
Measuring the Metabolic Evolution of Glioblastoma throughout Tumor Development, Regression, and Recurrence with Hyperpolarized Magnetic Resonance

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Resource Availability
2.1.1. Materials Availability
2.1.2. Data and Code Availability
2.2. Experimental Model and Subject Details
2.2.1. Cell Lines
2.2.2. Animals
2.3. Method Details
2.3.1. Experimental Overview
2.3.2. Tumor Radiotherapy
2.3.3. Anatomic Magnetic Resonance Imaging
2.3.4. Tumor Volume Measurements
2.3.5. Hyperpolarized Sample Preparation
2.3.6. 13C Magnetic Resonance Spectroscopy
2.3.7. Pyruvate-to-Lactate Measurements
2.3.8. Brain Sample Excision
2.3.9. Sample Preparation for Metabolite Extraction
2.3.10. Nuclear Magnetic Resonance Spectroscopy
2.3.11. Metabolite Pool Size Measurements
2.3.12. Sample Preparation for Immunohistochemistry
2.3.13. Immunohistochemistry
2.3.14. Protein Expression Measurements
2.4. Quantification and Statistical Analysis
2.4.1. Median Survival Time
2.4.2. In Vivo Tumor Volume Measurements
2.4.3. In Vivo Pyruvate-to-Lactate Measurements
2.4.4. Ex Vivo Metabolite Pool Size Measurements
2.4.5. Ex Vivo Protein Expression Measurements
3. Results
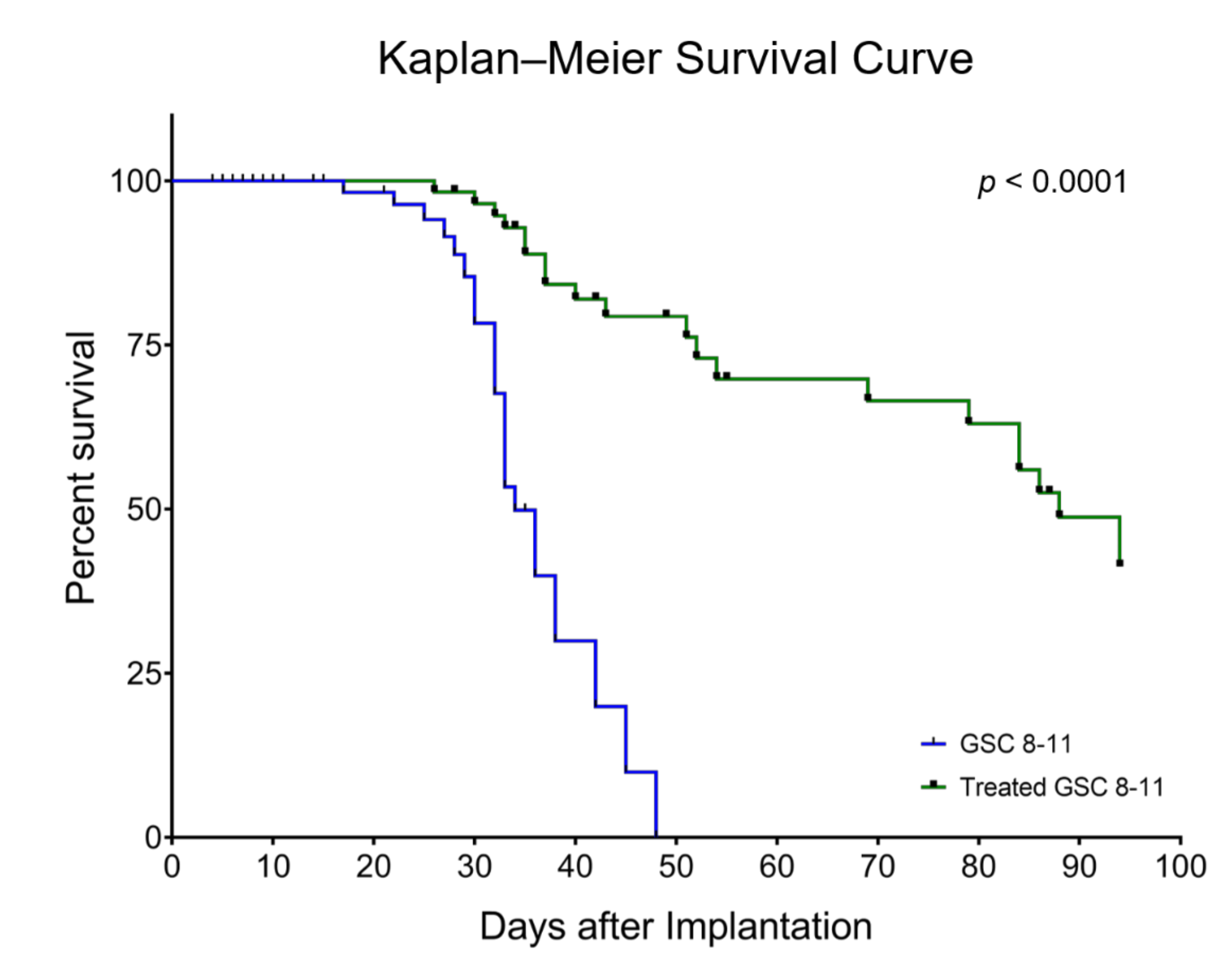
3.1. Radiotherapy Significantly Extends Survival of GSC 8-11 Tumor-Bearing Mice
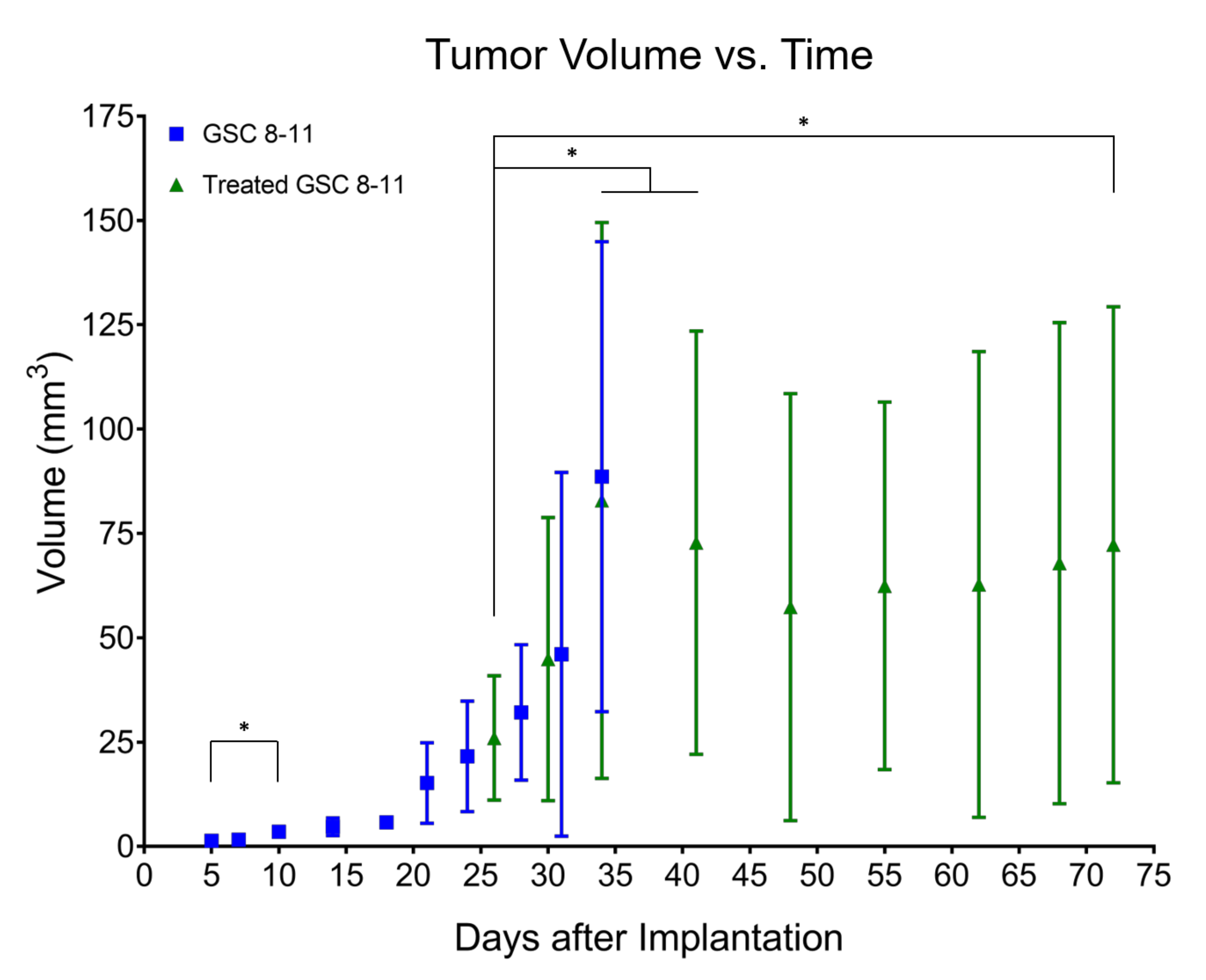
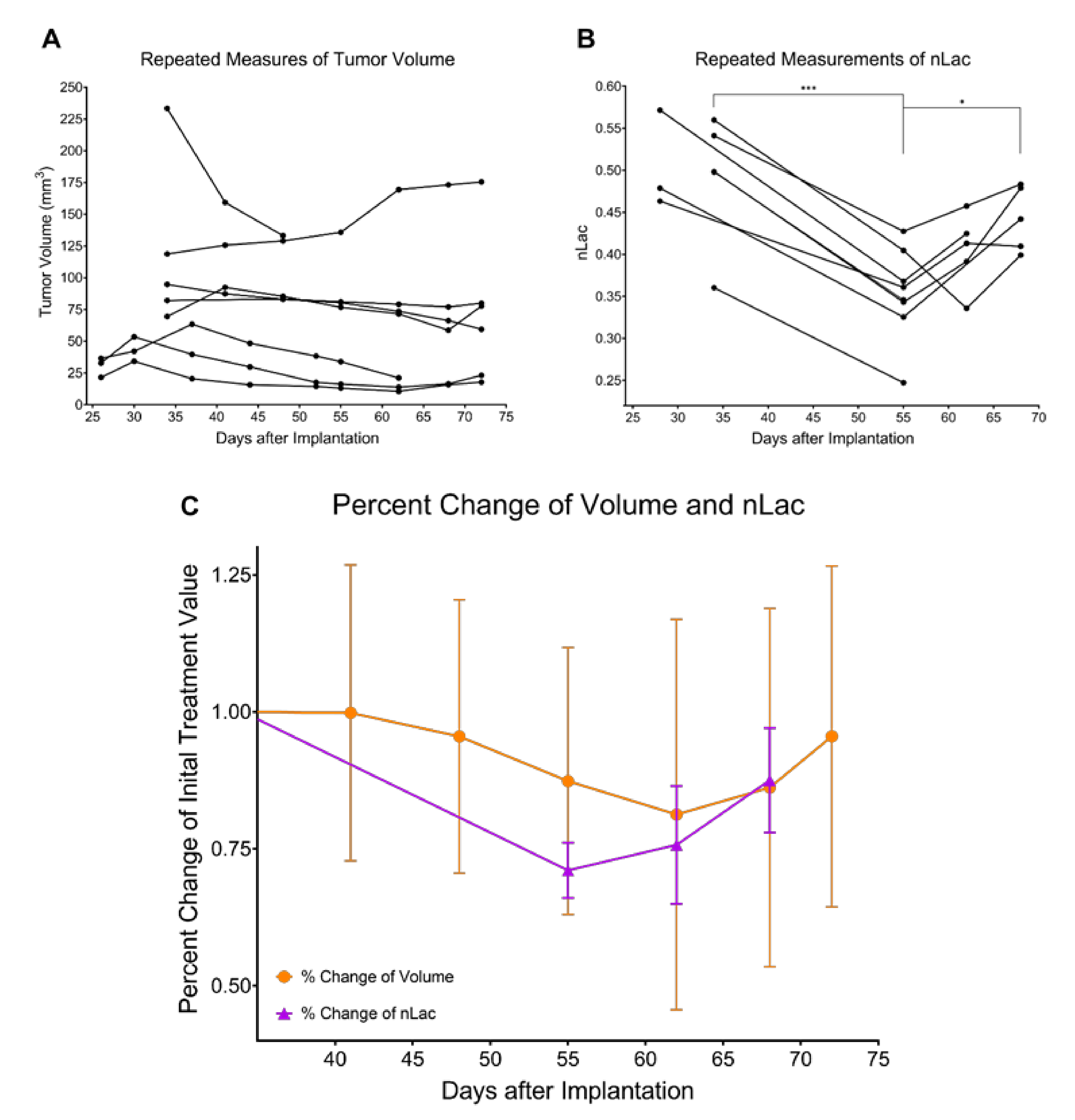
3.2. Tumor Volume Increases during Development but Does Not Significantly Change throughout Regression or Recurrence
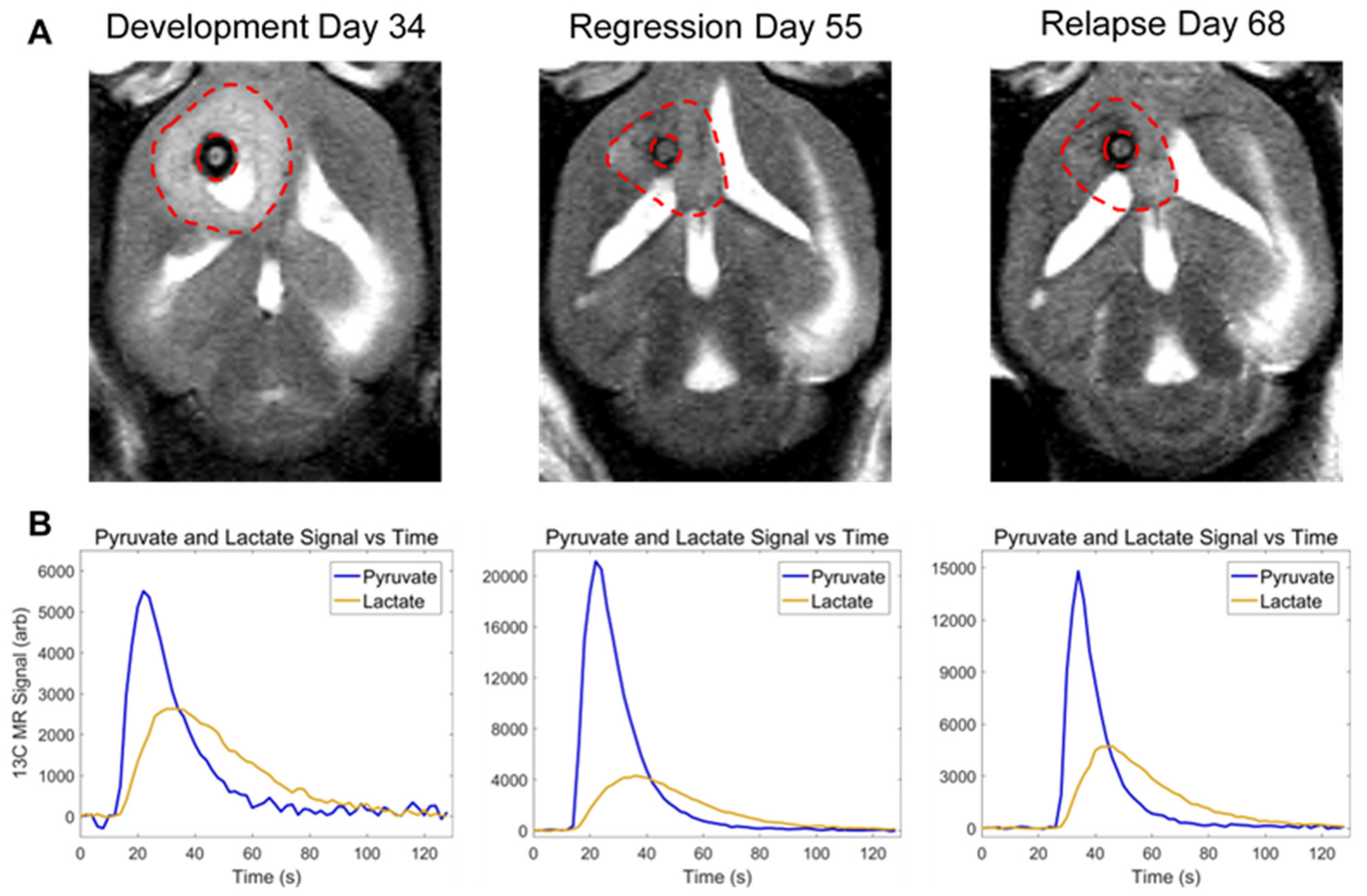
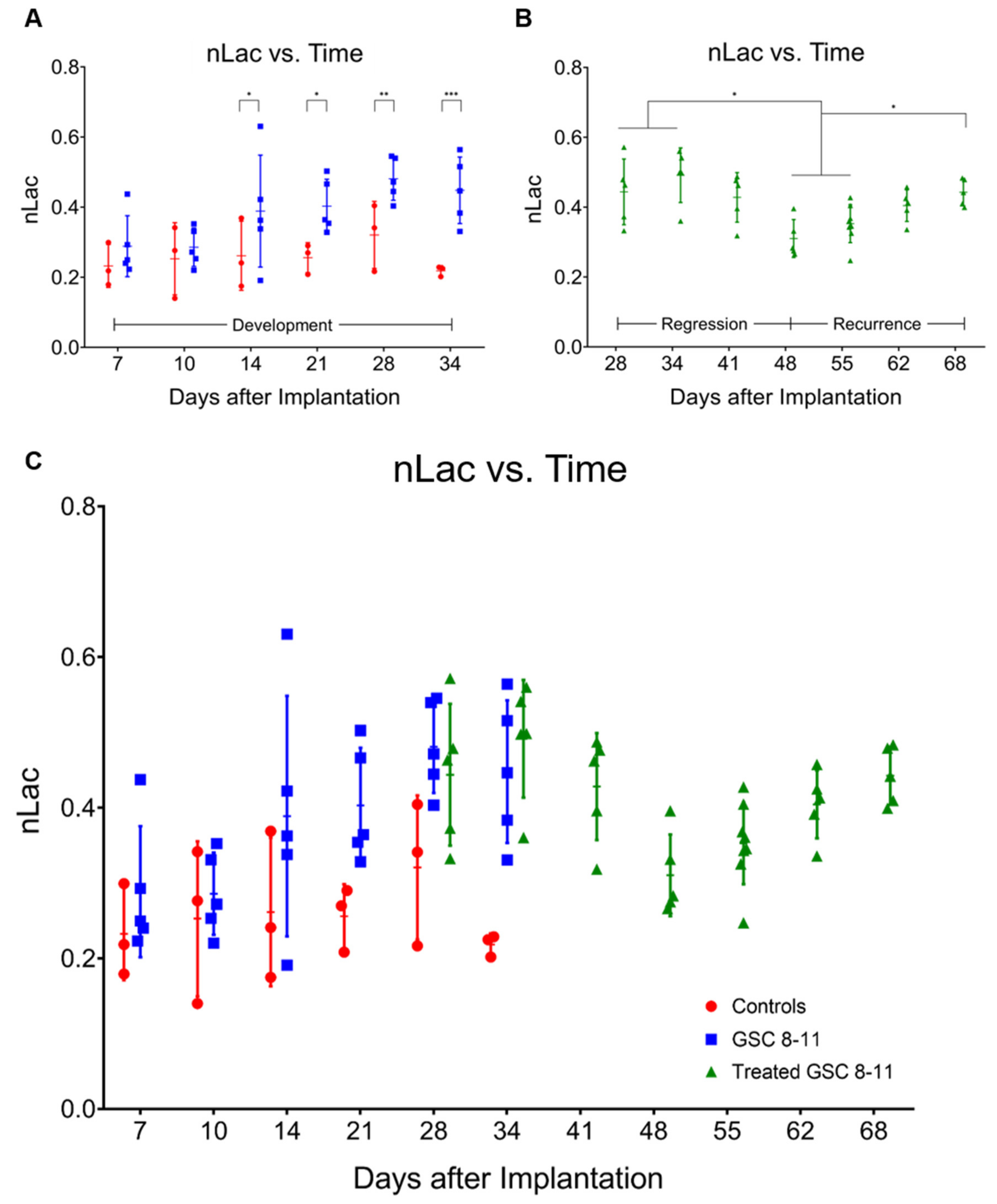
3.3. In Vivo Pyruvate-to-Lactate Conversion Is Significantly Altered throughout Tumor Development, Regression, and Recurrence
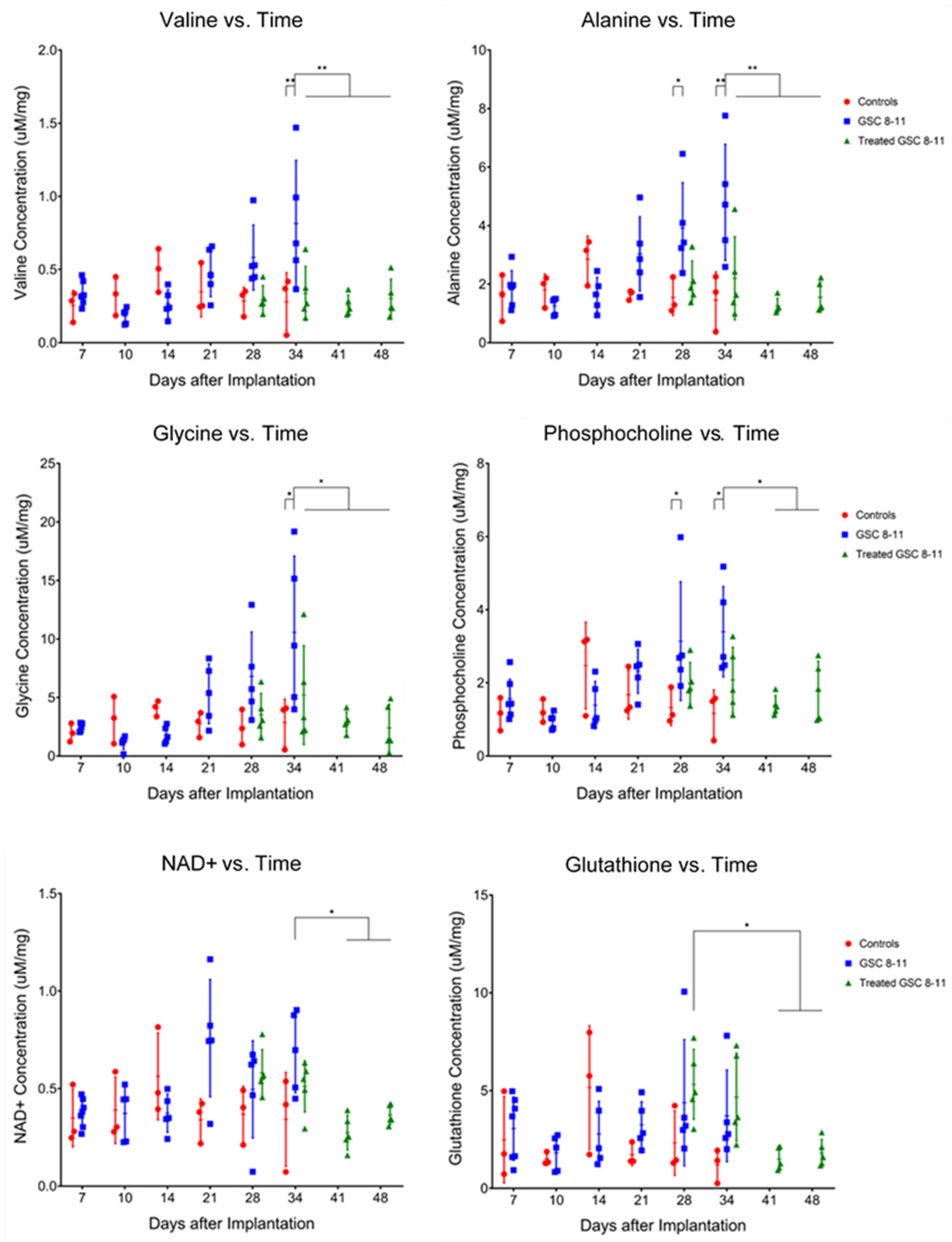
3.4. Ex Vivo Metabolite Pool Sizes Are Significantly Altered throughout Tumor Development and Regression
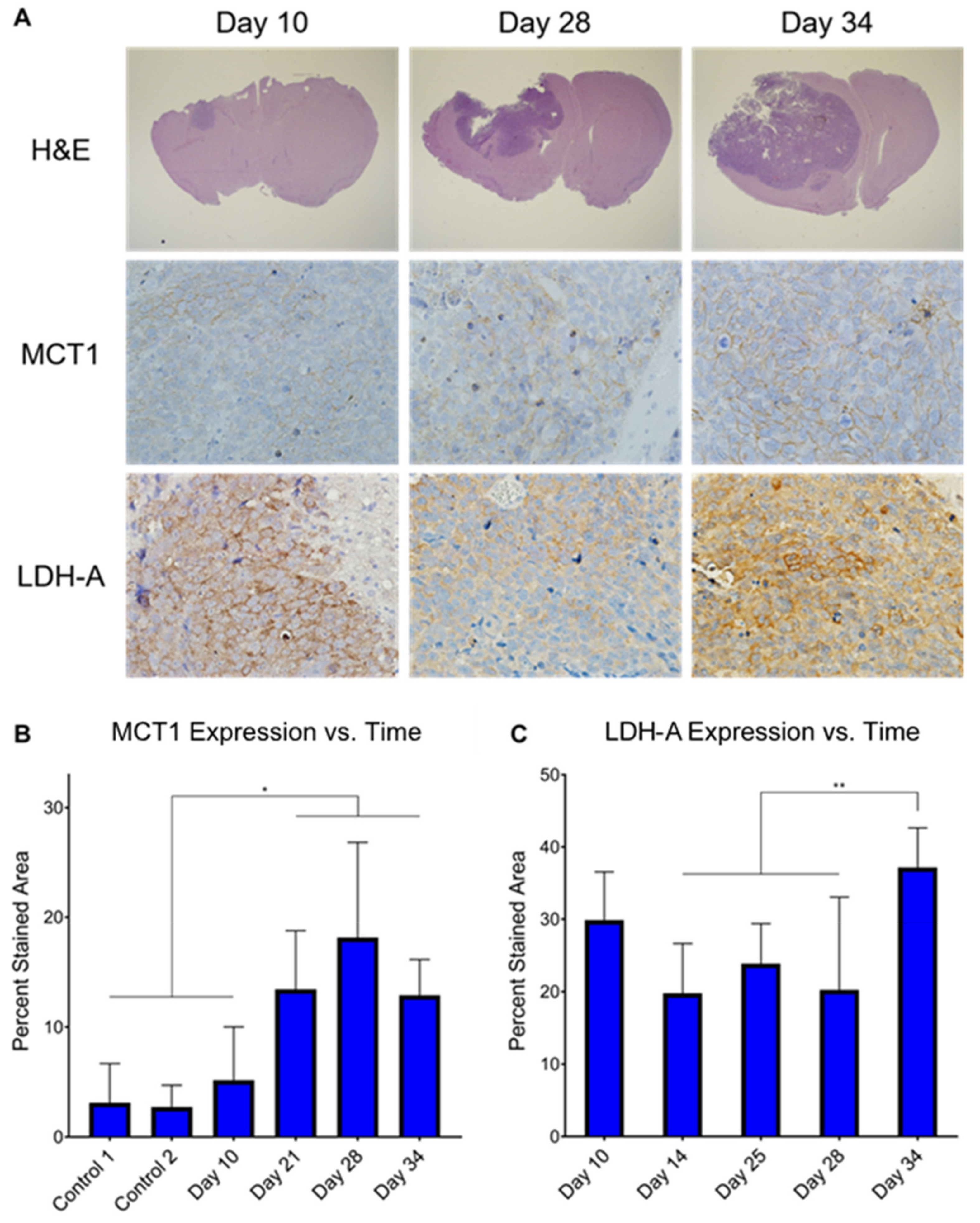
3.5. Ex Vivo MCT1 Expression Significantly Increases throughout Tumor Development
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; De Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro Oncol. 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzillo, T.; Hu, J.; Nguyen, L.; Whiting, N.; Lee, J.; Weygand, J.; Dutta, P.; Pudakalakatti, S.; Millward, N.Z.; Gammon, S.; et al. Interrogating Metabolism in Brain Cancer. Magn. Reson. Imaging Clin. N. Am. 2016, 24, 687–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Salzillo, T.; Sailasuta, N.; Lang, F.F.; Bhattacharya, P. Interrogating IDH Mutation in Brain Tumor. Top. Magn. Reson. Imaging 2017, 26, 27–32. [Google Scholar] [CrossRef]
- Kurhanewicz, J.; Vigneron, D.B.; Ardenkjaer-Larsen, J.H.; Bankson, J.A.; Brindle, K.; Cunningham, C.; Gallagher, F.A.; Keshari, K.R.; Kjaer, A.; Laustsen, C.; et al. Hyperpolarized 13C MRI: Path to Clinical Translation in Oncology. Neoplasia 2019, 21, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Salzillo, T.C.; Pudakalakatti, S.; Gammon, S.T.; Kaipparettu, B.A.; McAllister, F.; Wagner, S.; Frigo, D.E.; Logothetis, C.J.; Zacharias, N.M.; et al. Assessing Therapeutic Efficacy in Real-time by Hyperpolarized Magnetic Resonance Metabolic Imaging. Cells 2019, 8, 340. [Google Scholar] [CrossRef] [Green Version]
