
Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase

1
Cardiovascular Research Laboratory, Biochemical-Pharmacological Center, Philipps-University Marburg, Karl-von-Frisch-Strasse 1, D-35043 Marburg, Germany
2
Fachbereich Chemie, Philipps-University, D-35032 Marburg, Germany
3
Department of Heart Surgery, Campus Marburg, University Hospital of Giessen and Marburg, D-35043 Marburg, Germany
*
Author to whom correspondence should be addressed.
Cells 2021, 10(3), 514; https://doi.org/10.3390/cells10030514
Submission received: 20 January 2021
/
Revised: 22 February 2021
/
Accepted: 22 February 2021
/
Published: 28 February 2021
(This article belongs to the Collection Regulation of Eukaryotic Cytochrome c Oxidase)
{kind=link}
Abstract
:Cytochrome c oxidase (COX), the rate-limiting enzyme of mitochondrial respiration, is regulated by various mechanisms. Its regulation by ATP (adenosine triphosphate) appears of particular importance, since it evolved early during evolution and is still found in cyanobacteria, but not in other bacteria. Therefore the “allosteric ATP inhibition of COX” is described here in more detail. Most regulatory properties of COX are related to “supernumerary” subunits, which are largely absent in bacterial COX. The “allosteric ATP inhibition of COX” was also recently described in intact isolated rat heart mitochondria.
1. Introduction
There exists a basic difference in the regulation of ATP synthesis by oxidative phosphorylation (OxPhos) between bacteria (except cyanobacteria) and eukaryotic organisms. Aerobic bacteria grow and divide continuously under constant conditions. Higher organisms change the rates of ATP synthesis and consumption up to a factor of 10 depending on various inner and outer signals. This regulation is based on acquisition of additional regulatory protein subunits (supernumerary subunits) during evolution for the enzyme complexes of OxPhos, which in eukaryotes are located in special organelles, the mitochondria.
OxPhos includes the respiratory chain containing the enzyme complexes I (NADH: ubiquinone oxidoreductase), II (succinate dehydrogenase), III (cytochrome bc1 complex), IV (cytochrome c oxidase, COX), and V (ATP synthase). According to the Mitchell theory [1,2], a proton motive force Δp is involved in OxPhos as an energy-rich intermediate for the exergonic oxidation of the reducing equivalents NADH and flavin adenine dinucleotide (FADH2), consisting of a membrane potential ΔΨm and a pH gradient across the inner mitochondrial membrane (Δp = ΔΨm − 59·ΔpH (mV)). Δp consists mostly of ΔΨm and is generated in the respiratory chain at enzyme complexes I, III, and IV via the translocation of protons from the matrix into the intermembrane space. Degradation of ΔΨm occurs predominantly via ATP synthase and “uncouplers” of OxPhos or uncoupling proteins [3].
2. Cytochrome C Oxidase (COX)
Eukaryotes synthesize ATP mostly in mitochondria, with COX as the final and oxygen-accepting complex of the respiratory chain. COX represents the rate-limiting step of respiration in living cells [4,5]. In contrast, from application of the metabolic control analysis to isolated mitochondria [6,7,8], a 5–7-fold excess of COX capacity was measured over the amount required to support the endogenous respiration of isolated mitochondria [9,10,11]. Therefore, the rate-limiting and regulatory role of COX for respiration in living organisms was ignored for a long time.
With increasing complexity during evolution [12], the number of protein subunits in the COX complex increased from 2–4 in bacteria to >7 in the slime mold Dictyostelium discoideum, 11 in yeast, and 13 in mammals [13]. In eukaryotes the “catalytic” subunits I–III are encoded on mitochondrial DNA and synthesized in mitochondria. The additional “supernumerary” subunits are encoded on nuclear DNA and synthesized on cytoplasmic ribosomes [14]. For the transport of these subunits into mitochondria [15] and for the assembly into the 13-subunit COX complex of vertebrates, a complicated machinery is required [16].
