
15-Deoxy-Δ12,14-prostaglandin J2 Upregulates VEGF Expression via NRF2 and Heme Oxygenase-1 in Human Breast Cancer Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical and Biochemical Reagents
2.2. Human Tissue Samples
2.3. Immunohistochemistry
2.4. Measurement of 15d-PGJ2
2.5. Cell Culture
2.6. Preparation of Nuclear Proteins
2.7. Western Blot Analysis
2.8. Electrophoretic Mobility Shift Assay (EMSA) for Measuring Human NRF2-ARE Binding Activity
2.9. Immunofluorescent Analysis
2.10. Transient Transfection and the Luciferase Reporter Gene Assay
2.11. Chromatin Immunoprecipitation (ChIP) Assay
2.12. Enzyme-Linked Immunosorbent Assay (ELISA)
2.13. Aortic Ring Assay
2.14. Ab Initio Calculation
2.15. Immunoprecipitation
2.16. Tube Formation Assay
2.17. RNA Isolation and Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.18. Measurement of HO Activity
2.19. Illumina Gene Expression Microarray and Data Analysis
2.20. The Cancer Genome Atlas (TCGA) Data Analysis
2.21. Statistical Analysis
3. Results
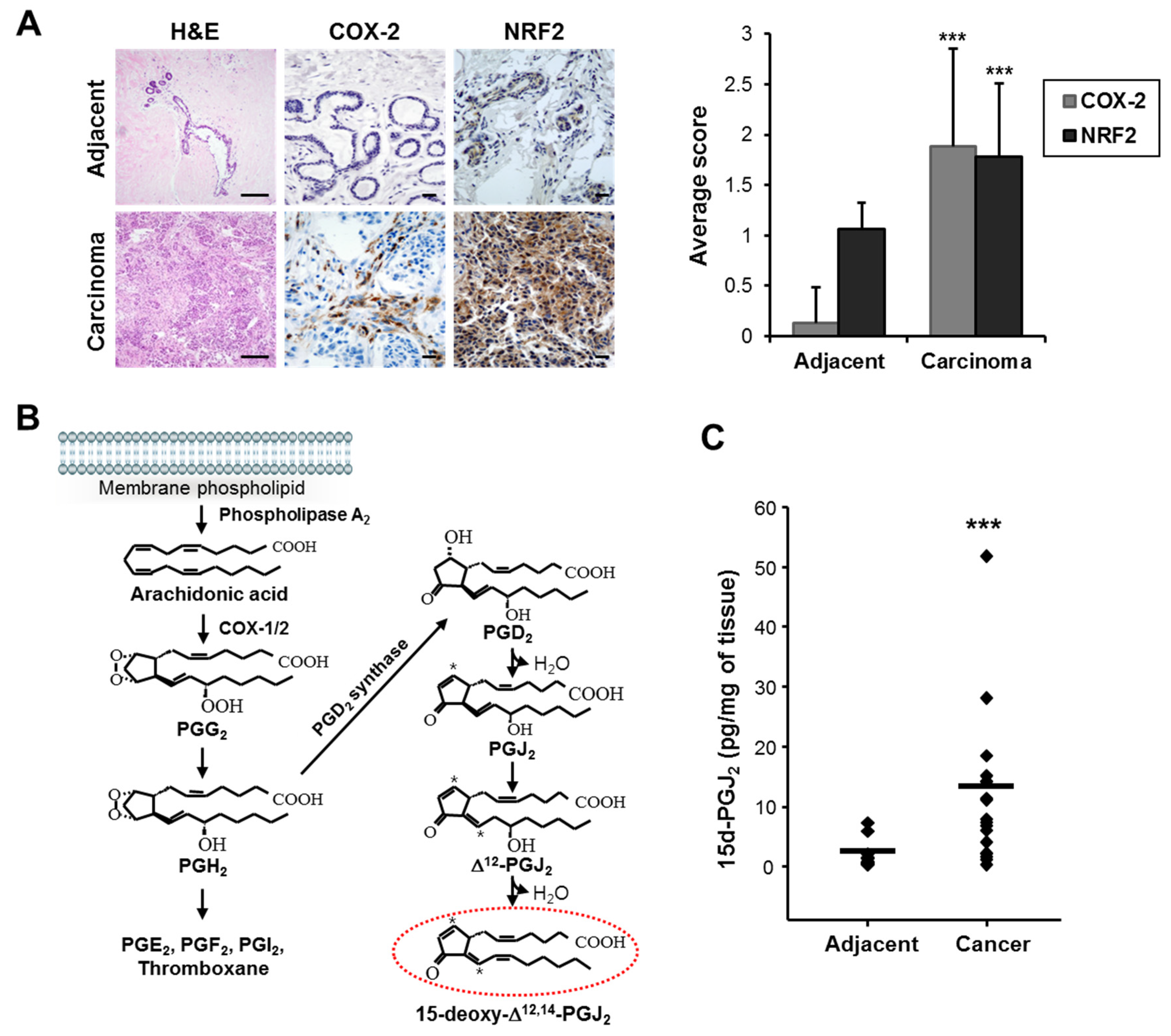
3.1. COX-2 Expression and 15-Deoxy-Δ12,14-prostaglandin J2 Production Are Elevated in Human Breast Cancer
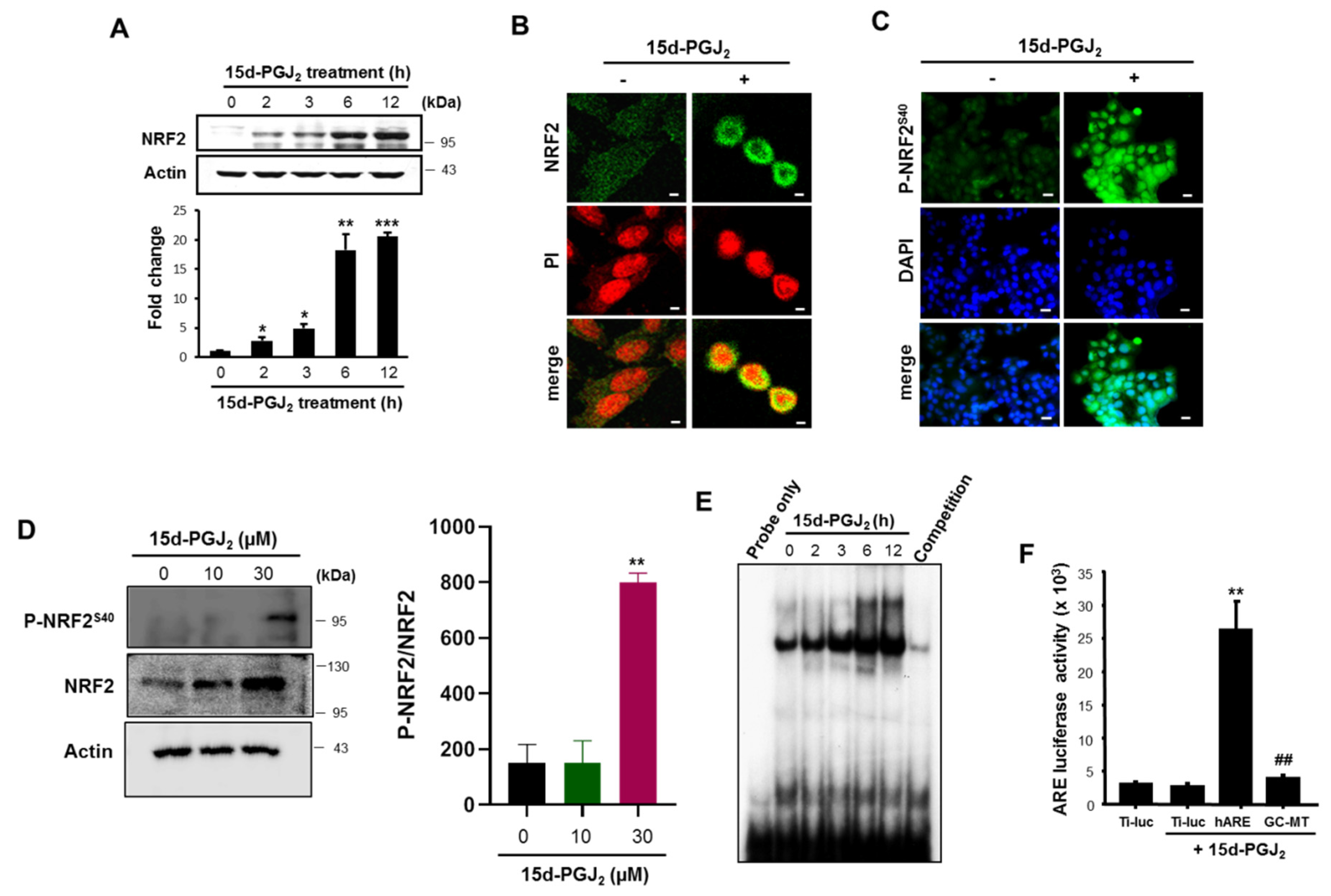
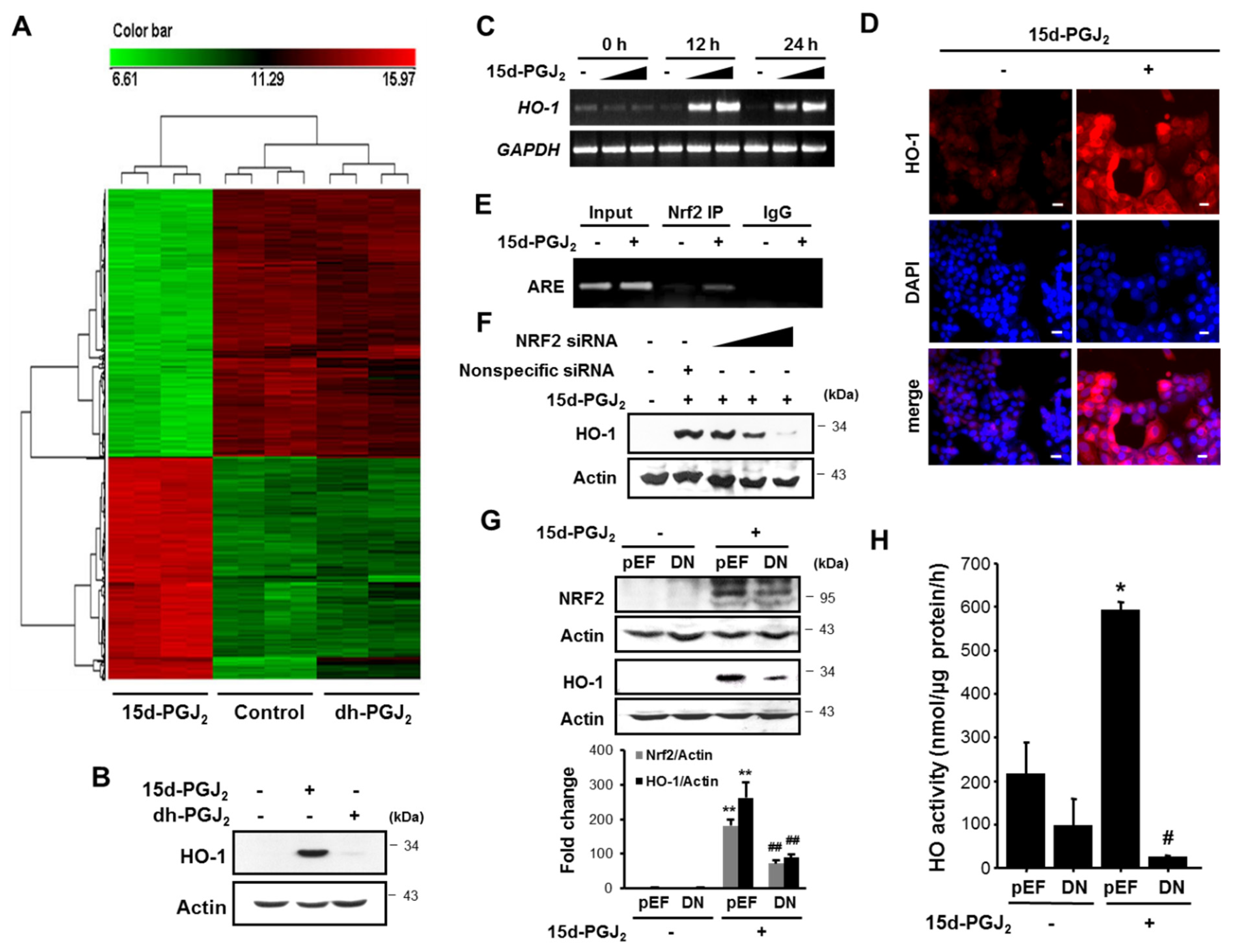
3.2. 15d-PGJ2 Increases the Nuclear Accumulation of NRF2 and Its Binding to ARE
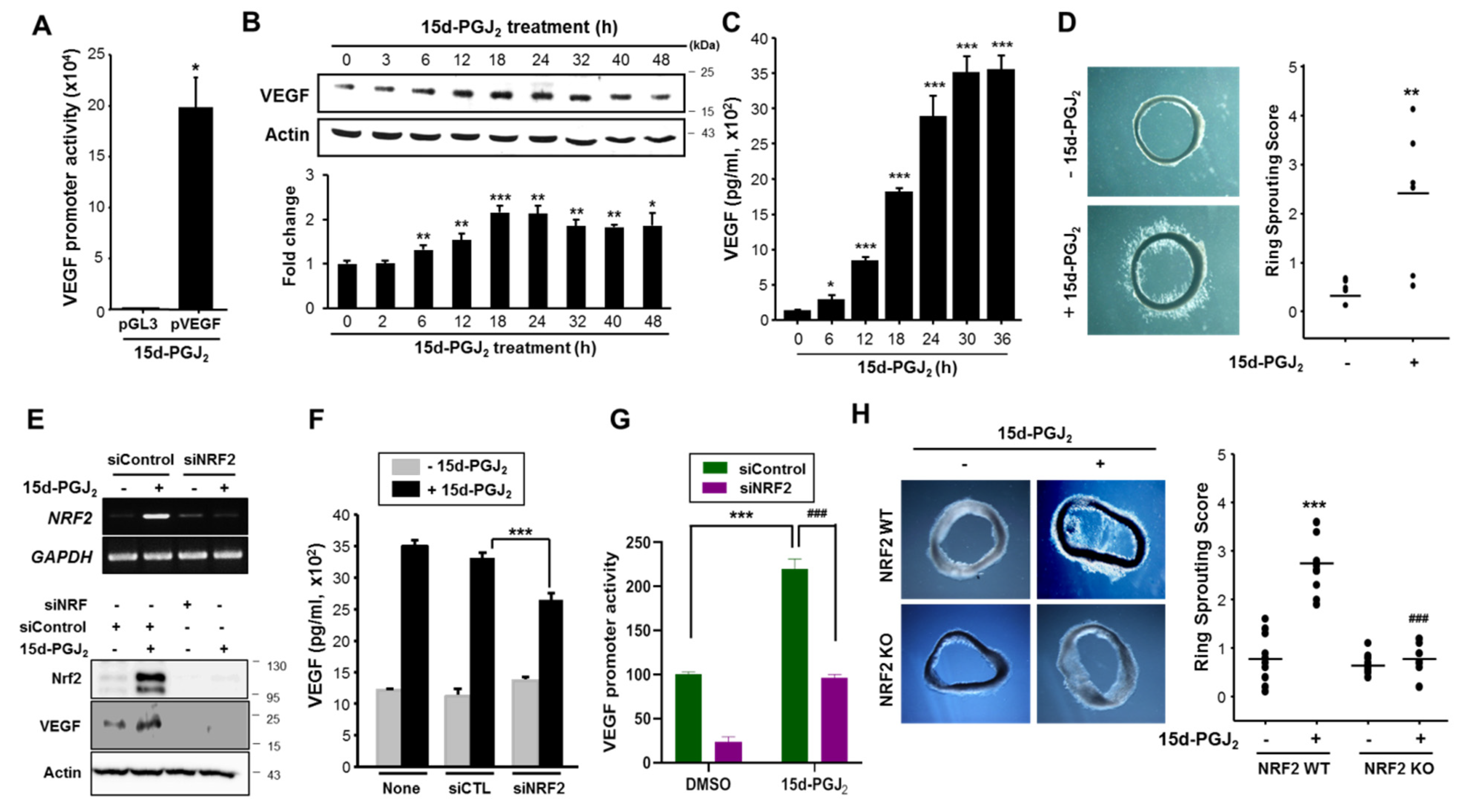
3.3. 15d-PGJ2 Induces VEGF Expression and Angiogenesis through NRF2 Activation
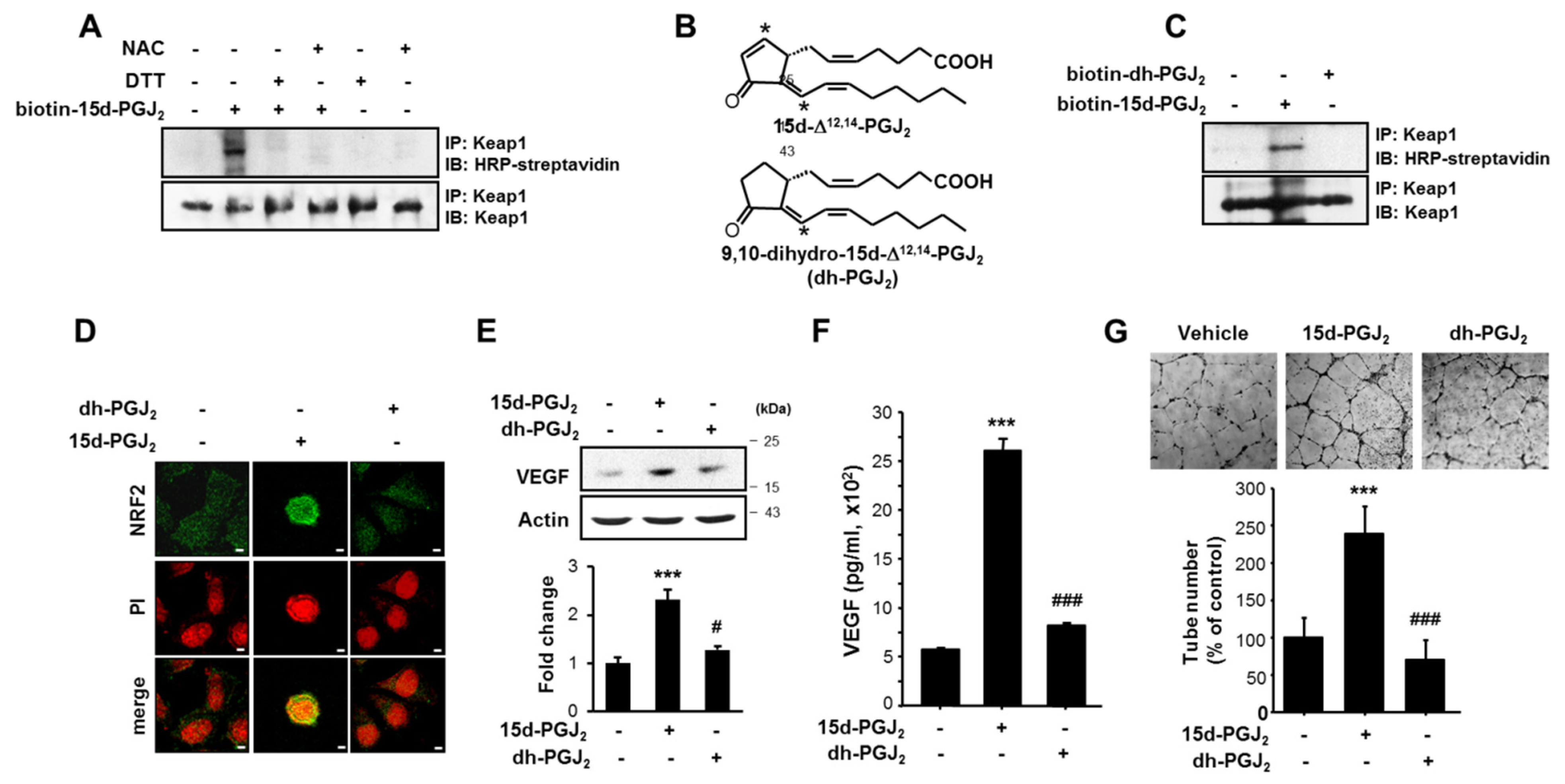
3.4. The α,β-Unsaturated Carbonyl Moiety Present in the Cyclopentenone Ring of 15d-PGJ2 Is Essential for Its Induction of NRF2 Activation, VEGF Upregulation, and Angiogenesis
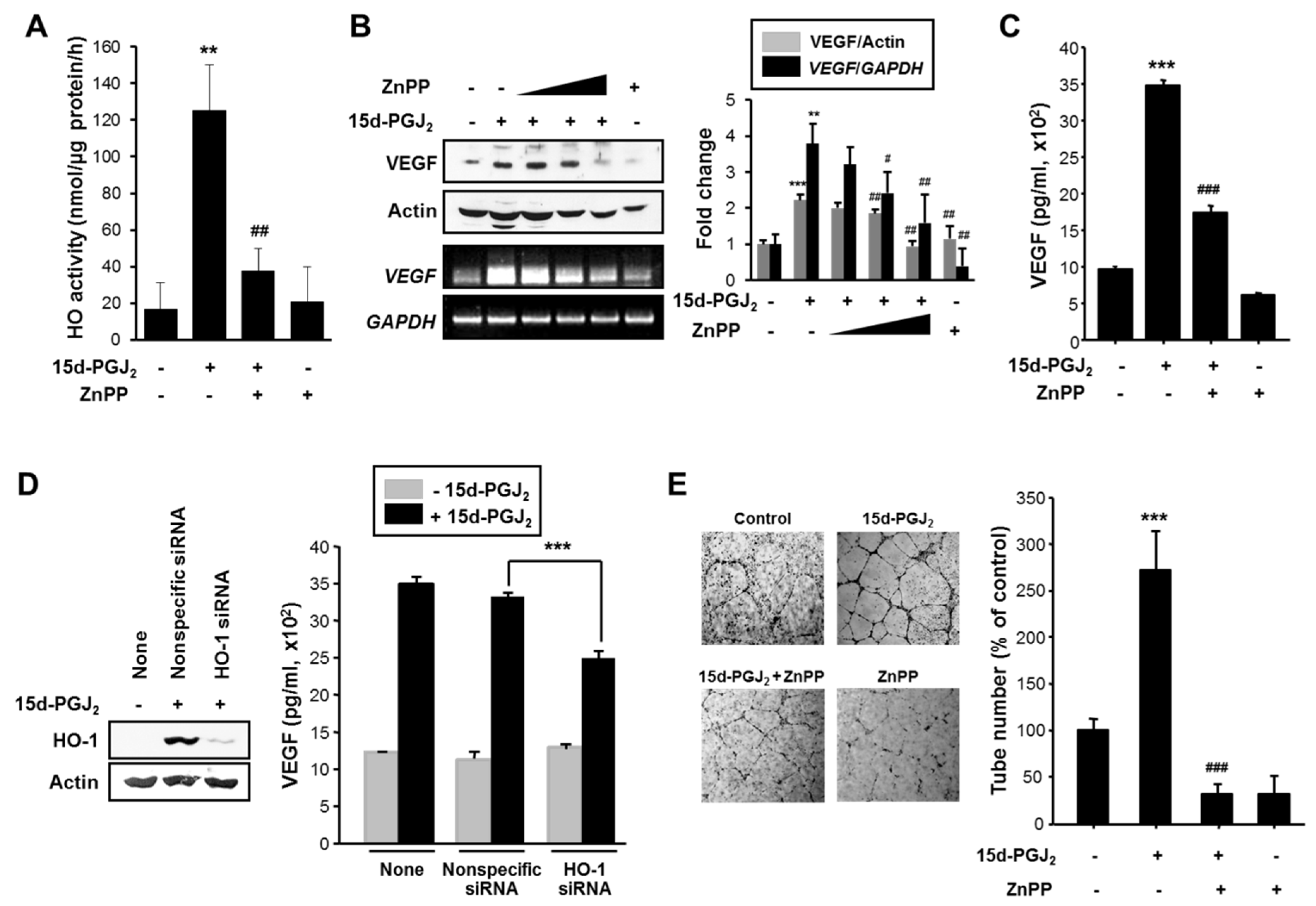
3.5. HO-1 Induced by 15d-PGJ2 Mediates VEGF Expression and Angiogenesis in Human Breast Cancer
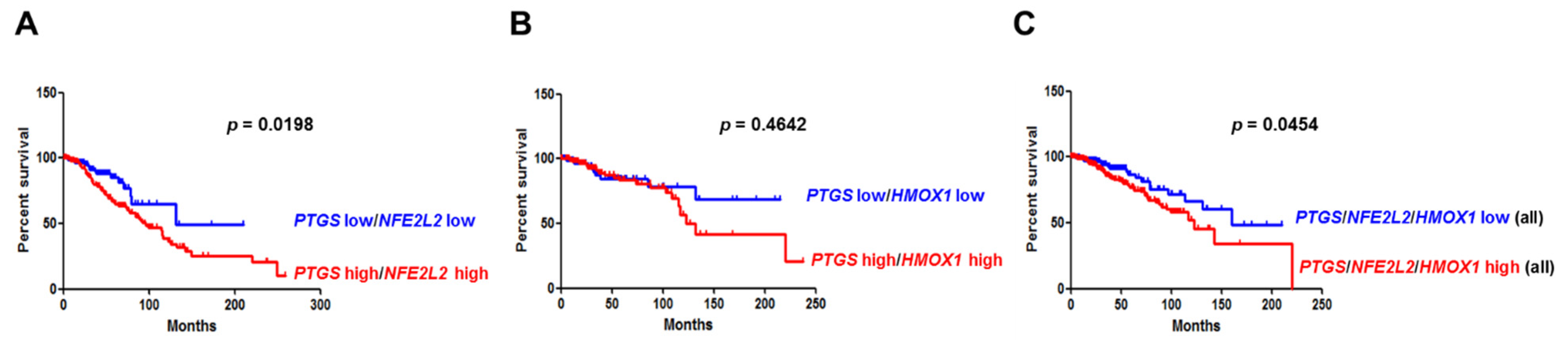
3.6. Overexpression of COX-2 and NRF2 or COX-2 and HO-1 Correlates with Poor Clinical Outcomes in Breast Cancer Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From metabolism to molecular therapy. Am. J. Respir. Cell Mol. Biol. 2009, 41, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Balla, J.; Otterbein, L.; Smith, R.N.; Brouard, S.; Lin, Y.; Csizmadia, E.; Sevigny, J.; Robson, S.C.; Vercellotti, G.; et al. Carbon monoxide generated by heme oxygenase-1 suppresses the rejection of mouse-to-rat cardiac transplants. J. Immunol. 2001, 166, 4185–4194. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Cadetg, P.; Ruf, R.; Mazzucchelli, L.; Ferrari, P.; Redaelli, C.A. Heme oxygenase-1 attenuates ischemia/reperfusion-induced apoptosis and improves survival in rat renal allografts. Kidney Int. 2003, 63, 1564–1573. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Qian, J.M. Cytoprotective role of heme oxygenase-1 in liver ischemia reperfusion injury. Int. J. Clin. Exp. Med. 2015, 8, 19867–19873. [Google Scholar]
