
Current Insights into Immunology and Novel Therapeutics of Atopic Dermatitis

 , ,
, ,  ,
,  , ,
, , 
Abstract
:1. Introduction
2. Pathophysiology of Atopic Dermatitis
3. Atopic Dermatitis Phenotypes
4. Factors Causing Atopic Dermatitis
5. Innate Immunity in Atopic Dermatitis
6. Adaptive Immunity in Atopic Dermatitis
7. Murine Models for Preclinical Studies of AD
8. Therapeutic Strategies in Atopic Dermatitis
8.1. First Line Therapies
8.2. Second Line Therapies
8.3. Third Line Therapies
8.3.1. Emerging Therapeutic Biologics for AD
8.3.2. Emerging Therapeutic Small Molecules for AD
8.3.3. Phototherapy
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seth, D.; Cheldize, K.; Brown, D.; Freeman, E.E. Global Burden of Skin Disease: Inequities and Innovations. Curr. Dermatol. Rep. 2017, 6, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Ring, J. Terminology of Allergic Phenomena. Superantigens Superallergens 2014, 100, 46–52. [Google Scholar] [CrossRef]
- Stander, S. Atopic dermatitis. N. Engl. J. Med. 2021, 384, 1136–1143. [Google Scholar] [CrossRef]
- Langan, S.M.; Irvine, A.D.; Weidinger, S. Atopic dermatitis. Lancet 2020, 396, 345–360. [Google Scholar] [CrossRef]
- Weidinger, S.; Novak, N. Atopic dermatitis. Lancet 2016, 387, 1109–1122. [Google Scholar] [CrossRef]
- Furue, M.; Ulzii, D.; Vu, Y.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T. Pathogenesis of Atopic Dermatitis: Current Paradigm. Iran. J. Immunol. 2019, 16, 97–107. [Google Scholar]
- Yang, E.J.; Sekhon, S.; Sanchez, I.M.; Beck, K.M.; Bhutani, T. Recent Developments in Atopic Dermatitis. Pediatrics 2018, 142, e20181102. [Google Scholar] [CrossRef] [Green Version]
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic dermatitis. Nat. Rev. Dis. Primers 2018, 4, 1. [Google Scholar] [CrossRef]
- Saunders, S.P.; Moran, T.; Floudas, A.; Wurlod, F.; Kaszlikowska, A.; Salimi, M.; Quinn, E.M.; Oliphant, C.J.; Núñez, G.; McManus, R.; et al. Spontaneous atopic dermatitis is mediated by innate immunity, with the secondary lung inflammation of the atopic march requiring adaptive immunity. J. Allergy Clin. Immunol. 2016, 137, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.T.; Abrams, M.; Tlougan, B.; Rademaker, A.; Paller, A.S. Treatment of Staphylococcus aureus Colonization in Atopic Dermatitis Decreases Disease Severity. Pediatrics 2009, 123, e808–e814. [Google Scholar] [CrossRef] [Green Version]
- Yaghmaie, P.; Koudelka, C.W.; Simpson, E.L. Mental health comorbidity in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2013, 131, 428–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strom, M.; Fishbein, A.; Paller, A.; Silverberg, J. Association between atopic dermatitis and attention deficit hyperactivity disorder in U.S. children and adults. Br. J. Dermatol. 2016, 175, 920–929. [Google Scholar] [CrossRef] [Green Version]
- Roduit, C.; Frei, R.; Depner, M.; Karvonen, A.M.; Renz, H.; Braun-Fahrländer, C.; Schmausser-Hechfellner, E.; Pekkanen, J.; Riedler, J.; Dalphin, J.-C.; et al. Phenotypes of Atopic Dermatitis Depending on the Timing of Onset and Progression in Childhood. JAMA Pediatr. 2017, 171, 655–662. [Google Scholar] [CrossRef]
- Nakajima, S.; Nomura, T.; Common, J.; Kabashima, K. Insights into atopic dermatitis gained from genetically defined mouse models. J. Allergy Clin. Immunol. 2019, 143, 13–25. [Google Scholar] [CrossRef]
- Cabanillas, B.; Brehler, A.-C.; Novak, N. Atopic dermatitis phenotypes and the need for personalized medicine. Curr. Opin. Allergy Clin. Immunol. 2017, 17, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, K.; Heitmiller, K.D.; Czarnowicki, T. An Update on the Pathophysiology of Atopic Dermatitis. Dermatol. Clin. 2017, 35, 317–326. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Waldman, A.; Ahluwalia, J.; Ong, P.Y.; Eichenfield, L. Atopic dermatitis: Pathogenesis. Semin. Cutan. Med. Surg. 2017, 36, 100–103. [Google Scholar] [CrossRef]
- Furue, M.; Chiba, T.; Tsuji, G.; Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Kadono, T. Atopic dermatitis: Immune deviation, barrier dysfunction, IgE autoreactivity and new therapies. Allergol. Int. 2017, 66, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; Strong, C.D.G.; Krueger, J.G.; et al. Progressive activation of TH2/TH22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Eyerich, K.; Novak, N. Immunology of atopic eczema: Overcoming the Th1/Th2 paradigm. Allergy 2013, 68, 974–982. [Google Scholar] [CrossRef]
- Werfel, T.; Allam, J.-P.; Biedermann, T.; Eyerich, K.; Gilles, S.; Guttman-Yassky, E.; Hoetzenecker, W.; Knol, E.; Simon, H.-U.; Wollenberg, A.; et al. Cellular and molecular immunologic mechanisms in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2016, 138, 336–349. [Google Scholar] [CrossRef] [Green Version]
- Nygaard, U.; Hvid, M.; Johansen, C.; Buchner, M.; Fölster-Holst, R.; Deleuran, M.; Vestergaard, C. TSLP, IL-31, IL-33 and sST2 are new biomarkers in endophenotypic profiling of adult and childhood atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Tokura, Y. Extrinsic and intrinsic types of atopic dermatitis. J. Dermatol. Sci. 2010, 58, 1–7. [Google Scholar] [CrossRef]
- Suárez-Fariñas, M.; Dhingra, N.; Gittler, J.; Shemer, A.; Cardinale, I.; Strong, C.D.G.; Krueger, J.G.; Guttman-Yassky, E. Intrinsic atopic dermatitis shows similar TH2 and higher TH17 immune activation compared with extrinsic atopic dermatitis. J. Allergy Clin. Immunol. 2013, 132, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daltro, S.R.T.; Meira, C.S.; Santos, I.P.; Dos Santos, R.R.; Soares, M.B.P. Mesenchymal Stem Cells and Atopic Dermatitis: A Review. Front. Cell Dev. Biol. 2020, 8, 326. [Google Scholar] [CrossRef]
- Grewe, M.; Walther, S.; Gyufko, K.; Czech, W.; Schöpf, E.; Krutmann, J. Analysis of the Cytokine Pattern Expressed In Situ in Inhalant Allergen Patch Test Reactions of Atopic Dermatitis Patients. J. Investig. Dermatol. 1995, 105, 407–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, K.P.; Mills, K. Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol. 2013, 34, 521–530. [Google Scholar] [CrossRef]
- Volz, T.; Nega, M.; Buschmann, J.; Kaesler, S.; Guenova, E.; Peschel, A.; Röcken, M.; Götz, F.; Biedermann, T. Natural Staphylococcus aureus—Derived peptidoglycan fragments activate NOD2 and act as potent costimulators of the innate immune system exclusively in the presence of TLR signals. FASEB J. 2010, 24, 4089–4102. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Honda, T.; Kabashima, K. Multipolarity of cytokine axes in the pathogenesis of atopic dermatitis in terms of age, race, species, disease stage and biomarkers. Int. Immunol. 2018, 30, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, L.C.; Rodriguez, E.; Stölzl, D.; Wehkamp, U.; Sun, J.; Gerdes, S.; Sarkar, M.; Hübenthal, M.; Zeng, C.; Uppala, R.; et al. Progression of acute-to-chronic atopic dermatitis is associated with quantitative rather than qualitative changes in cytokine responses. J. Allergy Clin. Immunol. 2020, 145, 1406–1415. [Google Scholar] [CrossRef] [Green Version]