3. Regulation of COX Activity by “Allosteric ATP-Inhibition”
By studying the kinetics of ferrocytochrome c oxidation in isolated COX or in mitochondria after solubilization in 1% Tween-20, full inhibition of oxygen uptake was found at high ATP/ADP ratios and low ferrocytochrome c concentrations [17]. The polarographically measured oxygen uptake at increasing ferrocytochrome c concentrations revealed a sigmoidal curve with a Hill coefficient of 2. At low ATP/ADP ratios, a normal hyperbolic curve was found. Half-maximal inhibition of activity was measured at ATP/ADP = 28 [18]. This “second mechanism of respiratory control” is independent of ΔΨm and presents a feedback inhibition of mitochondrial respiration by its final product ATP [19]. The sigmoidal curve (Hill coefficient = 2) indicates cooperativity of two substrate (ferrocytochrome c)-binding sites. These occur in the dimeric enzyme, as visualized in the first crystal structure of COX [20], since monomeric COX contains only one binding site for cytochrome c [21]. After pretreatment of solubilized mitochondria with a monoclonal antibody against subunit IV, the inhibition of COX activity at high ATP/ADP ratios was completely abolished, and further experiments indicated that ATP interacts with the matrix domain of the transmembraneous subunit IV [17]. The “allosteric ATP inhibition of COX” is absent in bacterial COX [22], which does not have the “supernumerary” subunit IV (except cyanobacteria [23]).
Regulation of COX activity by adenine nucleotides evolved early during evolution, since it occurs already in COX of cyanobacteria containing an aa3-type enzyme [24]. Purified COX from Synechocystis contains four protein subunits corresponding to subunits I, II, III, and IV of vertebrate COX. The deduced amino-acid sequence of “subunit IV” from Synechocystis sp. PCC6803 showed approximately 50% and 20% sequence identity to COX subunit IV from Saccharomyces cerevisiae and beef heart, respectively. After reconstitution in liposomes, intraliposomal ADP stimulated and ATP inhibited the oxidation of ferrocytochrome c by cyanobacteria [23].
The allosteric ATP inhibition of COX was recently also shown with intact isolated rat heart mitochondria [25]. In a previous study, it was suggested that cAMP-dependent phosphorylation of COX subunit I at Ser-441 switches on the allosteric ATP inhibition [26]. This site is located at the intermembrane space and is accessible to cytosolic calcium via the pore in the outer mitochondrial membrane. Ser-441 of COX subunit I from beef is the only amino acid of consensus sequences for cAMP-dependent phosphorylation, located at the intermembrane space [26]. Stress increases cytosolic calcium (>1 µM Ca2+) [27,28] and dephosphorylates this site via a Ca2+-activated protein phosphatase. It was suggested that the allosteric ATP inhibition of COX keeps the mitochondrial membrane potential (ΔΨm) at low values (120–140 mV) and prevents formation of high amounts of ROS (reactive oxygen species) [28,29,30]. ROS have been shown to be involved in the generation of numerous diseases [31,32,33,34].
4. Role of Calcium in Generation of Increased ROS and ΔΨm
The relationship between stress factors and the generation of ROS was unknown until recently [31,32]. We presented a regulatory mechanism explaining the formation of ROS in mitochondria [28], which includes the increase of cytosolic calcium by stress factors and the abolishment of the “allosteric ATP inhibition of COX” (see below).
Stress factors are known to increase the cytosolic calcium concentration [28,32,33], which, under resting conditions, is very low (<1 µM). Calcium represents the most important signal for mitochondrial activation [34], and this occurs via dephosphorylation of most mitochondrial proteins through calcium-dependent mitochondrial phosphatases [34]. The stress factors, which induce hyperpolarization of ΔΨm, include high glucose [35,36], stress hormones [37,38,39,40,41], and psychosocial stress [42,43,44,45,46]. Furthermore, high amounts of glutamate induce mitochondrial hyperpolarization, ROS generation, and enhanced oxygen consumption in murine neuronal HT22 cells [47].