- Liu, L.; Wu, Y.; Bian, C.; Nisar, M.F.; Wang, M.; Hu, X.; Diao, Q.; Nian, W.; Wang, E.; Xu, W.; et al. Heme oxygenase 1 facilitates cell proliferation via the B-Raf-ERK signaling pathway in melanoma. Cell Commun. Signal. 2019, 17, 3. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.X.; Yan, F.; Xue, Q.; Wu, G.J.; Qin, W.J.; Wang, F.L.; Qin, J.; Tian, C.J.; Yuan, J.L. Heme oxygenase-1 is a predictive biomarker for therapeutic targeting of advanced clear cell renal cell carcinoma treated with sorafenib or sunitinib. OncoTargets Ther. 2015, 8, 2081–2088. [Google Scholar]
- Schacter, B.A.; Kurz, P. Alterations in microsomal drug metabolism and heme oxygenase activity in isolated hepatic parenchymal and sinusoidal cells in Murphy-Sturm lymphosarcoma-bearing rats. Clin. Invest. Med. 1986, 9, 150–155. [Google Scholar] [PubMed]
- Maines, M.D.; Abrahamsson, P.A. Expression of heme oxygenase-1 (HSP32) in human prostate: Normal, hyperplastic, and tumor tissue distribution. Urology 1996, 47, 727–733. [Google Scholar] [CrossRef]
- Was, H.; Dulak, J.; Jozkowicz, A. Heme oxygenase-1 in tumor biology and therapy. Curr. Drug Targets 2010, 11, 1551–1570. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Sasahira, T.; Ohmori, H.; Fujii, K.; Kuniyasu, H. Inhibition of heme oxygenase-1 by zinc protoporphyrin IX reduces tumor growth of LL/2 lung cancer in C57BL mice. Int. J. Cancer 2007, 120, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Sunamura, M.; Duda, D.G.; Ghattas, M.H.; Lozonschi, L.; Motoi, F.; Yamauchi, J.; Matsuno, S.; Shibahara, S.; Abraham, N.G. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis 2003, 6, 15–24. [Google Scholar] [CrossRef]
- Liu, Z.M.; Chen, G.G.; Ng, E.K.; Leung, W.K.; Sung, J.J.; Chung, S.C. Upregulation of heme oxygenase-1 and p21 confers resistance to apoptosis in human gastric cancer cells. Oncogene 2004, 23, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Was, H.; Cichon, T.; Smolarczyk, R.; Rudnicka, D.; Stopa, M.; Chevalier, C.; Leger, J.J.; Lackowska, B.; Grochot, A.; Bojkowska, K.; et al. Overexpression of heme oxygenase-1 in murine melanoma: Increased proliferation and viability of tumor cells, decreased survival of mice. Am. J. Pathol. 2006, 169, 2181–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 induction in cancer progression: A matter of cell adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Surh, Y.J.; Na, H.K.; Park, J.M.; Lee, H.N.; Kim, W.; Yoon, I.S.; Kim, D.D. 15-Deoxy-Δ12,14-prostaglandin J2, an electrophilic lipid mediator of anti-inflammatory and pro-resolving signaling. Biochem. Pharmacol. 2011, 82, 1335–1351. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.; Rattan, R.; Singh, A.K.; Singh, I. The 15-deoxy-Δ12,14-prostaglandin J2 inhibits the inflammatory response in primary rat astrocytes via down-regulating multiple steps in phosphatidylinositol 3-kinase-Akt-NF-κB-p300 pathway independent of peroxisome proliferator-activated receptor gamma. J. Immunol. 2004, 173, 5196–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Kapahi, P.; Natoli, G.; Takahashi, T.; Chen, Y.; Karin, M.; Santoro, M.G. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature 2000, 403, 103–108. [Google Scholar] [CrossRef]