- Brindle, K. Watching tumours gasp and die with MRI: The promise of hyperpolarised13C MR spectroscopic imaging. Br. J. Radiol. 2012, 85, 697–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Lee, C.Y.; Soliman, H.; Geraghty, B.J.; Chen, A.P.; Connelly, K.A.; Endre, R.; Perks, W.J.; Heyn, C.; Black, S.E.; Cunningham, C.H. Lactate topography of the human brain using hyperpolarized 13C-MRI. NeuroImage 2020, 204, 116202. [Google Scholar] [CrossRef] [PubMed]
- Grist, J.; McLean, M.A.; Riemer, F.; Schulte, R.F.; Deen, S.S.; Zaccagna, F.; Woitek, R.; Daniels, C.J.; Kaggie, J.; Matys, T.; et al. Quantifying normal human brain metabolism using hyperpolarized [1–13C]pyruvate and magnetic resonance imaging. NeuroImage 2019, 189, 171–179. [Google Scholar] [CrossRef]
- Chung, B.T.; Chen, H.-Y.; Gordon, J.; Mammoli, D.; Sriram, R.; Autry, A.W.; Le Page, L.M.; Chaumeil, M.; Shin, P.; Slater, J.; et al. First hyperpolarized [2-13C]pyruvate MR studies of human brain metabolism. J. Magn. Reson. 2019, 309, 106617. [Google Scholar] [CrossRef] [PubMed]
- Miloushev, V.Z.; Granlund, K.L.; Boltyanskiy, R.; Lyashchenko, S.K.; DeAngelis, L.M.; Mellinghoff, I.K.; Brennan, C.; Tabar, V.; Yang, T.J.; Holodny, A.; et al. Metabolic Imaging of the Human Brain with Hyperpolarized 13C Pyruvate Demonstrates 13C Lactate Production in Brain Tumor Patients. Cancer Res. 2018, 78, 3755–3760. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.W.; Chen, H.-Y.; Autry, A.; Park, I.; Van Criekinge, M.; Mammoli, D.; Milshteyn, E.; Bok, R.; Xu, D.; Li, Y.; et al. Translation of Carbon-13 EPI for hyperpolarized MR molecular imaging of prostate and brain cancer patients. Magn. Reson. Med. 2019, 81, 2702–2709. [Google Scholar] [CrossRef]
- Park, I.; Larson, P.E.; Gordon, J.W.; Carvajal, L.; Chen, H.-Y.; Bok, R.; Van Criekinge, M.; Ferrone, M.; Slater, J.B.; Xu, D.; et al. Development of methods and feasibility of using hyperpolarized carbon-13 imaging data for evaluating brain metabolism in patient studies. Magn. Reson. Med. 2018, 80, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Mammoli, D.; Gordon, J.; Autry, A.; Larson, P.E.Z.; Li, Y.; Chen, H.-Y.; Chung, B.; Shin, P.; Van Criekinge, M.; Carvajal, L.; et al. Kinetic Modeling of Hyperpolarized Carbon-13 Pyruvate Metabolism in the Human Brain. IEEE Trans. Med Imaging 2020, 39, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.C.; Gordon, J.W.; Chen, H.; Autry, A.W.; Li, Y.; Olson, M.P.; Kurhanewicz, J.; Vigneron, D.B.; Larson, P.E.; Xu, D. Hyperpolarized 13 C MRI data acquisition and analysis in prostate and brain at University of California, San Francisco. NMR Biomed. 2021, 34, e4280. [Google Scholar] [CrossRef]
- Chaumeil, M.M.; Radoul, M.; Najac, C.; Eriksson, P.; Viswanath, P.; Blough, M.D.; Chesnelong, C.; Luchman, H.A.; Cairncross, J.G.; Ronen, S.M. Hyperpolarized 13 C MR imaging detects no lactate production in mutant IDH1 gliomas: Implications for diagnosis and response monitoring. NeuroImage: Clin. 2016, 12, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.M.; Josan, S.; Jang, T.; Merchant, M.; Yen, Y.-F.; Hurd, R.E.; Recht, L.; Spielman, D.M.; Mayer, D. Metabolite kinetics in C6 rat glioma model using magnetic resonance spectroscopic imaging of hyperpolarized [1-13C]pyruvate. Magn. Reson. Med. 2012, 68, 1886–1893. [Google Scholar] [CrossRef] [Green Version]