The “resting state” of mitochondrial respiration (see Figure 1) with high efficiency requires cardiolipin to stabilize the presumed weak interaction between the two monomers in dimeric COX [17]. The crystallized structure [20] contains, instead of ADP, 10 cholate molecules per monomer, which stabilize the non-physiological dimer [48]. The weak interaction between monomers in dimeric COX under physiological conditions is supported by observations of Kyoko Shinzawa-Itoh: “I have tried purifying cholate-free COX already. However, unfortunately, I have never succeeded in preparing COX samples enough for crystallization without using cholate” (personal communication to B.K., 2020). The high efficiency of dimeric COX is apparently due to allosteric ATP inhibition, which keeps ΔΨm at low values [28], thus preventing proton leak of the membrane at high ΔΨm [49], which increases with increasing ΔΨ [50] and causes slippage of proton pumping in COX [51]. In addition, it was suggested that the higher efficiency of the “resting state” is based on increased proton pumping in COX with a H+/e− stoichiometry of 2, instead of 1, which is thermodynamically possible [20]. Babcock and Wikström claimed that the H+/e− stoichiometry of COX is constant and always 1 [52] (however, see [53]). The additional proton pumping was proposed to be related to the H-channel, identified by the Yoshikawa group in the bovine heart enzyme [54,55,56], which is absent in bacteria [57]. An H+/e− stoichiometry of 2 in COX was in fact measured by the Lehninger group in isolated rat liver mitochondria [58,59,60,61].
The “active state” of mitochondrial respiration (without allosteric ATP inhibition) (see Figure 1) is characterized by maximal rates of COX activity and ATP synthesis. This state has partly elevated ΔΨm [30], accompanied by low efficiency. It is switched on under stress, including psychosocial stress [28], via increased cytosolic calcium concentrations, which dephosphorylate the dimeric COX resulting in monomeric COX [25], which is bound to NDUFA4 for stabilization [62]. These results exclude NDFUA4 as a permanent 14th subunit of COX. The activity of monomeric COX is supported by results obtained with purified COX. Removal of subunit VIb, which participates in the dimerization [26], increases COX activity [63]. In addition, without the allosteric ATP inhibition of COX, ΔΨm could rise above 130 mV, accompanied by proton leak [49] and slippage in COX [51], decreasing the efficiency of OxPhos. Although the respirasome (supramolecular structure I1III2IV1) contains only monomeric COX, the ordered cluster structure of the complete OxPhos system could allow a fast monomeric/dimeric configuration change. Thus, the establishment of “allosteric ATP control” mirrors the molecular composition [64].
In isolated mitochondria, supplemented with succinate, pyruvate, or glutamate, ADP stimulates oxygen consumption (state 3), whereas, after its conversion into ATP, respiration decreases (state 4) and ΔΨm increases to 180–200 mV. Moreover, isolated and reconstituted COX in liposomes could create a ΔΨ up to 225 mV [51]. The ratio of the rate of respiration at state 3 to state 4 was named “respiratory control”. In isolated mitochondria at state 4, 2% of total consumed oxygen was found to be converted into H2O2 [65]. This high amount of ROS (reactive oxygen species, mainly superoxide radical anion O2.− and H2O2) is deleterious for life and has been shown to be involved in the generation of numerous diseases [66,67,68,69,70]. In contrast, small amounts of ROS, produced by various oxidases [71,72], have signaling functions [31,73].
This high amount of ROS is produced in mitochondria mainly at complexes I–III [74,75], but not in COX, due to the unique structure of the oxygen-binding site in subunit I, composed of heme a3, CuB, and a tyrosyl-group, allowing simultaneous transfer of four electrons to O2, thus preventing the formation of intermediate ROS compounds [26]. It was found that mitochondrial ROS generation increases exponentially with increasing ΔΨm values above 140 mV [76,77,78]. Furthermore, high NADH/NAD+ ratios lead to the formation of O2− [73], which is rapidly converted into H2O2 by superoxide dismutases in the matrix and in the intermembrane space [79]. High and deleterious ΔΨm values above 140 mV are not necessary for the synthesis of ATP, because the ATP synthase is already saturated and maximal at 100–120 mV [80].
Fortunately, in living cells, mitochondrial ΔΨm values are normally below 140 mV (see [81]). However, under various stress conditions, an increase in ΔΨm to high and deleterious values was described. This mostly transient increase of ΔΨm was named mitochondrial “hyperpolarization” and is often followed by cell apoptosis [82,83].