- Millan, O.; Rico, D.; Peinado, H.; Zarich, N.; Stamatakis, K.; Perez-Sala, D.; Rojas, J.M.; Cano, A.; Bosca, L. Potentiation of tumor formation by topical administration of 15-deoxy-Δ12,14-prostaglandin J2 in a model of skin carcinogenesis. Carcinogenesis 2006, 27, 328–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinery, R.; Coffey, R.J.; Graves-Deal, R.; Kirkland, S.C.; Sanchez, S.C.; Zackert, W.E.; Oates, J.A.; Morrow, J.D. Prostaglandin J2 and 15-deoxy-Δ12,14-prostaglandin J2 induce proliferation of cyclooxygenase-depleted colorectal cancer cells. Cancer Res. 1999, 59, 2739–2746. [Google Scholar]
- Bussolati, B.; Mason, J.C. Dual role of VEGF-induced heme-oxygenase-1 in angiogenesis. Antioxid. Redox Signal. 2006, 8, 1153–1163. [Google Scholar] [CrossRef]
- Haslmayer, P.; Thalhammer, T.; Jager, W.; Aust, S.; Steiner, G.; Ensinger, C.; Obrist, P. The peroxisome proliferator-activated receptor gamma ligand 15-deoxy-Δ12,14-prostaglandin J2 induces vascular endothelial growth factor in the hormone-independent prostate cancer cell line PC 3 and the urinary bladder carcinoma cell line. Int. J. Oncol. 2002, 21, 915–920. [Google Scholar]
- Yamakawa, K.; Hosoi, M.; Koyama, H.; Tanaka, S.; Fukumoto, S.; Morii, H.; Nishizawa, Y. Peroxisome proliferator-activated receptor-gamma agonists increase vascular endothelial growth factor expression in human vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2000, 271, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Jozkowicz, A.; Huk, I.; Nigisch, A.; Weigel, G.; Weidinger, F.; Dulak, J. Effect of prostaglandin J2 on VEGF synthesis depends on the induction of heme oxygenase-1. Antioxid. Redox Signal. 2002, 4, 577–585. [Google Scholar] [CrossRef]
- Jozkowicz, A.; Dulak, J.; Piatkowska, E.; Placha, W.; Dembinska-Kiec, A. Ligands of peroxisome proliferator-activated receptor-gamma increase the generation of vascular endothelial growth factor in vascular smooth muscle cells and in macrophages. Acta Biochim. Pol. 2000, 47, 1147–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, M.; Itoh, H.; Tanaka, T.; Chun, T.H.; Doi, K.; Fukunaga, Y.; Sawada, N.; Yamshita, J.; Masatsugu, K.; Saito, T.; et al. Oxidized LDL regulates vascular endothelial growth factor expression in human macrophages and endothelial cells through activation of peroxisome proliferator-activated receptor-gamma. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 560–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.H.; Kim, D.H.; Na, H.K.; Surh, Y.J. Effects of cyclopentenone prostaglandins on the expression of heme oxygenase-1 in MCF-7 cells. Ann. NY Acad. Sci. 2004, 1030, 493–500. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimta, A.A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 activity in cancer development and progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [Green Version]