- Park, J.M.; Josan, S.; Jang, T.; Merchant, M.; Watkins, R.; Hurd, R.E.; Recht, L.D.; Mayer, D.; Spielman, D.M. Volumetric spiral chemical shift imaging of hyperpolarized [2- 13 c]pyruvate in a rat c6 glioma model. Magn. Reson. Med. 2016, 75, 973–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autry, A.W.; Hashizume, R.; James, C.D.; Larson, P.E.Z.; Vigneron, D.B.; Park, I. Measuring Tumor Metabolism in Pediatric Diffuse Intrinsic Pontine Glioma Using Hyperpolarized Carbon-13 MR Metabolic Imaging. Contrast Media Mol. Imaging 2018, 2018, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Mair, R.; Wright, A.J.; Ros, S.; Hu, D.-E.; Booth, T.; Kreis, F.; Rao, J.; Watts, C.; Brindle, K.M.; Booth, T.C. Metabolic Imaging Detects Low Levels of Glycolytic Activity That Vary with Levels of c-Myc Expression in Patient-Derived Xenograft Models of Glioblastoma. Cancer Res. 2018, 78, 5408–5418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, S.E.; Kettunen, M.; Cherukuri, M.K.; Mitchell, J.B.; Lizak, M.J.; Morris, H.D.; Matsumoto, S.; Koretsky, A.; Brindle, K.M. Detecting response of rat C6 glioma tumors to radiotherapy using hyperpolarized [1-13 C]pyruvate and 13 C magnetic resonance spectroscopic imaging. Magn. Reson. Med. 2011, 65, 557–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.; Bok, R.; Ozawa, T.; Phillips, J.J.; James, C.D.; Vigneron, D.B.; Ronen, S.M.; Nelson, S.J. Detection of early response to temozolomide treatment in brain tumors using hyperpolarized 13C MR metabolic imaging. J. Magn. Reson. Imaging 2011, 33, 1284–1290. [Google Scholar] [CrossRef] [Green Version]
- Datta, K.; Lauritzen, M.H.; Merchant, M.; Jang, T.; Liu, S.-C.; Hurd, R.; Recht, L.; Spielman, D.M. Reversed metabolic reprogramming as a measure of cancer treatment efficacy in rat C6 glioma model. PLoS ONE 2019, 14, e0225313. [Google Scholar] [CrossRef] [Green Version]
- Michel, K.A.; Zieliński, R.; Walker, C.M.; Le Roux, L.; Priebe, W.; Bankson, J.A.; Schellingerhout, D. Hyperpolarized Pyruvate MR Spectroscopy Depicts Glycolytic Inhibition in a Mouse Model of Glioma. Radiol. 2019, 293, 168–173. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Kiebish, M.A.; Marsh, J.; Shelton, L.M.; Huysentruyt, L.C.; Mukherjee, P. Metabolic management of brain cancer. Biochim. Biophys. Acta (BBA)—Bioenerg. 2011, 1807, 577–594. [Google Scholar] [CrossRef] [Green Version]
- Libby, C.; Tran, A.; Scott, S.E.; Griguer, C.; Hjelmeland, A.B. The pro-tumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells. Biochim. Biophys. Acta (BBA)—Bioenerg. 2018, 1869, 175–188. [Google Scholar] [CrossRef]
- Robertson-Tessi, M.; Gillies, R.J.; Gatenby, R.A.; Anderson, A. Impact of Metabolic Heterogeneity on Tumor Growth, Invasion, and Treatment Outcomes. Cancer Res. 2015, 75, 1567–1579. [Google Scholar] [CrossRef] [Green Version]
- Van Linde, M.E.; Brahm, C.G.; Hamer, P.D.W.; Reijneveld, J.C.; Bruynzeel, A.M.E.; Vandertop, W.P.; Van De Ven, P.M.; Wagemakers, M.; Van Der Weide, H.L.; Enting, R.H.; et al. Treatment outcome of patients with recurrent glioblastoma multiforme: A retrospective multicenter analysis. J. Neuro-Oncol. 2017, 135, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.-H.; Wang, Z.-F.; Pan, Z.-Y.; Péus, D.; Fernandez, J.D.; Pallud, J.; Li, Z.-Q. A Meta-Analysis of Survival Outcomes Following Reoperation in Recurrent Glioblastoma: Time to Consider the Timing of Reoperation. Front. Neurol. 2019, 10, 286. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, S.; Singh, S.; Alamsahebpour, A.; Burrell, K.; Li, M.; Karabork, M.; Ekinci, C.; Koch, E.; Solaroglu, I.; Chang, J.T.; et al. DICER governs characteristics of glioma stem cells and the resulting tumors in xenograft mouse models of glioblastoma. Oncotarget 2016, 7, 56431–56446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Barr, J.; Kong, L.-Y.; Wang, Y.; Wu, A.; Sharma, A.K.; Gumin, J.; Henry, V.; Colman, H.; Priebe, W.; et al. Glioblastoma Cancer-Initiating Cells Inhibit T-Cell Proliferation and Effector Responses by the Signal Transducers and Activators of Transcription 3 Pathway. Mol. Cancer Ther. 2010, 9, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Lal, S.; Lacroix, M.; Tofilon, P.; Fuller, G.; Sawaya, R.; Lang, F.F. An implantable guide-screw system for brain tumor studies in small animals. J. Neurosurg. 2000, 92, 326–333. [Google Scholar] [CrossRef]
- Fedorov, A.; Beichel, R.; Kalpathy-Cramer, J.; Finet, J.; Fillion-Robin, J.-C.; Pujol, S.; Bauer, C.; Jennings, D.; Fennessy, F.; Sonka, M.; et al. 3D Slicer as an image computing platform for the Quantitative Imaging Network. Magn. Reson. Imaging 2012, 30, 1323–1341. [Google Scholar] [CrossRef] [Green Version]
- Weygand, J.; Carter, S.E.; Salzillo, T.C.; Moussalli, M.; Dai, B.; Dutta, P.; Zuo, X.; Fleming, J.B.; Shureiqi, I.; Bhattacharya, P. Can an Organoid Recapitulate the Metabolome of its Parent Tissue? A Pilot NMR Spectroscopy Study. J. Cancer Prev. Curr. Res. 2017, 8, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Zacharias, N.M.; McCullough, C.; Shanmugavelandy, S.; Lee, J.; Lee, Y.; Dutta, P.; McHenry, J.; Nguyen, L.; Norton, W.; Jones, L.W.; et al. Metabolic Differences in Glutamine Utilization Lead to Metabolic Vulnerabilities in Prostate Cancer. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruifrok, A.C.; Johnston, D.A. Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol. 2001, 23, 291–299. [Google Scholar]
- Shin, P.J.; Zhu, Z.; Camarda, R.; Bok, R.A.; Zhou, A.; Kurhanewicz, J.; Goga, A.; Vigneron, D.B. Cancer recurrence monitoring using hyperpolarized [1-13C]pyruvate metabolic imaging in murine breast cancer model. Magn. Reson. Imaging 2017, 43, 105–109. [Google Scholar] [CrossRef]
- Taal, W.; Brandsma, D.; De Bruin, H.G.; Bromberg, J.E.C.; Swaak-Kragten, A.T.; Smitt, P.A.E.S.; Van Es, C.A.; Van Den Bent, M. Incidence of early pseudo-progression in a cohort of malignant glioma patients treated with chemoirradiation with temozolomide. Cancer 2008, 113, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Perez, M.R.; Lee, J.; Kang, Y.; Pratt, M.; Salzillo, T.; Weygand, J.; Zacharias, N.M.; Gammon, S.; Koay, E.J.; et al. Combining Hyperpolarized Real-Time Metabolic Imaging and NMR Spectroscopy To Identify Metabolic Biomarkers in Pancreatic Cancer. J. Proteome Res. 2019, 18, 2826–2834. [Google Scholar] [CrossRef]
- Rao, Y.; Gammon, S.; Zacharias, N.M.; Liu, T.; Salzillo, T.; Xi, Y.; Wang, J.; Bhattacharya, P.; Piwnica-Worms, D. Hyperpolarized [1-13C]pyruvate-to-[1-13C]lactate conversion is rate-limited by monocarboxylate transporter-1 in the plasma membrane. Proc. Natl. Acad. Sci. USA 2020, 117, 22378–22389. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, F.A.; Woitek, R.; McLean, M.A.; Gill, A.B.; Garcia, R.M.; Provenzano, E.; Riemer, F.; Kaggie, J.; Chhabra, A.; Ursprung, S.; et al. Imaging breast cancer using hyperpolarized carbon-13 MRI. Proc. Natl. Acad. Sci. USA 2020, 117, 2092–2098. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Ulasov, I.; Chen, L.; Harkins, L.E.; Wallenborg, K.; Hothi, P.; Rostad, S.; Hood, L.; Cobbs, C.S. TGFβ-Responsive HMOX1 Expression Is Associated with Stemness and Invasion in Glioblastoma Multiforme. Stem Cells 2016, 34, 2276–2289. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Smith, C.P.; Wilbur, R.R.; Cain, C.P.; Kallu, S.R.; Valasapalli, S.; Sahoo, A.; Guda, M.R.; Tsung, A.J.; Velpula, K.K. An overview of MCT1 and MCT4 in GBM: Small molecule transporters with large implications. Am. J. Cancer Res. 2018, 8, 1967–1976. [Google Scholar]
- Viswanath, P.; Najac, C.; Izquierdo-Garcia, J.; Pankov, A.; Hong, C.; Eriksson, P.; Costello, J.F.; Pieper, R.O.; Ronen, S.M. Mutant IDH1 expression is associated with down-regulation of monocarboxylate transporters. Oncotarget 2016, 7, 34942–34955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1andIDH2Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Froberg, M.K.; Gerhart, D.Z.; Enerson, B.E.; Manivel, C.; Guzman-Paz, M.; Seacotte, N.; Drewes, L.R. Expression of monocarboxylate transporter MCT1 in normal and neoplastic human CNS tissues. NeuroReport 2001, 12, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Gonçalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (MCTs) in gliomas: Expression and exploitation as therapeutic targets. Neuro-Oncol. 2012, 15, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Colen, C.B.; Shen, Y.; Ghoddoussi, F.; Yu, P.; Francis, T.B.; Koch, B.J.; Monterey, M.D.; Galloway, M.; Sloan, A.E.; Mathupala, S.P. Metabolic Targeting of Lactate Efflux by Malignant Glioma Inhibits Invasiveness and Induces Necrosis: An In Vivo Study. Neoplasia 2011, 13, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Granlund, K.L.; Tee, S.-S.; Vargas, H.A.; Lyashchenko, S.K.; Reznik, E.; Fine, S.; Laudone, V.; Eastham, J.A.; Touijer, K.A.; Reuter, V.E.; et al. Hyperpolarized MRI of Human Prostate Cancer Reveals Increased Lactate with Tumor Grade Driven by Monocarboxylate Transporter 1. Cell Metab. 2020, 31, 105–114.e3. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.; De Micheli, E.; Bricolo, A.; Ballini, C.; Fattori, M.; Venturi, C.; Pedata, F.; Tipton, K.F.; Della Corte, L. Extracellular Levels of Amino Acids and Choline in Human High Grade Gliomas: An Intraoperative Microdialysis Study. Neurochem. Res. 2004, 29, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Ananieva, E.A.; Wilkinson, A.C. Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Tönjes, M.; Barbus, S.; Park, Y.J.; Wang, W.; Schlotter, M.; Lindroth, A.M.; Pleier, S.V.; Bai, A.H.C.; Karra, D.; Piro, R.M.; et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat. Med. 2013, 19, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Suh, E.H.; Hackett, E.P.; Wynn, R.M.; Chuang, D.T.; Zhang, B.; Luo, W.; Sherry, A.D.; Park, J.M. In vivo assessment of increased oxidation of branched-chain amino acids in glioblastoma. Sci. Rep. 2019, 9, 340. [Google Scholar] [CrossRef] [Green Version]
- Serrao, E.M.; Kettunen, M.I.; Rodrigues, T.B.; Dzien, P.; Wright, A.J.; Gopinathan, A.; Gallagher, F.A.; Lewis, D.Y.; Frese, K.K.; Almeida, J.; et al. MRI with hyperpolarised [1-13C]pyruvate detects advanced pancreatic preneoplasia prior to invasive disease in a mouse model. Gut 2015, 65, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Dutta, P.; Pando, S.C.; Mascaro, M.; Riquelme, E.; Zoltan, M.; Zacharias, N.M.; Gammon, S.T.; Piwnica-Worms, D.; Pagel, M.D.; Sen, S.; et al. Early Detection of Pancreatic Intraepithelial Neoplasias (PanINs) in Transgenic Mouse Model by Hyperpolarized 13C Metabolic Magnetic Resonance Spectroscopy. Int. J. Mol. Sci. 2020, 21, 3722. [Google Scholar] [CrossRef]