Inhibition of ΔΨm by ATP was also measured directly in isolated rat liver mitochondria using a tetraphenyl phosphonium electrode [84]. Above 140 mV, the ROS production in mitochondria increases continuously [26,76,77]. While low ROS concentrations have regulatory functions in cells [31,73,75], high amounts, produced in mitochondria under stress [28], are known to cause multiple diseases [65,66,67,68,69] and to accelerate aging [72,85,86].
Allosteric ATP inhibition requires dimeric COX, based on its cooperativity of substrate action [25]. Monomeric COX, as isolated in nonionic detergents, is rather stable, while the dimer form is intrinsically unstable and dissociates into monomers at increased detergent concentration [87]. In isolated bovine heart mitochondria, more than 85% was found to be monomeric [88]. Bile acids stabilize the dimeric form [89] by exchanging bound ADP with cholate [90].
The feedback inhibition of COX by ATP, discovered 23 years ago, was not recognized by the scientific community, because it was generally not found in isolated mitochondria for two reasons: (i) measurements in mitochondria were usually not performed in the presence of ATP and high ATP/ADP ratios; (ii) the mechanism is switched off under stress conditions via Ca2+-activated dephosphorylation of COX. The phosphorylation site is located at the cytosolic side of COX subunit I [91], and it is essential for allosteric ATP inhibition.
After dephosphorylation of this site by a Ca2+-activated protein phosphatase, allosteric ATP inhibition is switched off. Re-phosphorylation by a cAMP-dependent protein kinase switches it on again [25]. These observations were made with the isolated enzyme, partly reconstituted in liposomes [28,90,93]. The reversible switching on and off of allosteric ATP inhibition was also shown recently with intact rat heart mitochondria [25]. In this study, a very low concentration of calcium (1–10 µM) was sufficient to switch off allosteric ATP inhibition. Various stress signals increase the cytosolic Ca2+ concentration and activate a Ca2+-dependent protein phosphatase, located at the intermembrane space, leading to dephosphorylation of COX with subsequent loss of allosteric ATP inhibition, increase in ΔΨm, and formation of ROS [28] (see Figure 1).
In conclusion, we suggest that allosteric ATP inhibition of COX is continuously changing in all mitochondria in order to optimize the amount and efficiency of energy (ATP) generation in eukaryotic cells according to the actual requirements (see Figure 1).
5. Regulation of COX via Reversible Phosphorylation
A further regulation of COX activity occurs via reversible phosphorylation of protein subunits [91,92,94,95]. Using mass spectrometry, 18 different phosphorylation sites were identified in COX subunits [91]. The specific functions of phosphorylation sites, however, were identified only in a few cases (see above). A review of phosphorylation sites was presented by Covian and Balaban [95] and Hüttemann et al. [96]. Selective protein kinase A (PKA)-dependent phosphorylation of subunits I, IV-1, and Vb was found under hypoxic stress in rabbit heart [97]. Interestingly, in cytochrome c, the reducing substrate of COX is also reversibly phosphorylated (at five positions), which influences the catalytic property of COX [96].
6. Regulation of COX via Expressing Supernumerary Subunit Isoforms
Regulation of COX activity via expressing isoforms of supernumerary subunits in the complex of 13 total subunits was first described in 1982 for subunits VIa, VIIa, and VIII [98]. Isoforms of supernumerary subunits occur in tissue-, developmental-, and species-specific forms. The identification of isoforms for subunits VIa, VIIa, and VIII occurred in skeletal muscle (H-isoform) and in non-skeletal muscle tissues (L-isoform), including smooth muscle [99], as shown by different runs in SDS-PAGE and by different N-terminal amino acid sequences [98]. The two genes for an isoform were published first in 1988 for subunits VIa (VIa-H = heart isoform, VIa-L = liver-isoform [100]). In most tissues, the liver isoform was found to occur (VIa-L, VIIa-L, VIIIa-L), which differs from the isoforms occurring in muscle tissue (VIa-H, VIIa-H, VIII-H), except in smooth muscle [99]), indicating different genes of these isoforms in skeletal muscle tissue.