- Sugaya, M. The Role of Th17-Related Cytokines in Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman-Yassky, E.; Lowes, M.A.; Fuentes-Duculan, J.; Zaba, L.C.; Cardinale, I.; Nograles, K.E.; Khatcherian, A.; Novitskaya, I.; Carucci, J.A.; Bergman, R.; et al. Low Expression of the IL-23/Th17 Pathway in Atopic Dermatitis Compared to Psoriasis. J. Immunol. 2008, 181, 7420–7427. [Google Scholar] [CrossRef] [PubMed]
- Toda, M.; Leung, D.Y.; Molet, S.; Boguniewicz, M.; Taha, R.; Christodoulopoulos, P.; Fukuda, T.; Elias, J.A.; Hamid, Q.A. Polarized in vivo expression of IL-11 and IL-17 between acute and chronic skin lesions. J. Allergy Clin. Immunol. 2003, 111, 875–881. [Google Scholar] [CrossRef]
- Rebane, A.; Zimmermann, M.; Aab, A.; Baurecht, H.; Koreck, A.; Karelson, M.; Abram, K.; Metsalu, T.; Pihlap, M.; Meyer, N.; et al. Mechanisms of IFN-γ–induced apoptosis of human skin keratinocytes in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2012, 129, 1297–1306. [Google Scholar] [CrossRef]
- Campione, E.; Lanna, C.; Diluvio, L.; Cannizzaro, M.V.; Grelli, S.; Galluzzo, M.; Talamonti, M.; Annicchiarico-Petruzzelli, M.; Mancini, M.; Melino, G.; et al. Skin immunity and its dysregulation in atopic dermatitis, hidradenitis suppurativa and vitiligo. Cell Cycle 2020, 19, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.J.; Estravís, M.; García-Sánchez, A.; Dávila, I.; Isidoro-García, M.; Sanz, C. Genetics and Epigenetics of Atopic Dermatitis: An Updated Systematic Review. Genes 2020, 11, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, K.C. An update on the genetics of atopic dermatitis: Scratching the surface in 2009. J. Allergy Clin. Immunol. 2010, 125, 16–29. [Google Scholar] [CrossRef]
- Strong, C.D.G.; Conlan, S.; Deming, C.B.; Cheng, J.; Sears, K.E.; Segre, J.A. A milieu of regulatory elements in the epidermal differentiation complex syntenic block: Implications for atopic dermatitis and psoriasis. Hum. Mol. Genet. 2010, 19, 1453–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, Y.-H.; Hwu, W.-L.; Chiang, B.-L. The Genetics of Atopic Dermatitis. Clin. Rev. Allergy Immunol. 2007, 33, 178–190. [Google Scholar] [CrossRef]
- Mu, Z.; Zhao, Y.; Liu, X.; Chang, C.; Zhang, J. Molecular Biology of Atopic Dermatitis. Clin. Rev. Allergy Immunol. 2014, 47, 193–218. [Google Scholar] [CrossRef]
- Totri, C.R.; Diaz, L.; Eichenfield, L.F. 2014 update on atopic dermatitis in children. Curr. Opin. Pediatr. 2014, 26, 466–471. [Google Scholar] [CrossRef]
- D’Auria, E.; Banderali, G.; Barberi, S.; Gualandri, L.; Pietra, B.; Riva, E.; Cerri, A. Atopic dermatitis: Recent insight on pathogenesis and novel therapeutic target. Asian Pac. J. Allergy Immunol. 2016, 34, 98–108. [Google Scholar] [CrossRef]
- Nakahara, T.; Kido-Nakahara, M.; Tsuji, G.; Furue, M. Basics and recent advances in the pathophysiology of atopic dermatitis. J. Dermatol. 2021, 48, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Wickett, R.R. Basics of skin structure. J. Cosmet. Sci. 2004, 55, 132–133. [Google Scholar] [PubMed]
- Yu, H.-S.; Kang, M.-J.; Kwon, J.-W.; Lee, S.-Y.; Lee, E.; Yang, S.-I.; Jung, Y.-H.; Hong, K.; Kim, Y.-J.; Lee, S.-H.; et al. Claudin-1 polymorphism modifies the effect of mold exposure on the development of atopic dermatitis and production of IgE. J. Allergy Clin. Immunol. 2015, 135, 827–830.e5. [Google Scholar] [CrossRef] [PubMed]
- Kubo, A.; Nagao, K.; Amagai, M. Epidermal barrier dysfunction and cutaneous sensitization in atopic diseases. J. Clin. Investig. 2012, 122, 440–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-J.; Lee, S.-H. Epidermal Permeability Barrier Defects and Barrier Repair Therapy in Atopic Dermatitis. Allergy Asthma Immunol. Res. 2014, 6, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Berroth, A.; Kühnl, J.; Kurschat, N.; Schwarz, A.; Stäb, F.; Schwarz, T.; Wenck, H.; Fölster-Holst, R.; Neufang, G. Role of fibroblasts in the pathogenesis of atopic dermatitis. J. Allergy Clin. Immunol. 2013, 131, 1547–1554. [Google Scholar] [CrossRef]
- Palmer, C.N.A.; Irvine, A.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.D.; et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006, 38, 441–446. [Google Scholar] [CrossRef]
- Leung, D.Y. Allergic skin diseases beyond TH2. Ann. Allergy Asthma Immunol. 2020, 124, 1. [Google Scholar] [CrossRef]
- Roekevisch, E.; Szegedi, K.; Hack, D.; Schram, M.; Res, P.; Bos, J.; Leeflang, M.; Luiten, R.; Kezic, S.; Spuls, P.; et al. Effect of immunosuppressive treatment on biomarkers in adult atopic dermatitis patients. J. Eur. Acad. Dermatol. Venereol. 2019, 34, 1545–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.; Relton, C.; Liao, H.; Zhao, Y.; Sandilands, A.; McLean, W.; Cordell, H.; Reynolds, N. Filaggrin haploinsufficiency is highly penetrant and is associated with increased severity of eczema: Further delineation of the skin phenotype in a prospective epidemiological study of 792 school children. Br. J. Dermatol. 2009, 161, 884–889. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.; Chen, H.; Koh, W.-P.; Common, J.; Van Bever, H.; McLean, W.H.I.; Lane, E.; Giam, Y.; Tang, M. Filaggrin mutations are associated with recurrent skin infection in Singaporean Chinese patients with atopic dermatitis. Br. J. Dermatol. 2011, 166, 200–203. [Google Scholar] [CrossRef]
- Howell, M.D.; Kim, B.E.; Gao, P.; Grant, A.V.; Boguniewicz, M.; DeBenedetto, A.; Schneider, L.; Beck, L.A.; Barnes, K.C.; Leung, D.Y. Cytokine modulation of atopic dermatitis filaggrin skin expression. J. Allergy Clin. Immunol. 2009, 124, R7–R12. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.-H.; Yoshida, T.; De Benedetto, A.; Beck, L.A. The cutaneous innate immune response in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2013, 131, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Kabashima, K. Reconciling innate and acquired immunity in atopic dermatitis. J. Allergy Clin. Immunol. 2020, 145, 1136–1137. [Google Scholar] [CrossRef]
- Pivarcsi, A.; Kemény, L.; Dobozy, A. Innate Immune Functions of the Keratinocytes. Acta Microbiol. Immunol. Hung. 2004, 51, 303–310. [Google Scholar] [CrossRef]
- Mrabet-Dahbi, S.; Maurer, M. Innate Immunity in Atopic Dermatitis. Curr. Probl. Dermatol. 2011, 41, 104–111. [Google Scholar] [CrossRef]
- Czarnowicki, T.; He, H.; Krueger, J.G.; Guttman-Yassky, E. Atopic dermatitis endotypes and implications for targeted therapeutics. J. Allergy Clin. Immunol. 2019, 143, 1–11. [Google Scholar] [CrossRef]
- Novak, N. An update on the role of human dendritic cells in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2012, 129, 879–886. [Google Scholar] [CrossRef]
- Kapitány, A.; Béke, G.; Nagy, G.; Doan-Xuan, Q.; Bacso, Z.; Gáspár, K.; Boros, G.; Dajnoki, Z.; Bíró, T.; Rajnavölgyi, É.; et al. CD1c+ Blood Dendritic Cells in Atopic Dermatitis are Premature and Can Produce Disease-specific Chemokines. Acta Derm. Venereol. 2017, 97, 325–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaeschen, A.S.; Nümm, T.J.; Herrmann, N.; Leib, N.; Maintz, L.; Sakai, T.; Wenzel, J.; Bieber, T. Jak1/2 inhibition impairs the development and function of inflammatory dendritic epidermal cells in atopic dermatitis. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef]
