAGAAGGGCAGGATACAGC |
| RPL19 | GGGCATAGGTAAGCGGAAGG | TCAGGTACAGGCTGTGATACA |
| Ubiquitin | AGTCCCTTCTCGGCGATTCT | GCATTGTCAAGTGACGATCACAGC |
| Mouse Primers | Forward Sequence | Reverse Sequence |
| E-Selectin | GGACACCACAAATCCCAGTCTG | TCGCAGGAGAACTCACAACTGG |
| Lamin | GAGCAAAGTGCGTGAGGAGTTC | CCTTGGAGTTGAGAAGAGCCTC |
| α-SMA | TGCTGACAGAGGCACCACTGAA | CAGTTGTACGTCCAGAGGCATAG |
| Fibronectin | CCCTATCTCTGATACCGTTGTCC | TGCCGCAACTACTGTGATTCGG |
| E-Cadh | GGTCATCAGTGTGCTCACCTCT | GCTGTTGTGCTCAAGCCTTCAC |
| N-Cadh | CCTCCAGAGTTTACTGCCATGAC | CCACCACTGATTCTGTATGCCG |
| Zeb1 | ATTCAGCTACTGTGAGCCCTGC | CATTCTGGTCCTCCACAGTGGA |
| Zeb2 | GCAGTGAGCATCGAAGAGTACC | GGCAAAAGCATCTGGAGTTCCAG |
| Twist1 | AGCGGGTCATGGCTAACG | AGCGGGTCATGGCTAACG |
| Gli | CCCATAGGGTCTCGGGGTCTCAAAC | GGAGGACCTGCGGCTGACTGTGTAA |
| Ptchd | TTGAATATGACTTCCTACTCCAACG | GTGGTACACTGAGGACATAGCAAAT |
| Cxcr2 | TGTCTGCTCCCTTCCATCTT | CCATTTCCTCTCCTCCAGCT |
| Pfkp | GGAAGCCAAATGGGACTGT | CGCACTACCGATGATGGTC |
| Fasn | CTGCGTGGCTATGATTATGG | AGGTTGCTGTCGTCTGTAGT |
| Hk2 | TGGGTTTCACCTTCTCGTTC | TTCACCAGGATGAGTCTGAC |
| Atp6 | TCCCAATCGTTGTAGCCATCA | AGACGGTTGTTGATTAGGCGT |
| Co I | ATCACTACCAGTGCTAGCCG | CCTCCAGCGGGATCAAAGAA |
| Rpl19 | ATGAGTATGCTCAGGCTACAGA | GCATTGGCCGATTTCATTGGTC |
| Ubiquitin | GCCCAGTGTTACCACCAAGAAG | GCTCTTTTTAGATACTGTGGTGAGGAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, S.; Kumaravel, S.; Banerjee, P.; White, T.K.; O’Brien, A.; Seelig, C.; Chauhan, R.; Ekser, B.; Bayless, K.J.; Alpini, G.; et al. Tumor Lymphatic Interactions Induce CXCR2-CXCL5 Axis and Alter Cellular Metabolism and Lymphangiogenic Pathways to Promote Cholangiocarcinoma. Cells 2021, 10, 3093. https://doi.org/10.3390/cells10113093
Roy S, Kumaravel S, Banerjee P, White TK, O’Brien A, Seelig C, Chauhan R, Ekser B, Bayless KJ, Alpini G, et al. Tumor Lymphatic Interactions Induce CXCR2-CXCL5 Axis and Alter Cellular Metabolism and Lymphangiogenic Pathways to Promote Cholangiocarcinoma. Cells. 2021; 10(11):3093. https://doi.org/10.3390/cells10113093
Chicago/Turabian StyleRoy, Sukanya, Subhashree Kumaravel, Priyanka Banerjee, Tori K. White, April O’Brien, Catherine Seelig, Rahul Chauhan, Burcin Ekser, Kayla J. Bayless, Gianfranco Alpini, and et al. 2021. "Tumor Lymphatic Interactions Induce CXCR2-CXCL5 Axis and Alter Cellular Metabolism and Lymphangiogenic Pathways to Promote Cholangiocarcinoma" Cells 10, no. 11: 3093. https://doi.org/10.3390/cells10113093