Meanwhile, six different isoforms were identified for supernumerary COX subunits [101]. These include subunit IV (IV-1 + IV-2 [102] + IV-3 [103]). Allosteric ATP inhibition of COX is active in most cell types which express subunit IV-1. Isoform subunit IV-2 was found to be expressed in human cell lines under hypoxia [102,104]. Moreover, in isolated astrocytes and cerebellar granule cells, subunit IV-2 is expressed under hypoxic conditions accompanied by an abolition of the allosteric inhibition of COX by ATP [105]. The third isoform for COX subunit IV (Coxfa4l3) was found to occur during spermatogenesis [103]. Further isoforms occur for subunit VIa (VIa-H + VIa-L), VIb (VIb-1 and VIb-2, which appears to be testis-specific [106]), VIIa (VIIa-H + VIIa-L + COX-7AR [107]), VIIb (VIIb-1 + VIIb-2 [108]), and VIII (VIII-H + VIII-L + VIII-3 [109]).
Interestingly, in the other complexes of OxPhos (complexes I, II, III, and V), no isoforms of supernumerary subunits were found.
7. Regulation of COX via Binding Small Metabolites, Proteins, and Ligands and Deacetylation of Subunits
Various metabolites have been described to bind to COX and change its activity. Copper (Cu) is an essential trace element required for the normal development of living organisms. Copper-dependent enzymes such as ceruloplasmin, superoxide dismutase SOD1 and SOD3, the group of metallothionein proteins, and COX are present at all stages of gametogenesis, as well as in the somatic cells of the testis. Due to its redox potential, copper is a cofactor in many enzymes responsible for important processes in cells. Copper is a very reactive element and, in its free state, it can trigger the production of large amounts of free radicals, which consequently lead to the damage of proteins and DNA [112]. Hepatic expression of the Cu chaperones antioxidant 1 copper chaperone and cytochrome c oxidase copper chaperone (COX17) was decreased during iron deficiency, while the expression of the genes of zinc metabolism was unaltered [113]. the anion-binding behavior of the magnesium/manganese (Mg/Mn) site in cytochrome c oxidase has a possible role in proton pumping. Due to its close proximity and a shared ligand, oxidized Cu(A) is spin-coupled to the Mn(II) ion. This new observation of anion binding at the Mg/Mn site is of interest in terms of accessibility of the buried site and its potential role in redox-dependent proton pumping [114]. Moreover, the buried Mg/Mn site in COX is important for water ligands. Possibly, it represents a pathway for the exit of protons or water produced during turnover [115]. Inhibition of COX is viewed as a primary mode of cytotoxic hydrogen sulfide (H2S). However, studies conducted over the last two decades unveiled multiple biological regulatory roles of H2S as an endogenously produced mammalian gaseous transmitter. H2S serves as a stimulator of electron transport in mammalian mitochondria by acting as an electron donor, with sulfide/quinone oxidoreductase (SQR) being the immediate electron acceptor, and it stimulates mitochondrial ATP production [116]. A conserved bile acid-binding site (BABS) was crystallographically defined in the membrane domain of mammalian COX and COX from Rhodobacter sphaeroides. Diverse amphipathic ligands were shown previously to bind to this site and affect the electron transfer equilibrium between heme a and a3 cofactors by blocking the K proton uptake path. Identified candidate ligands include steroids, nicotinamides, flavins, nucleotides, retinoic acid, and thyroid hormones, which are predicted to make key protein contacts with the residues involved in bile acid binding [117]. Deoxycholate binds with its carboxyl group at the entrance of the K path; thus, this conserved steroid binding site could reveal a regulatory site for steroids or structurally related molecules that act on the essential K proton path and enzyme activity [118]. Cholate can affect the splitting up of COX into two conformational states. Following the kinetics of cyanide binding to the oxidized enzyme, different fractions of “slow” and “fast” conformations are found. The structural relationships between the known cholate-binding site and the binuclear cytochrome a3-CuB site, as well as the variation in the occupancy of this binding site with cholate or nucleotides, may modify the reactivity of the oxidized binuclear center toward cyanide [119]. 3,5-Diidothyronine (3,5-T2) binds to COX subunit Va and abolishes allosteric ATP inhibition of COX [120]. Another metabolite is palmitate, which decreases the H+/e− stoichiometry of proton pumping of non-muscle COX [121]. The binding of proteins to COX, including voltage-dependent anion channel (VDAC), epidermal growth factor receptor (EGFR), HIG1 domain family member 1A (Higd1a), B-cell lymphoma 2 protein (BCL-2), a hepatitis B viral protein (HBx), mitochondrial nuclear retrograde regulator 1 (MNRR1), Coiled-coil-helix-coiled-coil-helix domain containing 2 protein (CHCHD2), amyloid-β protein, Nitric oxide synthase 1 (NOS-1), NDUFA4, and NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 4-like 2 (NDUFA4L2), was discussed in former publications [101,107].