- Nakajima, S.; Igyártó, B.Z.; Honda, T.; Egawa, G.; Otsuka, A.; Hara-Chikuma, M.; Watanabe, N.; Ziegler, S.F.; Tomura, M.; Inaba, K.; et al. Langerhans cells are critical in epicutaneous sensitization with protein antigen via thymic stromal lymphopoietin receptor signaling. J. Allergy Clin. Immunol. 2012, 129, 1048–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollenberg, A.; Kraft, S.; Hanau, D.; Bieber, T. Immunomorphological and Ultrastructural Characterization of Langerhans Cells and a Novel, Inflammatory Dendritic Epidermal Cell (IDEC) Population in Lesional Skin of Atopic Eczema. J. Investig. Dermatol. 1996, 106, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Wollenberg, A.; Günther, S.; Moderer, M.; Wetzel, S.; Wagner, M.; Towarowski, A.; Tuma, E.; Rothenfusser, S.; Endres, S.; Hartmann, G. Plasmacytoid Dendritic Cells: A New Cutaneous Dendritic Cell Subset with Distinct Role in Inflammatory Skin Diseases. J. Investig. Dermatol. 2002, 119, 1096–1102. [Google Scholar] [CrossRef] [Green Version]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.-C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.J.; et al. A role for IL-25 and IL-33–driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Castillo, J.M.; Galand, C.; Mashiko, S.; Bissonnette, R.; McGurk, A.; Ziegler, S.F.; Dong, C.; McKenzie, A.N.; Sarfati, M.; Geha, R.S. ILC2 activation by keratinocyte-derived IL-25 drives IL-13 production at sites of allergic skin inflammation. J. Allergy Clin. Immunol. 2020, 145, 1606–1614.e4. [Google Scholar] [CrossRef]
- Kim, B.S.; Siracusa, M.C.; Saenz, S.A.; Noti, M.; Monticelli, L.A.; Sonnenberg, G.F.; Hepworth, M.R.; Van Voorhees, A.S.; Comeau, M.R.; Artis, D. TSLP Elicits IL-33-Independent Innate Lymphoid Cell Responses to Promote Skin Inflammation. Sci. Transl. Med. 2013, 5, 170ra16. [Google Scholar] [CrossRef] [Green Version]
- Toyoshima, S.; Sakamoto-Sasaki, T.; Kurosawa, Y.; Hayama, K.; Matsuda, A.; Watanabe, Y.; Terui, T.; Gon, Y.; Matsumoto, K.; Okayama, Y. Mir103a-3p in extracellular vesicles from FcεRI-aggregated human mast cells enhances il-5 production by group 2 innate lymphoid cells. J. Allergy Clin. Immunol. 2021, 147, 1878–1891. [Google Scholar] [CrossRef]
- Kim, B.S.; Wang, K.; Siracusa, M.C.; Saenz, S.A.; Brestoff, J.; Monticelli, L.A.; Noti, M.; Wojno, E.D.T.; Fung, T.C.; Kubo, M.; et al. Basophils Promote Innate Lymphoid Cell Responses in Inflamed Skin. J. Immunol. 2014, 193, 3717–3725. [Google Scholar] [CrossRef] [Green Version]
- Mashiko, S.; Mehta, H.; Bissonnette, R.; Sarfati, M. Increased frequencies of basophils, type 2 innate lymphoid cells and Th2 cells in skin of patients with atopic dermatitis but not psoriasis. J. Dermatol. Sci. 2017, 88, 167–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natsume, C.; Aoki, N.; Aoyama, T.; Senda, K.; Matsui, M.; Ikegami, A.; Tanaka, K.; Azuma, Y.-T.; Fujita, T. Fucoxanthin Ameliorates Atopic Dermatitis Symptoms by Regulating Keratinocytes and Regulatory Innate Lymphoid Cells. Int. J. Mol. Sci. 2020, 21, 2180. [Google Scholar] [CrossRef] [Green Version]
- Czarnowicki, T.; Malajian, D.; Shemer, A.; Fuentes-Duculan, J.; Gonzalez, J.; Suárez-Fariñas, M.; Krueger, J.G.; Guttman-Yassky, E. Skin-homing and systemic T-cell subsets show higher activation in atopic dermatitis versus psoriasis. J. Allergy Clin. Immunol. 2015, 136, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Novak, N.; Koch, S.; Allam, J.-P.; Bieber, T. Dendritic cells: Bridging innate and adaptive immunity in atopic dermatitis. J. Allergy Clin. Immunol. 2010, 125, 50–59. [Google Scholar] [CrossRef]
- Roesner, L.M.; Werfel, T.; Heratizadeh, A. The adaptive immune system in atopic dermatitis and implications on therapy. Expert Rev. Clin. Immunol. 2016, 12, 787–796. [Google Scholar] [CrossRef]
- Campana, R.; Dzoro, S.; Mittermann, I.; Fedenko, E.; Elisyutina, O.; Khaitov, M.; Karaulov, A.; Valenta, R. Molecular aspects of allergens in atopic dermatitis. Curr. Opin. Allergy Clin. Immunol. 2017, 17, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Langeveld-Wildschut, E.G.; Thepen, T.; Bihari, I.C.; Reijsen, F.C.V.; De Vries, I.J.; Bruijnzeel, P.L.; A Bruijnzeel-Koomen, C. Evaluation of the atopy patch test and the cutaneous late-phase reaction as relevant models for the study of allergic inflammation in patients with atopic eczema. J. Allergy Clin. Immunol. 1996, 98, 1019–1027. [Google Scholar] [CrossRef]
- Mommert, S.; Gschwandtner, M.; Gutzmer, R.; Werfel, T. The Role of the Histamine H4 Receptor in Atopic Dermatitis. Curr. Allergy Asthma Rep. 2010, 11, 21–28. [Google Scholar] [CrossRef]
- Carlier, T.D.B.; Badloe, F.M.S.; Ring, J.; Gutermuth, J.; Krohn, I.K. Autoreactive T cells and their role in atopic dermatitis. J. Autoimmun. 2021, 120, 102634. [Google Scholar] [CrossRef]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Crawford, A.; MacLeod, M.; Schumacher, T.; Corlett, L.; Gray, D. Primary T Cell Expansion and Differentiation In Vivo Requires Antigen Presentation by B Cells. J. Immunol. 2006, 176, 3498–3506. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class Switch Recombination and Hypermutation Require Activation-Induced Cytidine Deaminase (AID), a Potential RNA Editing Enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Zan, H.; Pone, E.J.; Mai, T.; Casali, P. Immunoglobulin class-switch DNA recombination: Induction, targeting and beyond. Nat. Rev. Immunol. 2012, 12, 517–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, S.P.; Ma, E.G.M.; Aranda, C.J.; De Lafaille, M.A.C. Non-classical B Cell Memory of Allergic IgE Responses. Front. Immunol. 2019, 10, 715. [Google Scholar] [CrossRef] [PubMed]
- Weisel, F.; Shlomchik, M. Memory B Cells of Mice and Humans. Annu. Rev. Immunol. 2017, 35, 255–284. [Google Scholar] [CrossRef] [PubMed]
- Hofmaier, S.; Comberiati, P.; Matricardi, P.M. Immunoglobulin g in ige-mediated allergy and allergen-specific immunotherapy. Eur. Ann. Allergy Clin. Immunol. 2014, 46, 6–11. [Google Scholar] [PubMed]
- DiLillo, D.J.; Matsushita, T.; Tedder, T.F. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann. N. Y. Acad. Sci. 2010, 1183, 38–57. [Google Scholar] [CrossRef]
- Bouaziz, J.-D.; Yanaba, K.; Tedder, T.F. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol. Rev. 2008, 224, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Suga, H.; Sato, S. Il-10–producing regulatory b cells in skin diseases. J. Cutan. Immunol. Allergy 2019, 2, 68–74. [Google Scholar] [CrossRef]
- Azeem, M.; Kader, H.; Kerstan, A.; Hetta, H.F.; Serfling, E.; Goebeler, M.; Muhammad, K. Intricate Relationship Between Adaptive and Innate Immune System in Allergic Contact Dermatitis. Yale J. Biol. Med. 2020, 93, 699–709. [Google Scholar]
- Egbuniwe, I.; Karagiannis, S.N.; Nestle, F.O.; Lacy, K.E. Revisiting the role of B cells in skin immune surveillance. Trends Immunol. 2015, 36, 102–111. [Google Scholar] [CrossRef]
- Simon, D.; Hösli, S.; Kostylina, G.; Yawalkar, N.; Simon, H.-U. Anti-CD20 (rituximab) treatment improves atopic eczema. J. Allergy Clin. Immunol. 2008, 121, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, J.J.; Rijvers, L.; Arends, N.J.; Driessen, G.J.; Pasmans, S.G.; van Dongen, J.J.M.; de Jongste, J.C.; van Zelm, M.C. Ige-expressing memory b cells and plasmablasts are increased in blood of children with asthma, food allergy, and atopic dermatitis. Allergy 2018, 73, 1331–1336. [Google Scholar] [CrossRef] [Green Version]