- Jalbert, L.E.; Elkhaled, A.; Phillips, J.J.; Neill, E.; Williams, A.; Crane, J.C.; Olson, M.P.; Molinaro, A.M.; Berger, M.S.; Kurhanewicz, J.; et al. Metabolic Profiling of IDH Mutation and Malignant Progression in Infiltrating Glioma. Sci. Rep. 2017, 7, 44792. [Google Scholar] [CrossRef] [Green Version]
- Coloff, J.L.; Murphy, J.P.; Braun, C.R.; Harris, I.; Shelton, L.M.; Kami, K.; Gygi, S.P.; Selfors, L.M.; Brugge, J.S. Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell Metab. 2016, 23, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Lin, S.-H.; Wang, Y.; Chin, Y.E.; Kang, L.; Mi, J. Glutamic Pyruvate Transaminase GPT2 Promotes Tumorigenesis of Breast Cancer Cells by Activating Sonic Hedgehog Signaling. Theranostics 2017, 7, 3021–3033. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.-J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itkonen, H.M.; Gorad, S.S.; Duveau, D.Y.; Martin, S.; Barkovskaya, A.; Bathen, T.F.; Moestue, S.A.; Mills, I.G. Inhibition of O-GlcNAc transferase activity reprograms prostate cancer cell metabolism. Oncotarget 2016, 7, 12464–12476. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.; Amendola, C.R.; Hollinshead, K.E.R.; Yu, Q.; Yamamoto, K.; Encarnación-Rosado, J.; Rose, R.E.; LaRue, M.M.; Sohn, A.S.W.; Biancur, D.E.; et al. Selective Alanine Transporter Utilization Creates a Targetable Metabolic Niche in Pancreatic Cancer. Cancer Discov. 2020, 10, 1018–1037. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Nikiforov, M.A.; Chandriani, S.; O’Connell, B.; Petrenko, O.; Kotenko, I.; Beavis, A.; Sedivy, J.M.; Cole, M.D. A Functional Screen for Myc-Responsive Genes Reveals Serine Hydroxymethyltransferase, a Major Source of the One-Carbon Unit for Cell Metabolism. Mol. Cell. Biol. 2002, 22, 5793–5800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, V.; Daoud, E.; Hatanpaa, K.J.; Gao, A.; Zhang, S.; An, Z.; Ganji, S.K.; Raisanen, J.M.; Lewis, C.M.; Askari, P.; et al. Glycine by MR spectroscopy is an imaging biomarker of glioma aggressiveness. Neuro-Oncol. 2020, 22, 1018–1029. [Google Scholar] [CrossRef]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine Decarboxylase Activity Drives Non-Small Cell Lung Cancer Tumor-Initiating Cells and Tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedeschi, P.M.; Markert, E.K.; Gounder, M.; Lin, H.; Dvorzhinski, D.; Dolfi, S.C.; Chan, L.L.-Y.; Qiu, J.; DiPaola, R.S.; Hirshfield, K.M.; et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 2013, 4, e877. [Google Scholar] [CrossRef] [Green Version]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef] [Green Version]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [Green Version]
- Baltruskeviciene, E.; Kazbariene, B.; Aleknavicius, E.; Krikstaponiene, A.; Venceviciene, L.; Suziedelis, K.; Stratilatovas, E.; Didziapetriene, J. Changes of reduced glutathione and glutathione S-transferase levels in colorectal cancer patients undergoing treatment. Tumori J. 2018, 104, 375–380. [Google Scholar] [CrossRef]