Mitochondrial complexes are prone to sirtuin (Sirt)3-mediated deacetylation modification. COX subunit I was discovered as a new deacetylation target of Sirt3, indicating that the Sirt3/MT-CO1 axis is a promising therapy target of stress-related diseases [122].
8. Overexpression of COX Subunits during Ischemic Injury, Cancerogenesis and Regulation via Forming Supercomplexes
Upregulating the expression levels of mitochondrially encoded NADH dehydrogenase 1 (MT-ND1) and COX subunit I (MT-CO1) in tachypacing cardiomyocytes resulted in increased ATP content and superior cell viability, whereas the expression levels of NADH ubiquinone oxidoreductase core subunit 1 (NDUFS1) and COX subunit VIc (COXVIc) had no effect [123]. Cytochrome c oxidase subunit Va (COXVa) is involved in maintaining normal mitochondrial function. Newly established transgenic mice with systemic COXVa overexpression resulted in the improvement of spatial recognition, memory and hippocampal synaptic plasticity, recovery of hippocampal CA1 dendrites, and activation of the BDNF/ERK1/2 (brain-derived neurotrophic factor/extracellular signal-regulated kinase) signaling pathway in vivo. COXVa in the hippocampus plays a vital role in aging-related cognitive deterioration via BDNF/ERK1/2 regulation, suggesting that COXVa may be a potential target for antisenescence drugs [124]. COXVa overexpression protects cortical neurons from hypoxic ischemic injury in neonatal rats associated with triosephosphate isomerase upregulation [125]. COXVb subunit is linked directly to stress regulatory networks. Copper/zinc superoxide dismutases possess the ability to enhance copper sulfate stress response and induction of messenger RNA (mRNA) transcription levels and microRNA [126]. In human hepatocellular carcinoma (HCC) tissues, 11 out of the 13 mitochondrial DNA (mtDNA)-encoded genes exhibited decreased mRNA levels and five genes displayed decreased protein levels, including the cytochrome B and MT-CO2 genes. After overexpression of mitomiR-181a-5p, cytochrome B and MT-CO2 levels were reduced in HCC cells, and the ΔΨm maintained by the electron transport chain (ETC) was decreased [127]. A systematic study of the overexpression of proteins and genes in tumor cells resulted in various publications. Somatic mutations within mitochondrial DNA-encoded MT-CO1 are frequent in various cancer types. Reactive oxygen species generated in cells overexpressing MT-CO1 variants acted as key effectors mediating differential expression of apoptosis and DNA damage pathway-related genes [128]. Overexpression of COXVIb1 protected against ischemia/reperfusion-induced neuronal injury in rat hippocampal neurons and relieved hypoxia/reoxygenation injury of neonatal rat cardiomyocytes [129,130]. Overexpression of COXVIIa1 suppressed cell proliferation and colony formation ability, as well as promoted cell apoptosis, in human non-small-cell lung cancer cells. COXVIIa1 holds a key position in regulating the development and progression of lung cancer by affecting autophagy [131]. In some reports, an increased correlation between the overexpression of COX subunit Va and/or Vb and the growth rate of various tumors was found [132,133,134,135,136,137,138]. The molecular basis for this relationship, however, remained unsolved.