- Sicher, K.; Waterhouse, J.A. Evaluation of TNM classification of carcinoma of the breast. Br. J. Cancer 1973, 28, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Bang, H.Y.; Park, S.A.; Saeidi, S.; Na, H.K.; Surh, Y.J. Docosahexaenoic acid induces expression of heme oxygenase-1 and NAD(P)H:quinone oxidoreductase through activation of Nrf2 in human mammary epithelial cells. Molecules 2017, 22, 969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keum, Y.S.; Yu, S.; Chang, P.P.; Yuan, X.; Kim, J.H.; Xu, C.; Han, J.; Agarwal, A.; Kong, A.N. Mechanism of action of sulforaphane: Inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Res. 2006, 66, 8804–8813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, J.K.; Shin, Y.K.; Kim, S.H.; Surh, Y.J. Resveratrol inhibits phorbol ester-induced expression of COX-2 and activation of NF-κB in mouse skin by blocking IκB kinase activity. Carcinogenesis 2006, 27, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Foresti, R.; Intaglietta, M.; Winslow, R.M. NO-mediated activation of heme oxygenase: Endogenous cytoprotection against oxidative stress to endothelium. Am. J. Physiol. 1996, 270, H107–H114. [Google Scholar] [CrossRef]
- Goel, M.K.; Khanna, P.; Kishore, J. Understanding survival analysis: Kaplan-Meier estimate. Int. J. Ayurveda Res. 2010, 1, 274–278. [Google Scholar] [PubMed] [Green Version]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.H.; Surh, Y.J. 15-Deoxy-Δ12,14-prostaglandin J2 as a potential endogenous regulator of redox-sensitive transcription factors. Biochem. Pharmacol. 2006, 72, 1516–1528. [Google Scholar] [CrossRef] [PubMed]
- Okuda, A.; Imagawa, M.; Maeda, Y.; Sakai, M.; Muramatsu, M. Structural and functional analysis of an enhancer GPEI having a phorbol 12-O-tetradecanoate 13-acetate responsive element-like sequence found in the rat glutathione transferase P gene. J. Biol. Chem. 1989, 264, 16919–16926. [Google Scholar] [CrossRef]
- Favreau, L.V.; Pickett, C.B. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. Identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J. Biol. Chem. 1991, 266, 4556–4561. [Google Scholar] [CrossRef]
- Li, Y.; Jaiswal, A.K. Regulation of human NAD(P)H:quinone oxidoreductase gene. Role of AP1 binding site contained within human antioxidant response element. J. Biol. Chem. 1992, 267, 15097–15104. [Google Scholar] [CrossRef]