- Tanei, R.; Hasegawa, Y.; Sawabe, M. Abundant immunoglobulin E-positive cells in skin lesions support an allergic etiology of atopic dermatitis in the elderly. J. Eur. Acad. Dermatol. Venereol. 2012, 27, 952–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Tanaka, T.; Shi, W.; Matsumoto, M.; Minami, M.; Kashiwamura, S.-I.; Nakanishi, K.; Yoshida, N.; Kishimoto, T.; Akira, S. Essential role of Stat6 in IL-4 signalling. Nat. Cell Biol. 1996, 380, 627–630. [Google Scholar] [CrossRef]
- Turqueti-Neves, A.; Otte, M.; Prazeres da Costa, O.; Höpken, U.E.; Lipp, M.; Buch, T.; Voehringer, D. B-cell-intrinsic stat6 signaling controls germinal center formation. Eur. J. Immunol. 2014, 44, 2130–2138. [Google Scholar] [CrossRef] [PubMed]
- Haase, P.; Mokada-Gopal, L.; Radtke, D.; Voehringer, D. Modulation of the humoral immune response by constitutively active STAT6 expression in murine B cells. Eur. J. Immunol. 2020, 50, 558–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czarnowicki, T.; Gonzalez, J.; Bonifacio, K.M.; Shemer, A.; Xiangyu, P.; Kunjravia, N.; Malajian, D.; Fuentes-Duculan, J.; Esaki, H.; Noda, S.; et al. Diverse activation and differentiation of multiple B-cell subsets in patients with atopic dermatitis but not in patients with psoriasis. J. Allergy Clin. Immunol. 2016, 137, 118–129.e5. [Google Scholar] [CrossRef]
- Valenta, R.; Steiner, R.; Seiberler, S.; Maurer, D.; Sperr, W.R.; Valent, P.; Spitzauer, S.; Kapiotis, S.; Smolen, J.; Stingl, G. Immunoglobulin E Response to Human Proteins in Atopic Patients. J. Investig. Dermatol. 1996, 107, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeller, S.; Rhyner, C.; Meyer, N.; Schmid-Grendelmeier, P.; Akdis, C.A.; Crameri, R. Exploring the repertoire of ige-binding self-antigens associated with atopic eczema. J. Allergy Clin. Immunol. 2009, 124, 271–277, 278–285. [Google Scholar] [CrossRef]
- Schmid-Grendelmeier, P.; Flückiger, S.; Disch, R.; Trautmann, A.; Wüthrich, B.; Blaser, K.; Scheynius, A.; Crameri, R. Ige-mediated and t cell-mediated autoimmunity against manganese superoxide dismutase in atopic dermatitis. J. Allergy Clin. Immunol. 2005, 115, 1068–1075. [Google Scholar] [CrossRef]
- Rivera, A.; Chen, C.-C.; Ron, N.; Dougherty, J.P.; Ron, Y. Role of B cells as antigen-presenting cells in vivo revisited: Antigen-specific B cells are essential for T cell expansion in lymph nodes and for systemic T cell responses to low antigen concentrations. Int. Immunol. 2001, 13, 1583–1593. [Google Scholar] [CrossRef] [Green Version]
- Yanaba, K.; Kamata, M.; Asano, Y.; Tada, Y.; Sugaya, M.; Kadono, T.; Tedder, T.F.; Sato, S. CD19 expression in B cells regulates atopic dermatitis in a mouse model. Am. J. Pathol. 2013, 182, 2214–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerman, I.; Mitchell, D.C.; Richardson, C.T. Human cutaneous B cells: What do we really know? Ann. Transl. Med. 2021, 9, 440. [Google Scholar] [CrossRef]
- Wilson, R.P.; McGettigan, S.E.; Dang, V.D.; Kumar, A.; Cancro, M.P.; Nikbakht, N.; Stohl, W.; Debes, G.F. IgM Plasma Cells Reside in Healthy Skin and Accumulate with Chronic Inflammation. J. Investig. Dermatol. 2019, 139, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Geherin, S.A.; Fintushel, S.R.; Lee, M.; Wilson, R.P.; Patel, R.T.; Alt, C.; Young, A.J.; Hay, J.B.; Debes, G.F. The Skin, a Novel Niche for Recirculating B Cells. J. Immunol. 2012, 188, 6027–6035. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Chong, B.; Mirchandani, N.; Brinster, N.K.; Yamanaka, K.-I.; Dowgiert, R.K.; Kupper, T.S. The Vast Majority of CLA+ T Cells Are Resident in Normal Skin. J. Immunol. 2006, 176, 4431–4439. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Liu, Z.; Yuan, H.; Zhao, X.; Zou, Y.; Zheng, J.; Pan, M. Autoreactive B Cell Differentiation in Diffuse Ectopic Lymphoid-Like Structures of Inflamed Pemphigus Lesions. J. Investig. Dermatol. 2020, 140, 309–318.e8. [Google Scholar] [CrossRef]
- Li, J.; Shen, C.; Liu, Y.; Li, Y.; Sun, L.; Jiao, L.; Jiao, W.; Xiao, J.; Shen, C.; Qi, H.; et al. Impaired Function of CD5+CD19+CD1dhi B10 Cells on IgE Secretion in an Atopic Dermatitis-Like Mouse Model. PLoS ONE 2015, 10, e0132173. [Google Scholar] [CrossRef]
- Liu, B.-S.; Cao, Y.; Huizinga, T.W.; Hafler, D.A.; Toes, R. TLR-mediated STAT3 and ERK activation controls IL-10 secretion by human B cells. Eur. J. Immunol. 2014, 44, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xin, H.; Zhang, W.; Yazaki, P.J.; Zhang, Z.; Le, K.; Li, W.; Lee, H.; Kwak, L.; Forman, S.; et al. CD5 Binds to Interleukin-6 and Induces a Feed-Forward Loop with the Transcription Factor STAT3 in B Cells to Promote Cancer. Immunity 2016, 44, 913–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, P.A.; Yassin-Noreña, L.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. CD19+CD24hiCD38hi B Cells Exhibit Regulatory Capacity in Healthy Individuals but Are Functionally Impaired in Systemic Lupus Erythematosus Patients. Immunity 2010, 32, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, Y.; Yanaba, K.; Hayashi, M.; Chiba, M.; Ishiuji, Y.; Ishiji, T.; Nakagawa, H. IL-10-producing regulatory B cells are decreased in patients with atopic dermatitis and are inversely correlated with disease severity. J. Dermatol. Sci. 2017, 86, e26. [Google Scholar] [CrossRef]
- Gu, Y.; Li, K.; Sun, J.; Zhang, J. Characterization of CD19+CD24hiCD38hi B cells in Chinese adult patients with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 2863–2870. [Google Scholar] [CrossRef]
- Yoshihara, Y.; Ishiuji, Y.; Yoshizaki, A.; Kurita, M.; Hayashi, M.; Ishiji, T.; Nakagawa, H.; Asahina, A.; Yanaba, K. IL-10–Producing Regulatory B Cells Are Decreased in Patients with Atopic Dermatitis. J. Investig. Dermatol. 2019, 139, 475–478. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Serfling, E.; Berberich-Siebelt, F.; Chuvpilo, S.; Jankevics, E.; Klein-Hessling, S.; Twardzik, T.; Avots, A. The role of NF-AT transcription factors in T cell activation and differentiation. Biochim. Biophys. Acta Bioenerg. 2000, 1498, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Hock, M.; Vaeth, M.; Rudolf, R.; Patra, A.K.; Pham, D.A.T.; Muhammad, K.; Pusch, T.; Bopp, T.; Schmitt, E.; Rost, R.; et al. NFATc1 Induction in Peripheral T and B Lymphocytes. J. Immunol. 2013, 190, 2345–2353. [Google Scholar] [CrossRef]
- Muhammad, K.; Rudolf, R.; Pham, D.A.T.; Klein-Hessling, S.; Takata, K.; Matsushita, N.; Ellenrieder, V.; Kondo, E.; Serfling, E. Induction of Short NFATc1/αA Isoform Interferes with Peripheral B Cell Differentiation. Front. Immunol. 2018, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Deb, J.; Patra, A.K.; Pham, D.A.T.; Chen, W.; Vaeth, M.; Berberich-Siebelt, F.; Klein-Hessling, S.; Lamperti, E.D.; Reifenberg, K.; et al. NFATc1 affects mouse splenic B cell function by controlling the calcineurin-NFAT signaling network. J. Exp. Med. 2011, 208, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Alrefai, H.; Muhammad, K.; Rudolf, R.; Pham, D.A.; Klein-Hessling, S.; Patra, A.K.; Avots, A.; Bukur, V.; Sahin, U.; Tenzer, S.; et al. NFATc1 supports imiquimod-induced skin inflammation by suppressing il-10 synthesis in b cells. Nat. Commun. 2016, 7, 11724. [Google Scholar] [CrossRef] [Green Version]