Similar to the association of two COX monomers to the dimer, the complexes of the respiratory chain (CI–CIV) can associate to multiple complexes called “supercomplexes”. A well-known supercomplex is the “respirasome” (I1III2IV1). Their properties were described in review articles [139,140,141,142]. The links among mitochondrial dynamics, cristae remodeling, and supercomplex formation are presented therein: how mitochondrial structure can regulate bioenergetics, as well as the impact of mitochondrial dynamics and cristae shape on oxidative metabolism, the respiratory efficiency, and redox state. For example, CI (complex I) forms a supercomplex with CIII2 and CIV (SC I1+III2+IV1), known as the respirasome), as well as with CIII2 alone (SC I + III2). CIII2 forms a supercomplex with CIV (SC III2 + IV1), and CV alone forms dimers (CV2).
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemiosmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol. Rev. 1966, 41, 445–502. [Google Scholar] [CrossRef] [PubMed]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial uncoupling: A key controller of biological processes in physiology and diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villani, G.; Attardi, G. In vivo control of respiration by cytochrome c oxidase in wildtype and mitochondrial DNA mutation-carrying human cells. Proc. Natl. Acad. Sci. USA 1997, 94, 1166–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villani, G.; Attardi, G. In vivo control of respiration by cytochrome c oxidase in human cells. Free Radic. Biol. Med. 2000, 29, 202–210. [Google Scholar] [CrossRef]
- Kacser, H.; Burns, J.A. The control of flux. Symp. Soc. Exp. Biol. 1973, 27, 65–104. [Google Scholar] [CrossRef]
- Heinrich, R.; Rapoport, T.A. A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur. J. Biochem. 1974, 42, 89–95. [Google Scholar] [CrossRef]
- Fell, D. Understanding the control of metabolism. In Frontiers in Metabolism; Portland Press: London, UK, 1997; Volume 2. [Google Scholar]
- Tager, J.M.; Wanders, R.J.A.; Groen, A.K.; Kunz, W.; Bohnensack, R.; Küster, U.; Letko, G.; Böhme, G.; Duszynski, J.; Woijtczak, L. Control of mitochondrial respiration. FEBS Lett. 1981, 1–9. [Google Scholar]
- Letellier, T.; Malgat, M.; Mazat, J.P. Control of oxidative phosphorylation in rat muscle mitochondria: Implications for mitochondrial myopathies. Biochim. Biophys. Acta 1993, 1141, 58–64. [Google Scholar] [CrossRef]
- Letellier, T.; Heinrich, R.; Malgat, M.; Mazat, J.P. The kinetic basis of threshold effects observed in mitochondrial diseases: A systemic approach. Biochem. J. 1994, 302, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Pierron, D.; Wildman, D.E.; Hüttemann, M.; Markondapatnaikuni, G.C.; Aras, S.; Grossman, L.I. Cytochrome c oxidase: Evolution of control via nuclear subunit addition. Biochim. Biophys. Acta 2012, 1817, 590–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadenbach, B.; Jarausch, J.; Hartmann, R.; Merle, P. Separation of mammalian cytochrome c oxidase into 13 poly-peptides by a sodium dodecyl sulfate-gel electrophoretic procedure. Anal. Biochem. 1983, 129, 517–521. [Google Scholar] [CrossRef]
- Hundt, E.; Trapp, M.; Kadenbach, B. Biosynthesis of cytochrome c oxidase in isolated hepatocytes. FEBS Lett. 1980, 115, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Fielden, L.F.; Stojanovski, D. Mitochondrial protein transport in health and disease. Semin. Cell Dev. Biol. 2018, 76, 142–153. [Google Scholar] [CrossRef]
- Timón-Gómez, A.; Nývltová, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.; Kadenbach, B. Priority Paper. Cell respiration is controlled by ATP, an allosteric inhibitor of cytochrome c oxidase. Eur. J. Biochem. 1997, 249, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.; Kadenbach, B. The intramitochondrial ATP/ADP-ratio controls cytochrome c oxidase activity allosterically. FEBS Lett. 1999, 443, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Kadenbach, B.; Arnold, S. A second mechanism of respiratory control. FEBS Lett. 1999, 447, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yoshikawa, S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science 1996, 272, 1136–1144. [Google Scholar] [CrossRef]