- Prestera, T.; Talalay, P.; Alam, J.; Ahn, Y.I.; Lee, P.J.; Choi, A.M. Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: Regulation by upstream antioxidant-responsive elements (ARE). Mol. Med. 1995, 1, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, R.T.; Wartman, M.A.; Bailey, H.H.; Gipp, J.J. Constitutive and β-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. J. Biol. Chem. 1997, 272, 7445–7454. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Kong, A.N. Dietary chemopreventive compounds and ARE/EpRE signaling. Free Radic. Biol. Med. 2004, 36, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Vreman, H.J.; Schulz, S.; Kalish, F.S.; Pierce, N.W.; Stevenson, D.K. In vitro inhibition of heme oxygenase isoenzymes by metalloporphyrins. J. Perinatol. 2011, 31 (Suppl. S1), S35–S41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakowska, M.; Dulak, J.; Jozkowicz, A. Heme oxygenase-1—More than the cytoprotection. Postepy Biochem. 2015, 61, 147–158. [Google Scholar]
- Kim, W.; Lee, H.N.; Jang, J.H.; Kim, S.H.; Lee, Y.H.; Hahn, Y.I.; Ngo, H.K.; Choi, Y.; Joe, Y.; Chung, H.T.; et al. 15-Deoxy-Δ12,14-prostaglandin J2 exerts proresolving effects through nuclear factor E2-related factor 2-induced expression of CD36 and heme oxygenase-1. Antioxid. Redox Signal. 2017, 27, 1412–1431. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Song, N.Y.; Kim, E.H.; Na, H.K.; Joe, Y.; Chung, H.T.; Surh, Y.J. 15-Deoxy-Δ12,14-prostaglandin J2 induces p53 expression through Nrf2-mediated upregulation of heme oxygenase-1 in human breast cancer cells. Free Radic. Res. 2014, 48, 1018–1027. [Google Scholar] [CrossRef]
- Na, H.K.; Surh, Y.J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic. Biol. Med. 2014, 67, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Numazawa, S.; Ishikawa, M.; Yoshida, A.; Tanaka, S.; Yoshida, T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am. J. Physiol. Cell Physiol. 2003, 285, C334–C342. [Google Scholar] [CrossRef] [Green Version]
- Shibata, T. 15-Deoxy-Δ12,14-prostaglandin J2 as an electrophilic mediator. Biosci. Biotechnol. Biochem. 2015, 79, 1044–1049. [Google Scholar] [CrossRef] [Green Version]
- Vunta, H.; Davis, F.; Palempalli, U.D.; Bhat, D.; Arner, R.J.; Thompson, J.T.; Peterson, D.G.; Reddy, C.C.; Prabhu, K.S. The anti-inflammatory effects of selenium are mediated through 15-deoxy-Δ12,14-prostaglandin J2 in macrophages. J. Biol. Chem. 2007, 282, 17964–17973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straus, D.S.; Pascual, G.; Li, M.; Welch, J.S.; Ricote, M.; Hsiang, C.H.; Sengchanthalangsy, L.L.; Ghosh, G.; Glass, C.K. 15-Deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc. Natl. Acad. Sci. USA 2000, 97, 4844–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cernuda-Morollon, E.; Pineda-Molina, E.; Canada, F.J.; Perez-Sala, D. 15-Deoxy-Δ12,14-prostaglandin J2 inhibition of NF-κB-DNA binding through covalent modification of the p50 subunit. J. Biol. Chem. 2001, 276, 35530–35536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Kim, E.H.; Na, H.K.; Sun, Y.; Surh, Y.J. 15-Deoxy-Δ12,14-prostaglandin J2 stabilizes, but functionally inactivates p53 by binding to the cysteine 277 residue. Oncogene 2010, 29, 2560–2576. [Google Scholar] [CrossRef] [Green Version]