- Gran, F.; Kerstan, A.; Serfling, E.; Goebeler, M.; Muhammad, K. Current developments in the immunology of psoriasis. Yale J. Biol. Med. 2020, 93, 97–110. [Google Scholar]
- Picker, L.J.; Michie, S.A.; Rott, L.S.; Butcher, E.C. A unique phenotype of skin-associated lymphocytes in humans. Preferential expression of the HECA-452 epitope by benign and malignant T cells at cutaneous sites. Am. J. Pathol. 1990, 136, 1053–1068. [Google Scholar]
- Akdis, M.; Trautmann, A.; Klunker, S.; Blaser, K.; Akdis, C.A. Cytokine network and dysregulated apoptosis in atopic dermatitis. Acta Odontol. Scand. 2001, 59, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, N.; Benfeitas, R.; Katayama, S.; Bruhn, S.; Andersson, A.; Wikberg, G.; Lundeberg, L.; Lindvall, J.M.; Greco, D.; Kere, J.; et al. Epigenetic alterations in skin homing CD4+CLA+ T cells of atopic dermatitis patients. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef]
- Vennegaard, M.T.; Bonefeld, C.M.; Hagedorn, P.H.; Bangsgaard, N.; Løvendorf, M.B.; Ødum, N.; Woetmann, A.; Geisler, C.; Skov, L. Allergic contact dermatitis induces upregulation of identical microRNAs in humans and mice. Contact Dermat. 2012, 67, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, J.; Zhang, X.; Zhang, Y.; Qin, Z.-l.; Wang, H.; Luo, X.-y. Application of topical phosphodiesterase 4 inhibitors in mild to moderate atopic dermatitis: A systematic review and meta-analysis. JAMA Dermatol. 2019, 155, 585–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestergaard, C.; Yoneyama, H.; Murai, M.; Nakamura, K.; Tamaki, K.; Terashima, Y.; Imai, T.; Yoshie, O.; Irimura, T.; Mizutani, H.; et al. Overproduction of Th2-specific chemokines in NC/Nga mice exhibiting atopic dermatitis–like lesions. J. Clin. Investig. 1999, 104, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Czarnowicki, T.; Gonzalez, J.; Shemer, A.; Malajian, D.; Xu, H.; Zheng, X.; Khattri, S.; Gilleaudeau, P.; Sullivan-Whalen, M.; Suárez-Fariñas, M.; et al. Severe atopic dermatitis is characterized by selective expansion of circulating TH2/TC2 and TH22/TC22, but not TH17/TC17, cells within the skin-homing T-cell population. J. Allergy Clin. Immunol. 2015, 136, 104–115.e7. [Google Scholar] [CrossRef]
- Szegedi, K.D.; Kremer, A.E.; Kezic, S.; Teunissen, M.B.M.; Bos, J.D.; Luiten, R.M.; Res, P.C.; Middelkamp-Hup, M.A. Increased frequencies of IL-31-producing T cells are found in chronic atopic dermatitis skin. Exp. Dermatol. 2012, 21, 431–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neis, M.M.; Peters, B.; Dreuw, A.; Wenzel, J.; Bieber, T.; Mauch, C.; Krieg, T.; Stanzel, S.; Heinrich, P.C.; Merk, H.F. Enhanced expression levels of IL-31 correlate with IL-4 and IL-13 in atopic and allergic contact dermatitis. J. Allergy Clin. Immunol. 2006, 118, 930–937. [Google Scholar] [CrossRef]
- Takamori, A.; Nambu, A.; Sato, K.; Yamaguchi, S.; Matsuda, K.; Numata, T.; Sugawara, T.; Yoshizaki, T.; Arae, K.; Morita, H.; et al. IL-31 is crucial for induction of pruritus, but not inflammation, in contact hypersensitivity. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Boniface, K.; Guignouard, E.; Pedretti, N.; Garcia, M.; Delwail, A.; Bernard, F.; Nau, F.; Guillet, G.; Dagregorio, G.; Yssel, H.; et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin. Exp. Immunol. 2007, 150, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Boniface, K.; Bernard, F.-X.; Garcia, M.; Gurney, A.L.; Lecron, J.-C.; Morel, F. IL-22 Inhibits Epidermal Differentiation and Induces Proinflammatory Gene Expression and Migration of Human Keratinocytes. J. Immunol. 2005, 174, 3695–3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, P.; Pavel, A.B.; Khattri, S.; Leonard, A.; Malik, K.; Rose, S.; On, S.J.; Vekaria, A.S.; Traidl-Hoffmann, C.; Singer, G.K.; et al. Baseline IL-22 expression in patients with atopic dermatitis stratifies tissue responses to fezakinumab. J. Allergy Clin. Immunol. 2019, 143, 142–154. [Google Scholar] [CrossRef]
- Agrawal, R.; Woodfolk, J.A. Skin Barrier Defects in Atopic Dermatitis. Curr. Allergy Asthma Rep. 2014, 14, 1–11. [Google Scholar] [CrossRef]
- El Ghalbzouri, A.; Hensbergen, P.; Gibbs, S.; Kempenaar, J.; Van Der Schors, R.; Ponec, M. Fibroblasts facilitate re-epithelialization in wounded human skin equivalents. Lab. Investig. 2004, 84, 102–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, R.M.; Krueger, J.; Yourish, D.; Granelli-Piperno, A.; Murphy, D.P.; May, L.T.; Kupper, T.S.; Sehgal, P.B.; Gottlieb, A.B. Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proc. Natl. Acad. Sci. USA 1989, 86, 6367–6371. [Google Scholar] [CrossRef] [Green Version]
- Löwa, A.; Graff, P.; Kaessmeyer, S.; Hedtrich, S. Fibroblasts from atopic dermatitis patients trigger inflammatory processes and hyperproliferation in human skin equivalents. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 262. [Google Scholar] [CrossRef]
- Chan, B.C.L.; Lam, C.W.K.; Tam, L.-S.; Wong, C.K. IL33: Roles in Allergic Inflammation and Therapeutic Perspectives. Front. Immunol. 2019, 10, 364. [Google Scholar] [CrossRef] [Green Version]
- Smola, H.; Thiekötter, G.; Fusenig, N.E. Mutual induction of growth factor gene expression by epidermal-dermal cell interaction. J. Cell Biol. 1993, 122, 417–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas-Szabowski, N.; Shimotoyodome, A.; Fusenig, N. Keratinocyte growth regulation in fibroblast cocultures via a double paracrine mechanism. J. Cell Sci. 1999, 112, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; Gallo, R.L. The Role of the Skin Microbiome in Atopic Dermatitis. Curr. Allergy Asthma Rep. 2015, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Buddenkotte, J.; Steinhoff, M. Pathophysiology and therapy of pruritus in allergic and atopic diseases. Allergy 2010, 65, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.-H.; Oh, S.Y.; Lu, J.; Lou, H.; Myers, A.C.; Zhu, Z.; Zheng, T. TRPA1-Dependent Pruritus in IL-13–Induced Chronic Atopic Dermatitis. J. Immunol. 2013, 191, 5371–5382. [Google Scholar] [CrossRef] [Green Version]
- Indra, A.K. Epidermal TSLP: A trigger factor for pathogenesis of atopic dermatitis. Expert Rev. Proteom. 2013, 10, 309–311. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.Y. New Insights into Atopic Dermatitis: Role of Skin Barrier and Immune Dysregulation. Allergol. Int. 2013, 62, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Martel, B.C.; Lovato, P.; Bäumer, W.; Olivry, T. Translational Animal Models of Atopic Dermatitis for Preclinical Studies. Yale J. Biol. Med. 2017, 90, 389–402. [Google Scholar]
- Kabashima, K.; Nomura, T. Revisiting murine models for atopic dermatitis and psoriasis with multipolar cytokine axes. Curr. Opin. Immunol. 2017, 48, 99–107. [Google Scholar] [CrossRef]
- Guerrero-Aspizua, S.; Carretero, M.; Conti, C.J.; Del Río, M. The importance of immunity in the development of reliable animal models for psoriasis and atopic dermatitis. Immunol. Cell Biol. 2020, 98, 626–638. [Google Scholar] [CrossRef]