- Mila-Kierzenkowska, C.; Kedziora-Kornatowska, K.; Woźniak, A.; Drewa, T.; Woźniak, B.; Drewa, S.; Krzyzyńska-Malinowska, E.; Makarewicz, R. The effect of brachytherapy on antioxidant status and lipid peroxidation in patients with cancer of the uterine cervix. Cell. Mol. Biol. Lett. 2004, 9, 511–518. [Google Scholar]
- EKennedy, P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Van Der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta (BBA)—Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Wehrl, H.F.; Schwab, J.; Hasenbach, K.; Reischl, G.; Tabatabai, G.; Quintanilla-Martinez, L.; Jiru, F.; Chughtai, K.; Kiss, A.; Cay, F.; et al. Multimodal Elucidation of Choline Metabolism in a Murine Glioma Model Using Magnetic Resonance Spectroscopy and 11C-Choline Positron Emission Tomography. Cancer Res. 2013, 73, 1470–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Righi, V.; Roda, J.M.; Paz, J.; Mucci, A.; Tugnoli, V.; Rodriguez-Tarduchy, G.; Barrios, L.; Schenetti, L.; Cerdan, S.; García-Martín, M.L. 1H HR-MAS and genomic analysis of human tumor biopsies discriminate between high and low grade astrocytomas. NMR Biomed. 2009, 22, 629–637. [Google Scholar] [CrossRef]
- Hattingen, E.; Bähr, O.; Rieger, J.; Blasel, S.; Steinbach, J.; Pilatus, U. Phospholipid Metabolites in Recurrent Glioblastoma: In Vivo Markers Detect Different Tumor Phenotypes before and under Antiangiogenic Therapy. PLoS ONE 2013, 8, e56439. [Google Scholar] [CrossRef] [Green Version]
- Gillies, R.J.; Barry, J.A.; Ross, B.D. In Vitro andin Vivo13C and31P NMR analyses of phosphocholine metabolism in rat glioma cells. Magn. Reson. Med. 1994, 32, 310–318. [Google Scholar] [CrossRef]
- Katz-Brull, R.; Seger, D.; Rivenson-Segal, D.; Rushkin, E.; Degani, H. Metabolic markers of breast cancer: Enhanced choline metabolism and reduced choline-ether-phospholipid synthesis. Cancer Res. 2002, 62, 1966–1970. [Google Scholar]
- Glunde, K.; Jacobs, M.A.; Bhujwalla, Z.M. Choline metabolism in cancer: Implications for diagnosis and therapy. Expert Rev. Mol. Diagn. 2006, 6, 821–829. [Google Scholar] [CrossRef]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jimenez-Garcia, M.P.; Peinado-Serrano, J.; Carnero, A. NAMPT overexpression induces cancer stemness and defines a novel tumor signature for glioma prognosis. Oncotarget 2017, 8, 99514–99530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, B.; Young, D.A.; Lu, Z.-H.; Wang, T.; Meier, T.I.; Shepard, R.L.; Roth, K.; Zhai, Y.; Huss, K.; Kuo, M.-S.; et al. Pharmacological Inhibition of Nicotinamide Phosphoribosyltransferase (NAMPT), an Enzyme Essential for NAD+ Biosynthesis, in Human Cancer Cells. J. Biol. Chem. 2013, 288, 3500–3511. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.; Roulston, A.; Bélec, L.; Billot, X.; Marcellus, R.; Beédard, D.; Bernier, C.; Branchaud, S.; Chan, H.; Dairi, K.; et al. The Small Molecule GMX1778 Is a Potent Inhibitor of NAD + Biosynthesis: Strategy for Enhanced Therapy in Nicotinic Acid Phosphoribosyltransferase 1-Deficient Tumors. Mol. Cell. Biol. 2009, 29, 5872–5888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Hasan, K.; Alvarado, E.; Yuan, H.; Wu, H.; Chen, W.Y. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene 2010, 30, 907–921. [Google Scholar] [CrossRef] [Green Version]