- Shibata, T.; Yamada, T.; Ishii, T.; Kumazawa, S.; Nakamura, H.; Masutani, H.; Yodoi, J.; Uchida, K. Thioredoxin as a molecular target of cyclopentenone prostaglandins. J. Biol. Chem. 2003, 278, 26046–26054. [Google Scholar] [CrossRef] [Green Version]
- Perez-Sala, D.; Cernuda-Morollon, E.; Canada, F.J. Molecular basis for the direct inhibition of AP-1 DNA binding by 15-deoxy-Δ12,14-prostaglandin J2. J. Biol. Chem. 2003, 278, 51251–51260. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Gomez, F.J.; Cernuda-Morollon, E.; Stamatakis, K.; Perez-Sala, D. Protein thiol modification by 15-deoxy-Δ12,14-prostaglandin J2 addition in mesangial cells: Role in the inhibition of pro-inflammatory genes. Mol. Pharmacol. 2004, 66, 1349–1358. [Google Scholar] [CrossRef] [Green Version]
- Oliva, J.L.; Perez-Sala, D.; Castrillo, A.; Martinez, N.; Canada, F.J.; Bosca, L.; Rojas, J.M. The cyclopentenone 15-deoxy-Δ12,14-prostaglandin J2 binds to and activates H-Ras. Proc. Natl. Acad. Sci. USA 2003, 100, 4772–4777. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Saeidi, S.; Cho, N.C.; Kim, S.H.; Lee, H.B.; Han, W.; Noh, D.Y.; Surh, Y.J. Interaction of Nrf2 with dimeric STAT3 induces IL-23 expression: Implications for breast cancer progression. Cancer Lett. 2020, 500, 147–160. [Google Scholar] [CrossRef]
- Kim, K.; Park, J.M.; Kim, N.J.; Kim, S.J.; Moon, H.; An, H.; Lee, J.; Park, H.J.; Surh, Y.J.; Suh, Y.G. and Structural analysis of new Nrf2 activators by mechanism-based chemical transformation identification of 15-deoxy-Δ12,14-prostaglandin J2. ChemBioChem 2016, 17, 1900–1904. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, T.; Kamiya, N.; Shiki, S.; Kodama, T.S.; Kakizuka, A.; Jingami, H. α,β-unsaturated ketone is a core moiety of natural ligands for covalent binding to peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2005, 280, 14145–14153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, C.; Cheng, W.; Li, Q.; Han, Z.; Wang, X.; Jin, J.; Zou, J.; Liu, Z.; Zhou, Z.; Zhao, W.; et al. Heme oxygenase-1 retards hepatocellular carcinoma progression through the microRNA pathway. Oncol. Rep. 2016, 36, 2715–2722. [Google Scholar] [CrossRef]
- Tertil, M.; Golda, S.; Skrzypek, K.; Florczyk, U.; Weglarczyk, K.; Kotlinowski, J.; Maleszewska, M.; Czauderna, S.; Pichon, C.; Kieda, C.; et al. Nrf2-heme oxygenase-1 axis in mucoepidermoid carcinoma of the lung: Antitumoral effects associated with down-regulation of matrix metalloproteinases. Free Radic. Biol. Med. 2015, 89, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Barikbin, R.; Neureiter, D.; Wirth, J.; Erhardt, A.; Schwinge, D.; Kluwe, J.; Schramm, C.; Tiegs, G.; Sass, G. Induction of heme oxygenase 1 prevents progression of liver fibrosis in Mdr2 knockout