- Jin, H.; He, R.; Oyoshi, M.; Geha, R.S. Animal models of atopic dermatitis. J. Investig. Dermatol. 2009, 129, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Hener, P.; Zhang, Z.; Kato, S.; Metzger, D.; Chambon, P. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc. Natl. Acad. Sci. USA 2006, 103, 11736–11741. [Google Scholar] [CrossRef] [Green Version]
- Mestas, J.; Hughes, C.C.W. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Saini, S.; Pansare, M. New Insights and Treatments in Atopic Dermatitis. Pediatr. Clin. N. Am. 2019, 66, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Eichenfield, L.F.; Tom, W.L.; Chamlin, S.L.; Feldman, S.R.; Hanifin, J.M.; Simpson, E.L.; Berger, T.G.; Bergman, J.N.; Cohen, D.E.; Cooper, K.D.; et al. Guidelines of care for the management of atopic dermatitis: Section Diagnosis and assessment of atopic dermatitis. J. Am. Acad. Dermatol. 2014, 70, 338–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichenfield, L.F.; Tom, W.L.; Berger, T.G.; Krol, A.; Paller, A.S.; Schwarzenberger, K.; Bergman, J.N.; Chamlin, S.L.; Cohen, D.E.; Cooper, K.D.; et al. Guidelines of care for the management of atopic dermatitis: Section 2. Management and treatment of atopic dermatitis with topical therapies. J. Am. Acad. Dermatol. 2014, 71, 116–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidbury, R.; Tom, W.L.; Bergman, J.N.; Cooper, K.D.; Silverman, R.A.; Berger, T.G.; Chamlin, S.L.; Cohen, D.E.; Cordoro, K.M.; Davis, D.M.; et al. Guidelines of care for the management of atopic dermatitis: Section 4. Prevention of disease flares and use of adjunctive therapies and approaches. J. Am. Acad. Dermatol. 2014, 71, 1218–1233. [Google Scholar] [CrossRef] [Green Version]
- Sidbury, R.; Davis, D.M.; Cohen, D.E.; Cordoro, K.M.; Berger, T.G.; Bergman, J.N.; Chamlin, S.L.; Cooper, K.D.; Feldman, S.R.; Hanifin, J.M.; et al. Guidelines of care for the management of atopic dermatitis: Section 3. Management and treatment with phototherapy and systemic agents. J. Am. Acad. Dermatol. 2014, 71, 327–349. [Google Scholar] [CrossRef] [Green Version]
- Ersser, S.; Farasat, H.; Jackson, K.; Dennis, H.; Sheppard, Z.; More, A. A service evaluation of the Eczema Education Programe: An analysis of child, parent and service impact outcomes. Br. J. Dermatol. 2013, 169, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Maliyar, K.; Sibbald, C.; Pope, E.; Sibbald, R.G. Diagnosis and Management of Atopic Dermatitis: A Review. Adv. Ski. Wound Care 2018, 31, 538–550. [Google Scholar] [CrossRef]
- LeBovidge, J.S.; Elverson, W.; Timmons, K.G.; Hawryluk, E.; Rea, C.; Lee, M.; Schneider, L.C. Multidisciplinary interventions in the management of atopic dermatitis. J. Allergy Clin. Immunol. 2016, 138, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sher, L.G.; Chang, J.; Patel, I.B.; Balkrishnan, R.; Fleischer, A.B., Jr. Relieving the Pruritus of Atopic Dermatitis: A Meta-analysis. Acta Derm. Venereol. 2012, 92, 455–461. [Google Scholar] [CrossRef]
- Lucky, A.W.; Leach, A.D.; Laskarzewski, P.; Wenck, H. Use of an Emollient as a Steroid-Sparing Agent in the Treatment of Mild to Moderate Atopic Dermatitis in Children. Pediatr. Dermatol. 1997, 14, 321–324. [Google Scholar] [CrossRef]
- Hoare, C.; Po, A.L.W.; Williams, H. Williams Systematic review of treatments for atopic eczema. Health Technol. Assess. 2000, 4, 1–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, J.; Von Kobyletzki, L.; Svensson, Å.; Apfelbacher, C. Efficacy and tolerability of proactive treatment with topical corticosteroids and calcineurin inhibitors for atopic eczema: Systematic review and meta-analysis of randomized controlled trials. Br. J. Dermatol. 2010, 164, 415–428. [Google Scholar] [CrossRef]
- Kang, S.; Lucky, A.W.; Pariser, D.; Lawrence, I.; Hanifin, J.M. Long-term safety and efficacy of tacrolimus ointment for the treatment of atopic dermatitis in children. J. Am. Acad. Dermatol. 2001, 44, S58–S64. [Google Scholar] [CrossRef] [PubMed]
- Reitamo, S.; Wollenberg, A.; Schöpf, E.; Perrot, J.L.; Marks, R.; Ruzicka, T.; Christophers, E.; Kapp, A.; Lahfa, M.; Rubins, A.; et al. Safety and Efficacy of 1 Year of Tacrolimus Ointment Monotherapy in Adults with Atopic Dermatitis. Arch. Dermatol. 2000, 136, 999–1006. [Google Scholar] [CrossRef] [Green Version]
- Lübbe, J.; Friedlander, S.F.; Cribier, B.; Morren, M.-A.; García-Díez, A.; Gelmetti, C.; Hofmann, H.; Houwing, R.H.; Kownacki, S.; Langley, R.G.B.; et al. Safety, Efficacy, and Dosage of 1% Pimecrolimus Cream for the Treatment of Atopic Dermatitis in Daily Practice. Am. J. Clin. Dermatol. 2006, 7, 121–131. [Google Scholar] [CrossRef]
- Margolis, D.J.; Abuabara, K.; Hoffstad, O.J.; Wan, J.; Raimondo, D.; Bilker, W.B. Association Between Malignancy and Topical Use of Pimecrolimus. JAMA Dermatol. 2015, 151, 594–599. [Google Scholar] [CrossRef] [Green Version]
- Pelucchi, C.; Chatenoud, L.; Turati, F.; Galeone, C.; Moja, L.; Bach, J.F.; La Vecchia, C. Probiotics supplementation during pregnancy or infancy for the prevention of atopic dermatitis: A meta-analysis. Epidemiology 2012, 23, 402–414. [Google Scholar] [CrossRef]
- Doege, K.; Grajecki, D.; Zyriax, B.-C.; Detinkina, E.; Zu Eulenburg, C.; Bühling, K.J. Impact of maternal supplementation with probiotics during pregnancy on atopic eczema in childhood—A meta-analysis. Br. J. Nutr. 2011, 107, 1–6. [Google Scholar] [CrossRef]
- Panduru, M.; Panduru, N.M.; Sălăvăstru, C.M.; Tiplica, G.-S. Probiotics and primary prevention of atopic dermatitis: A meta-analysis of randomized controlled studies. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 232–242. [Google Scholar] [CrossRef]
- Klein, P.A.; Clark, R.A.F. An Evidence-Based Review of the Efficacy of Antihistamines in Relieving Pruritus in Atopic Dermatitis. Arch. Dermatol. 1999, 135, 1522–1525. [Google Scholar] [CrossRef] [PubMed]
- Noh, A.L.S.M.; Yang, M.; Lee, J.-M.; Park, H.; Lee, D.-S.; Yim, M. Phosphodiesterase 3 and 4 negatively regulate receptor activator of nuclear factor-kappaB ligand-mediated osteoclast formation by prostaglandin E2. Biol. Pharm. Bull. 2009, 32, 1844–1848. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, J.L.; Íñiguez, M.A.; Muñoz-Fernández, M.; Fresno, M. Effect of phosphodiesterase 4 inhibitors on NFAT-dependent cyclooxygenase-2 expression in human T lymphocytes. Cell. Signal. 2004, 16, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Weinberg, J.M. AN-2728, a PDE4 inhibitor for the potential topical treatment of psoriasis and atopic dermatitis. Curr. Opin. Investig. Drugs 2009, 10, 1236–1242. [Google Scholar]
- Beck, L.A.; Thaçi, D.; Hamilton, J.D.; Graham, N.M.; Bieber, T.; Rocklin, R.; Ming, J.E.; Ren, H.; Kao, R.; Simpson, E.; et al. Dupilumab Treatment in Adults with Moderate-to-Severe Atopic Dermatitis. N. Engl. J. Med. 2014, 371, 130–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gooderham, M.J.; Hong, H.C.-H.; Eshtiaghi, P.; Papp, K.A. Dupilumab: A review of its use in the treatment of atopic dermatitis. J. Am. Acad. Dermatol. 2018, 78, S28–S36. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Blauvelt, A.; Eichenfield, L.F.; Paller, A.S.; Armstrong, A.W.; Drew, J.; Gopalan, R.; Simpson, E.L. Efficacy and safety of lebrikizumab, a high-affinity interleukin 13 inhibitor, in adults with moderate to severe atopic dermatitis: A phase 2b randomized clinical trial. JAMA Dermatol. 2020, 156, 411–420. [Google Scholar] [CrossRef] [Green Version]