- Muruganandham, M.; Alfieri, A.A.; Matei, C.; Chen, Y.; Sukenick, G.; Schemainda, I.; Hasmann, M.; Saltz, L.; Koutcher, J.A. Metabolic Signatures Associated with a NAD Synthesis Inhibitor–Induced Tumor Apoptosis Identified by 1H-Decoupled-31P Magnetic Resonance Spectroscopy. Clin. Cancer Res. 2005, 11, 3503–3513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, X.-M.; Bi, T.-Q.; Liao, X.-H.; Zhang, D.-J.; Long, H.-L.; Li, H.-J.; Zhao, W. Overexpression of Nampt in gastric cancer and chemopotentiating effects of the Nampt inhibitor FK866 in combination with fluorouracil. Oncol. Rep. 2011, 26, 1251–1257. [Google Scholar] [CrossRef] [Green Version]
- Michel, K.A.; Ragavan, M.; Walker, C.M.; Merritt, M.E.; Lai, S.Y.; Bankson, J.A. Comparison of selective excitation and multi-echo chemical shift encoding for imaging of hyperpolarized [1-13C]pyruvate. J. Magn. Reson. 2021, 325, 106927. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Aggarwal, R.; Bok, R.A.; Ohliger, M.A.; Zhu, Z.; Lee, P.; Gordon, J.W.; van Criekinge, M.; Carvajal, L.; Slater, J.B.; et al. Hyperpolarized 13C-pyruvate MRI detects real-time metabolic flux in prostate cancer metastases to bone and liver: A clinical feasibility study. Prostate Cancer Prostatic Dis. 2020, 23, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Tumor Development | Tumor Regression | Potential Pathway |
|---|---|---|---|
| Valine | U34 > C34, q = 0.0072 | T34 < U34, q = 0.0061 | BCAA Catabolism |
| T41 < U34, q = 0.0013 | |||
| T48 < U34, q = 0.0027 | |||
| Alanine | U28 > C28, q = 0.0366 U34 > C34, q = 0.0027 | T34 < U34, q = 0.0072 | Glutamine Anaplerosis |
| T41 < U28, q = 0.0063 | |||
| T41 < U34, q = 0.0004 | |||
| T48 < U28, q = 0.0149 | |||
| T48 < U34, q = 0.0011 | |||
| Glycine | U34 > C34, q = 0.0106 | T34 < U34, q = 0.0457 | Glycine Cleavage, Folate Cycle |
| T41 < U34, q = 0.0034 | |||
| T48 < U34, q = 0.0021 | |||
| Phosphocholine | U28 > C28, q = 0.0491 U34 > C34, q = 0.0144 | T41 < U28, q = 0.0284 | Kennedy Pathway, Choline Cycle |
| T41 < U34, q = 0.0106 | |||
| T48 < U34, q = 0.0457 | |||
| Glycero-phosphocholine | U34 > C34, q = 0.0343 | ||
| Phosphoethanolamine | T41 < U28, q = 0.0154 | ||
| T41 < U34, q = 0.0496 | |||
| T48 < U28, q = 0.0401 | |||
| Glutathione | T41 > T28, q = 0.0328 | Trans-Sulphuration Pathway | |
| T48 > T28, q = 0.0491 | |||
| NAD+ | T41 < U34, q = 0.0106 | Energy Metabolism | |
| T48 < U34, q = 0.0496 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salzillo, T.C.; Mawoneke, V.; Weygand, J.; Shetty, A.; Gumin, J.; Zacharias, N.M.; Gammon, S.T.; Piwnica-Worms, D.; Fuller, G.N.; Logothetis, C.J.; et al. Measuring the Metabolic Evolution of Glioblastoma throughout Tumor Development, Regression, and Recurrence with Hyperpolarized Magnetic Resonance. Cells 2021, 10, 2621. https://doi.org/10.3390/cells10102621
Salzillo TC, Mawoneke V, Weygand J, Shetty A, Gumin J, Zacharias NM, Gammon ST, Piwnica-Worms D, Fuller GN, Logothetis CJ, et al. Measuring the Metabolic Evolution of Glioblastoma throughout Tumor Development, Regression, and Recurrence with Hyperpolarized Magnetic Resonance. Cells. 2021; 10(10):2621. https://doi.org/10.3390/cells10102621
Chicago/Turabian StyleSalzillo, Travis C., Vimbai Mawoneke, Joseph Weygand, Akaanksh Shetty, Joy Gumin, Niki M. Zacharias, Seth T. Gammon, David Piwnica-Worms, Gregory N. Fuller, Christopher J. Logothetis, and et al. 2021. "Measuring the Metabolic Evolution of Glioblastoma throughout Tumor Development, Regression, and Recurrence with Hyperpolarized Magnetic Resonance" Cells 10, no. 10: 2621. https://doi.org/10.3390/cells10102621