mice. Hepatology 2012, 55, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Marelli, G.; Erreni, M.; Anselmo, A.; Taverniti, V.; Guglielmetti, S.; Mantovani, A.; Allavena, P. Heme-oxygenase-1 production by intestinal CX3CR1+ macrophages helps to resolve inflammation and prevents carcinogenesis. Cancer Res. 2017, 77, 4472–4485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandini, N.A.; Alonso, E.N.; Fermento, M.E.; Mascaro, M.; Abba, M.C.; Colo, G.P.; Arevalo, J.; Ferronato, M.J.; Guevara, J.A.; Nunez, M.; et al. Heme oxygenase-1 has an antitumor role in breast cancer. Antioxid. Redox Signal. 2019, 30, 2030–2049. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.J.; Bae, J.S.; Jamiyandorj, U.; Park, H.S.; Kwon, K.S.; Jung, S.H.; Youn, H.J.; Lee, H.; Park, B.H.; Chung, M.J.; et al. Expression of nerve growth factor and heme oxygenase-1 predict poor survival of breast carcinoma patients. BMC Cancer 2013, 13, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, P.; Basu, A.; Datta, D.; Gasser, M.; Waaga-Gasser, A.M.; Pal, S. The heme oxygenase-1 protein is overexpressed in human renal cancer cells following activation of the Ras-Raf-ERK pathway and mediates anti-apoptotic signal. J. Biol. Chem. 2011, 286, 33580–33590. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HO-1/CO system in tumor growth, angiogenesis and metabolism—Targeting HO-1 as an anti-tumor therapy. Vascul. Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef]
- Huang, J.; Guo, P.; Ma, D.; Lin, X.; Fang, Q.; Wang, J. Overexpression of heme oxygenase-1 induced by constitutively activated NF-kappaB as a potential therapeutic target for activated B-cell-like diffuse large B-cell lymphoma. Int. J. Oncol. 2016, 49, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Tan, Q.; Wang, H.; Hu, Y.; Hu, M.; Li, X.; Aodengqimuge; Ma, Y.; Wei, C.; Song, L. Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer Sci. 2015, 106, 1023–1032. [Google Scholar] [CrossRef]
- Furfaro, A.L.; Piras, S.; Passalacqua, M.; Domenicotti, C.; Parodi, A.; Fenoglio, D.; Pronzato, M.A.; Marinari, U.M.; Moretta, L.; Traverso, N.; et al. HO-1 up-regulation: A key point in high-risk neuroblastoma resistance to bortezomib. Biochim. Biophys. Acta 2014, 1842, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Dennery, P.A. Signaling function of heme oxygenase proteins. Antioxid. Redox Signal. 2014, 20, 1743–1753. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Lv, H.; Li, J.; Che, Y.; Xu, B.; Tao, Z.; Jiang, W. Roles of Nrf2/HO-1 and HIF-1α/VEGF in lung tissue injury and repair following cerebral ischemia/reperfusion injury. J. Cell Physiol. 2019, 234, 7695–7707. [Google Scholar] [CrossRef] [PubMed]