- Deleanu, D.; Nedelea, I. Biological therapies for atopic dermatitis: An update (Review). Exp. Ther. Med. 2018, 17, 1061–1067. [Google Scholar] [CrossRef] [Green Version]
- Wollenberg, A.; Blauvelt, A.; Guttman-Yassky, E.; Worm, M.; Lynde, C.; Lacour, J.P.; Spelman, L.; Katoh, N.; Saeki, H.; Poulin, Y.; et al. Tralokinumab for moderate-to-severe atopic dermatitis: Results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase iii trials (ecztra 1 and ecztra 2). Br. J. Dermatol. 2021, 184, 437–449. [Google Scholar] [CrossRef]
- Kabashima, K.; Matsumura, T.; Komazaki, H.; Kawashima, M. Trial of Nemolizumab and Topical Agents for Atopic Dermatitis with Pruritus. N. Engl. J. Med. 2020, 383, 141–150. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Brunner, P.; Neumann, A.U.; Khattri, S.; Pavel, A.B.; Malik, K.; Singer, G.K.; Baum, D.; Gilleaudeau, P.; Sullivan-Whalen, M.; et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: A randomized, double-blind, phase 2a trial. J. Am. Acad. Dermatol. 2018, 78, 872–881.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratchataswan, T.; Banzon, T.M.; Thyssen, J.P.; Weidinger, S.; Guttman-Yassky, E.; Phipatanakul, W. Biologics for Treatment of Atopic Dermatitis: Current Status and Future Prospect. J. Allergy Clin. Immunol. Pract. 2021, 9, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Khattri, S.; Brunner, P.; Garcet, S.; Finney, R.; Cohen, S.R.; Oliva, M.; Dutt, R.; Fuentes-Duculan, J.; Zheng, X.; Li, X.; et al. Efficacy and safety of ustekinumab treatment in adults with moderate-to-severe atopic dermatitis. Exp. Dermatol. 2017, 26, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Xu, L.; Qiao, J.; Fang, H. A systematic review of ustekinumab in the treatment of atopic dermatitis. J. Dermatol. Treat. 2018, 29, 539–541. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Gutowska-Owsiak, D.; Hardman, C.S.; Westmoreland, M.; MacKenzie, T.; Cifuentes, L.; Waithe, D.; Lloyd-Lavery, A.; Marquette, A.; Londei, M.; et al. Proof-of-concept clinical trial of etokimab shows a key role for IL-33 in atopic dermatitis pathogenesis. Sci. Transl. Med. 2019, 11, eaax2945. [Google Scholar] [CrossRef]
- Chan, S.; Cornelius, V.; Cro, S.; Harper, J.I.; Lack, G. Treatment effect of omalizumab on severe pediatric atopic dermatitis: The adapt randomized clinical trial. JAMA Pediatr. 2020, 174, 29–37. [Google Scholar] [CrossRef]
- Boguniewicz, M. Biologics for Atopic Dermatitis. Immunol. Allergy Clin. N. Am. 2020, 40, 593–607. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Pavel, A.B.; Zhou, L.; Estrada, Y.D.; Zhang, N.; Xu, H.; Peng, X.; Wen, H.-C.; Govas, P.; Gudi, G.; et al. GBR 830, an anti-OX40, improves skin gene signatures and clinical scores in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 144, 482–493. [Google Scholar] [CrossRef] [Green Version]
- Simpson, E.L.; Parnes, J.R.; She, D.; Crouch, S.; Rees, W.; Mo, M.; van der Merwe, R. Tezepelumab, an anti–thymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: A randomized phase 2a clinical trial. J. Am. Acad. Dermatol. 2019, 80, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Parnes, J.R.; Sullivan, J.T.; Chen, L.; Dias, C. Pharmacokinetics, Safety, and Tolerability of Tezepelumab (AMG 157) in Healthy and Atopic Dermatitis Adult Subjects. Clin. Pharmacol. Ther. 2019, 106, 441–449. [Google Scholar] [CrossRef]
- Nakajima, S.; Kabata, H.; Kabashima, K.; Asano, K. Anti-TSLP antibodies: Targeting a master regulator of type 2 immune responses. Allergol. Int. 2020, 69, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Sroka-Tomaszewska, J.; Trzeciak, M. Molecular Mechanisms of Atopic Dermatitis Pathogenesis. Int. J. Mol. Sci. 2021, 22, 4130. [Google Scholar] [CrossRef] [PubMed]
- Bieber, T.; Thyssen, J.P.; Reich, K.; Simpson, E.L.; Katoh, N.; Torrelo, A.; De Bruin-Weller, M.; Thaci, D.; Bissonnette, R.; Gooderham, M.; et al. Pooled safety analysis of baricitinib in adult patients with atopic dermatitis from 8 randomized clinical trials. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Nezamololama, N.; Fieldhouse, K.; Metzger, K.; Gooderham, M. Emerging systemic JAK inhibitors in the treatment of atopic dermatitis: A review of abrocitinib, baricitinib, and upadacitinib. Drugs Context 2020, 9, 1–7. [Google Scholar] [CrossRef]
- Simpson, E.L.; Sinclair, R.; Forman, S.; Wollenberg, A.; Aschoff, R.; Cork, M.; Bieber, T.; Thyssen, J.P.; Yosipovitch, G.; Flohr, C.; et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet 2020, 396, 255–266. [Google Scholar] [CrossRef]
- Ferreira, S.; Guttman-Yassky, E.; Torres, T. Selective JAK1 Inhibitors for the Treatment of Atopic Dermatitis: Focus on Upadacitinib and Abrocitinib. Am. J. Clin. Dermatol. 2020, 21, 783–798. [Google Scholar] [CrossRef]
- Pavel, A.B.; Song, T.; Kim, H.-J.; Del Duca, E.; Krueger, J.G.; Dubin, C.; Peng, X.; Xu, H.; Zhang, N.; Estrada, Y.D.; et al. Oral Janus kinase/SYK inhibition (ASN002) suppresses inflammation and improves epidermal barrier markers in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 144, 1011–1024. [Google Scholar] [CrossRef] [Green Version]
- Cartron, A.M.; Nguyen, T.H.; Roh, Y.S.; Kwatra, M.M.; Kwatra, S.G. Janus kinase inhibitors for atopic dermatitis: A promising treatment modality. Clin. Exp. Dermatol. 2021. [Google Scholar] [CrossRef]
- Werfel, T.; Layton, G.; Yeadon, M.; Whitlock, L.; Osterloh, I.; Jimenez, P.; Liu, W.; Lynch, V.; Asher, A.; Tsianakas, A.; et al. Efficacy and safety of the histamine H4 receptor antagonist ZPL-3893787 in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 1830–1837. [Google Scholar] [CrossRef] [PubMed]
- Ständer, S.; Spellman, M.C.; Kwon, P.; Yosipovitch, G. The NK1 receptor antagonist serlopitant for treatment of chronic pruritus. Expert Opin. Investig. Drugs 2019, 28, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Andrade, A.; Kuah, C.Y.; Martin-Lopez, J.E.; Chua, S.; Shpadaruk, V.; Sanclemente, G.; Franco, J.V. Interventions for chronic pruritus of unknown origin. Cochrane Database Syst. Rev. 2020, 1, CD013128. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.; Xiao, C.; Kaden, A.; Brzezynski, J.; Mohrman, M.; Wang, J.; Smieszek, S.; Przychodzen, B.; Ständer, S.; Polymeropoulos, C.; et al. Neurokinin-1 receptor antagonist tradipitant has mixed effects on itch in atopic dermatitis: Results from EPIONE, a randomized clinical trial. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 338–340. [Google Scholar] [CrossRef]
- Saporito, R.C.; Cohen, D.J. Apremilast Use for Moderate-to-Severe Atopic Dermatitis in Pediatric Patients. Case Rep. Dermatol. 2016, 8, 179–184. [Google Scholar] [CrossRef]
- Samrao, A.; Berry, T.M.; Goreshi, R.; Simpson, E.L. A Pilot Study of an Oral Phosphodiesterase Inhibitor (Apremilast) for Atopic Dermatitis in Adults. Arch. Dermatol. 2012, 148, 890–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Koo, J.; Berger, T. UVB phototherapy and skin cancer risk: A review of the literature. Int. J. Dermatol. 2005, 44, 355–360. [Google Scholar] [CrossRef]