- Dulak, J.; Loboda, A.; Zagorska, A.; Jozkowicz, A. Complex role of heme oxygenase-1 in angiogenesis. Antioxid. Redox Signal. 2004, 6, 858–866. [Google Scholar] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in signaling and disease: Beyond discovery and development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Jozkowicz, A.; Huk, I.; Nigisch, A.; Weigel, G.; Dietrich, W.; Motterlini, R.; Dulak, J. Heme oxygenase and angiogenic activity of endothelial cells: Stimulation by carbon monoxide and inhibition by tin protoporphyrin-IX. Antioxid. Redox Signal. 2003, 5, 155–162. [Google Scholar] [CrossRef]
- Sun, D.; Guo, K.; Rusche, J.J.; Hurley, L.H. Facilitation of a structural transition in the polypurine/polypyrimidine tract within the proximal promoter region of the human VEGF gene by the presence of potassium and G-quadruplex-interactive agents. Nucleic Acids Res. 2005, 33, 6070–6080. [Google Scholar] [CrossRef]
- Ryuto, M.; Ono, M.; Izumi, H.; Yoshida, S.; Weich, H.A.; Kohno, K.; Kuwano, M. Induction of vascular endothelial growth factor by tumor necrosis factor α in human glioma cells. Possible roles of SP-1. J. Biol. Chem. 1996, 271, 28220–28228. [Google Scholar] [CrossRef] [Green Version]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed. Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef] [Green Version]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The role of Nrf2. Curr. Cancer Drug Targets 2018, 18, 538–557. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Ansari, K.M.; Sung, Y.M.; He, G.; Fischer, S.M. Prostaglandin receptor EP2 is responsible for cyclooxygenase-2 induction by prostaglandin E2 in mouse skin. Carcinogenesis 2007, 28, 2063–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.H.; Na, H.K.; Kim, D.H.; Park, S.A.; Kim, H.N.; Song, N.Y.; Surh, Y.J. 15-Deoxy-Δ12,14-prostaglandin J2 induces COX-2 expression through Akt-driven AP-1 activation in human breast cancer cells: A potential role of ROS. Carcinogenesis 2008, 29, 688–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offenbacher, S.; Odle, B.M.; Van Dyke, T.E. The use of crevicular fluid prostaglandin E2 levels as a predictor of periodontal attachment loss. J. Periodontal Res. 1986, 21, 101–112. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.-H.; Kim, S.-J.; Na, H.-K.; Han, W.; Kim, N.-J.; Suh, Y.-G.; Surh, Y.-J. 15-Deoxy-Δ12,14-prostaglandin J2 Upregulates VEGF Expression via NRF2 and Heme Oxygenase-1 in Human Breast Cancer Cells. Cells 2021, 10, 526. https://doi.org/10.3390/cells10030526
Kim E-H, Kim S-J, Na H-K, Han W, Kim N-J, Suh Y-G, Surh Y-J. 15-Deoxy-Δ12,14-prostaglandin J2 Upregulates VEGF Expression via NRF2 and Heme Oxygenase-1 in Human Breast Cancer Cells. Cells. 2021; 10(3):526. https://doi.org/10.3390/cells10030526
Chicago/Turabian StyleKim, Eun-Hee, Su-Jung Kim, Hye-Kyung Na, Wonshik Han, Nam-Jung Kim, Young-Ger Suh, and Young-Joon Surh. 2021. "15-Deoxy-Δ12,14-prostaglandin J2 Upregulates VEGF Expression via NRF2 and Heme Oxygenase-1 in Human Breast Cancer Cells" Cells 10, no. 3: 526. https://doi.org/10.3390/cells10030526