- Der-Petrossian, M.; Seeber, A.; Hönigsmann, H.; Tanew, A. Half-side comparison study on the efficacy of 8-methoxypsoralen bath-PUVA versus narrow-band ultraviolet B phototherapy in patients with severe chronic atopic dermatitis. Br. J. Dermatol. 2000, 142, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Patrizi, A.; Raone, B.; Ravaioli, G.M. Management of atopic dermatitis: Safety and efficacy of phototherapy. Clin. Cosmet. Investig. Dermatol. 2015, 8, 511–520. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
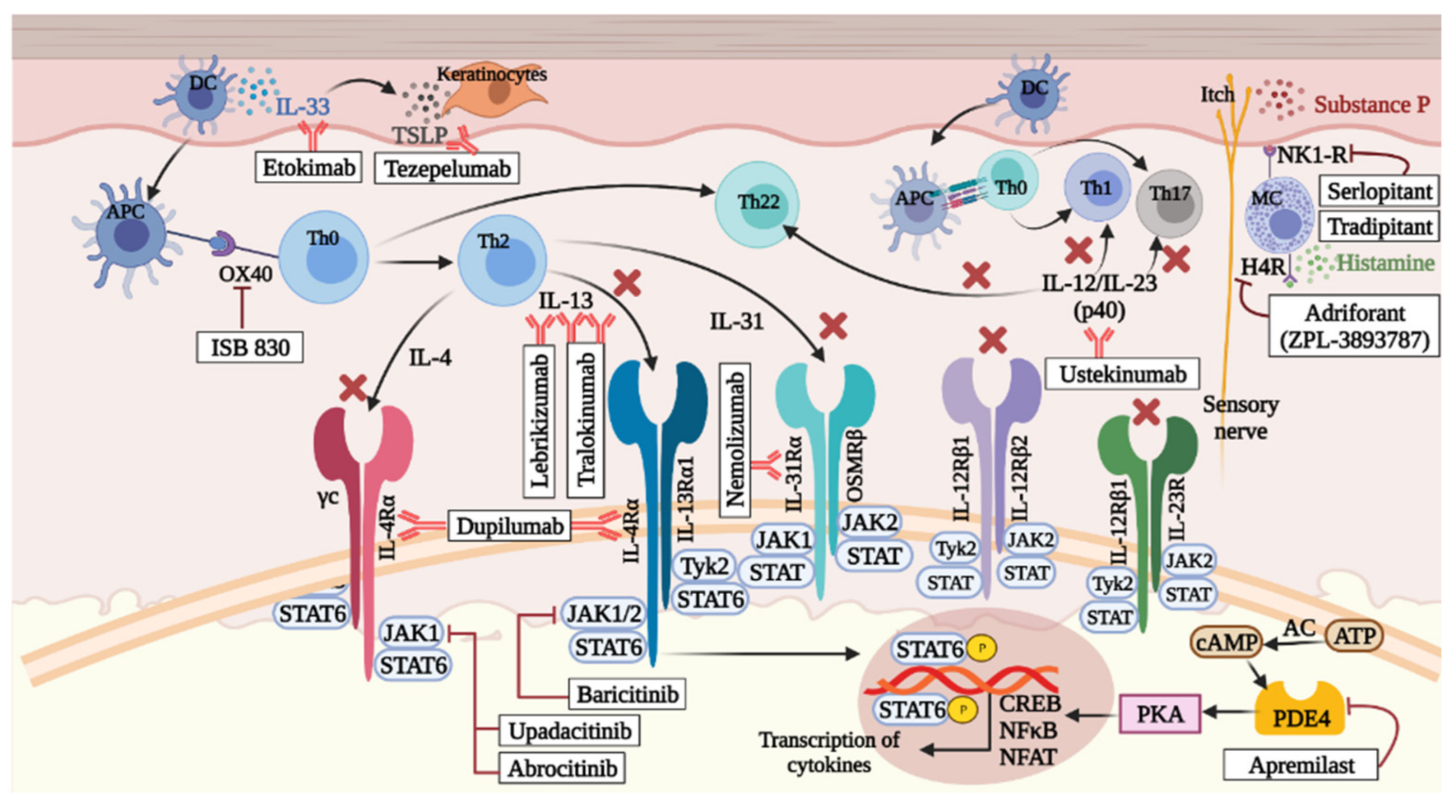
| Therapeutic Target | Drug Name | Mechanism of Action | AD Severity | Route of Administration | Status | Trial Identifier |
|---|---|---|---|---|---|---|
| IL-4Rα | Dupilumab | Inhibits the IL-4/IL-13 signaling pathway by blocking the shared IL-4 receptor-α subunit, thus reducing the Th2 response. | Moderate to severe | Subcutaneous injections | FDA approved in 2017 | Phase II NCT03861455 NCT03346434 Phase III NCT02612454 NCT01949311 Phase IV NCT03667014 NCT03389893 NCT03293030 NCT04447417 NCT04033367 |
| IL-13 | Lebrikizumab | Inhibits IL-13 by preventing the formation of the IL-13Rα1/IL-4Rα heterodimer. | Moderate to severe | Subcutaneous injections | Completed | Phase III NCT04146363 NCT04178967 NCT04392154 NCT04250350 NCT04250337 |
| IL-13 | Tralokinumab | Binds with high affinity to IL-13, preventing its interaction with its receptors and succeeding signaling pathways. | Moderate to severe | Subcutaneous injections | Completed | Phase III NCT03587805 NCT03761537 NCT03526861 |
| IL-31 | Nemolizumab | Inhibits IL-31 by binding to its receptor α subunit (IL-31α). | Moderate to severe | Subcutaneous injections | Completed | Phase II NCT03921411 NCT04365387 Phase III NCT03985943 NCT03989349 NCT03989206 |
| IL-22 | Fezakinumab | Blocks IL-22 involved in skin hyperplasia and skin barrier dysfunction. | Moderate to severe | Intravenous injections | Completed | NCT01941537 |
| IL-12/IL-23 | Ustekinumab | Blocks IL-12 and IL-23 by targeting their common p40 subunit, thus inhibiting the Th1 and Th17/Th22 responses | Moderate to severe | Subcutaneous injections | Completed | NCT01806662 |
| IL-33 | Etokimab | Blocks IL-33 and hence effectively inhibits neutrophil migration. | Moderate to severe | Intravenous injections | Ongoing | NCT03533751 |
| IgE | Omalizumab | Binds to the Fc (CH3 domain) of IgE molecules and prevents its interaction with its high affinity receptor FcεRI on mast cells and basophils, inhibiting the process of mast degranulation and release of inflammatory mediators. | Moderate to severe pediatric | Intravenous injections | Completed | NCT02300701 |
| OX40 | ISB 830 | Binds to OX40 (CD135), a co-stimulatory molecule expressed on T cells, and prevents its interaction with OX40L (CD252) expressed on activated antigen presenting cells, thus failing to potentiate the T cell responses triggered through T cell receptors. | Moderate to severe | Intravenous injections | Completed | Phase IIb NCT03568162 |
| TSLP | Tezepelumab | Binds to TSLP and prevents its interaction with its receptor hence blocking all downstream events associated with immune-modulating proteins and Th2 cytokines. | Moderate to severe | Subcutaneous injections | Completed | Phase IIb NCT02525094 |
| Therapeutic Target | Drug Name | Mechanism of Action | AD Severity | Route of Administration | Current Status of Study | Trial Identifier |
|---|---|---|---|---|---|---|
| JAK1/2 | Baricitinib | Inhibits JAK1 and JAK2 in the JAK-STAT signaling pathway, thus exerting an immunomodulatory and anti-proliferative effects. This is achieved by inhibiting immune cell function by detaching cytokine effects from the cells and | Moderate to severe | Oral | Ongoing phase III trials | Phase II NCT02576938 phase III NCT03334396 NCT03334422 NCT03428100 NCT03435081 NCT03733301 NCT03334435 NCT03559270 |
| JAK1 | Upadacitinib | Selectively inhibits JAK1 of the JAK-STAT pathway, thus blocking downstream cellular processes contributing to inflammatory conditions. | Moderate to severe | Oral | Ongoing phase III trials | Phase II NCT02925117 Phase III NCT03569293 NCT03568318 NCT03661138 NCT04195698 NCT03607422 NCT03738397 |
| JAK1 | Abrocitinib | Selectively inhibits JAK1 of the JAK-STAT signaling pathway, thus preventing activation of JAK1 containing heterodimeric receptors and thereby inhibiting downstream Th2 differentiation and itch while avoiding undesirable effects of JAK2 inhibition such as neutropenia and anemia. | Moderate to severe | Oral | Ongoing phase III trials | Phase II NCT02780167 Phase III NCT04345367 NCT03422822 NCT03627767 NCT03796676 NCT03720470 NCT03575871 NCT03349060 |
| JAK/SYK | Gusacitinib (ASN002) | Targets a broad range of cytokine axes through dual inhibition of JAK and SYK, a non-receptor tyrosine kinase that inhibits terminal differentiation of keratinocytes and is involved in regulation of B cell and dendritic cell. differentiation and Th17/IL-17 signaling | Moderate to severe | Oral | Completed phase II trials | Phase Ib NCT03139981 Phase II NCT03728504 NCT03654755 NCT03531957 |
| H4R | Adriforant (ZPL-3893787) | Highly potent;selectively inhibits histamine H4 receptor, thus blocking associated chemotactic and inflammatory responses in immune cells such as eosinophils, dendritic cells, and mast cells. | Moderate to severe | Oral | Completed | Phase II NCT02424253 |
| NK1-R | Serlopitant | Binds to neurokinin 1 receptor (NK1R) expressed on keratinocytes, mast cells, and fibroblasts and prevents interaction of its ligand, substance P (SP). This blocks the itch signals transmitted through the non-histaminergic pathway. | Chronic pruritus | Oral | Terminated | Phase II NCT03841331 NCT03343639 NCT02975206 NCT02196324 NCT01951274 NCT03282591 NCT00835718 NCT00290563 Phase III NCT03540160 NCT03677401 NCT03546816 |
| NK1-R | Tradipitant (VLY-686) | Inhibits the NK1 receptor and blocks SP- mediated itch signals. | AD with significant pruritus | Oral | Completed | Phase II NCT02004041 Phase III NCT03568331 |
| PDE4 | Apremilast | Inhibits phosphodiesterase4 (PDE4) hence preventing the hydrolysis of cAMP. This results in an increase in cAMP levels in immune and non-immune cells and helps to modulate the expression of various pro-inflammatory mediators. | Moderate to severe | Oral | Completed | Phase II NCT00931242 NCT02087943 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kader, H.A.; Azeem, M.; Jwayed, S.A.; Al-Shehhi, A.; Tabassum, A.; Ayoub, M.A.; Hetta, H.F.; Waheed, Y.; Iratni, R.; Al-Dhaheri, A.; et al. Current Insights into Immunology and Novel Therapeutics of Atopic Dermatitis. Cells 2021, 10, 1392. https://doi.org/10.3390/cells10061392
Kader HA, Azeem M, Jwayed SA, Al-Shehhi A, Tabassum A, Ayoub MA, Hetta HF, Waheed Y, Iratni R, Al-Dhaheri A, et al. Current Insights into Immunology and Novel Therapeutics of Atopic Dermatitis. Cells. 2021; 10(6):1392. https://doi.org/10.3390/cells10061392
Chicago/Turabian StyleKader, Hidaya A., Muhammad Azeem, Suhib A. Jwayed, Aaesha Al-Shehhi, Attia Tabassum, Mohammed Akli Ayoub, Helal F. Hetta, Yasir Waheed, Rabah Iratni, Ahmed Al-Dhaheri, and et al. 2021. "Current Insights into Immunology and Novel Therapeutics of Atopic Dermatitis" Cells 10, no. 6: 1392. https://doi.org/10.3390/cells10061392