- Osuda, Y.; Shinzawa-Itoh, K.; Tani, K.; Maeda, S.; Yoshikawa, S.; Tsukihara, T.; Gerle, C. Two dimensional crystallization of monomeric bovine cytochrome c oxidase with bound cytochrome c in reconstituted lipid membranes. Microscopy (Oxf.) 2016, 65, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follmann, K.; Arnold, S.; Ferguson-Miller, S.; Kadenbach, B. Cytochrome c oxidase activity from eucaryotes but not from procaryotes is allosterically inhibited by ATP. Biochem. Mol. Biol. Intern. 1998, 45, 1047–1055. [Google Scholar]
- Alge, D.; Wastyn, M.; Mayer, C.; Jungwirth, C.; Zimmermann, U.; Zoder, R.; Fromwald, S.; Peschek., G.A. Allosteric properties of cyanobacterial cytochrome c oxidase: Inhibition of the coupled enzyme by ATP and stimulation by ADP. IUBMB Life 1999, 48, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Peschek, G.A. Cytochrome oxidase and cta operon of cyanobacteria. Biochim. Biophys. Acta 1996, 1275, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Ramzan, R.; Rhiel, A.; Weber, P.; Kadenbach, B.; Vogt, S. Reversible dimerization of cytochrome c oxidase regulates mitochondrial respiration. Mitochondrion 2019, 49, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, B.; Bender, E.; Arnold, S.; Hüttemann, M.; Lee, I.; Kadenbach, B. Cytochrome c oxidase and the regulation of oxidative phosphorylation. ChemBioChem 2001, 2, 392–403. [Google Scholar] [CrossRef]
- Robb-Gaspers, L.D.; Rutter, G.A.; Denton, R.M.; Rizzuto, R.; Thomas, A.P. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998, 17, 4987–5000. [Google Scholar] [CrossRef] [Green Version]
- Ramzan, R.; Vogt, S.; Kadenbach, B. Stress-mediated generation of deleterious ROS in healthy individuals—Role of cytochrome c oxidase. J. Mol. Med. (Berl.) 2020, 98, 651–657. [Google Scholar] [CrossRef] [Green Version]
- Kadenbach, B.; Hüttemann, M.; Arnold, S.; Lee, I.; Mühlenbein, N.; Bender, E. Mitochondrial energy metabolism is regulated via nuclear-coded subunits of cytochrome c oxidase. Free Radic. Biol. Med. 2000, 29, 211–221. [Google Scholar] [CrossRef]
- Kadenbach, B.; Ramzan, R.; Wen, L.; Vogt, S. New extension of the Mitchell Theory for oxidative phosphorylation in mitochondria of living organisms. Biochim. Biophys. Acta 2010, 1800, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Kausar, S.; Wang, F.; Cui, H. The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedlic, F.; Muravyeva, M.; Sepac, A.; Sedlic, M.; Williams, A.M.; Yang, M.; Bai, X.; Bosnjak, Z.J. Targeted modification of mitochondrial ROS production converts high glucose-induced cytotoxicity to cytoprotection: Effects on anesthetic preconditioning. J. Cell Physiol. 2017, 232, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Gerencser, A.A. Metabolic activation-driven mitochondrial hyperpolarization predicts insulin secretion in human pancreatic beta-cells. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 817–828. [Google Scholar] [CrossRef]
- Capellino, S.; Claus, M.; Watzl, C. Regulation of natural killer cell activity by glucocorticoids, serotonin, dopamine, and epinephrine. Cell. Mol. Immunol. 2020, 17, 705–711. [Google Scholar] [CrossRef]
- Vincent, A.M.; Olzmann, J.A.; Brownlee, M.; Sivitz, W.I.; Russell, J.W. Uncoupling proteins prevent glucose-induced neuronal oxidative stress and programmed cell death. Diabetes 2004, 53, 726–734. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W.; Force, T. Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Investig. 2005, 115, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Kass, G.E.; Orenius, S. Calcium signaling and cytotoxicity. Environ. Health Perspect. 1999, 107, 25–35. [Google Scholar]
- Sher, L.D.; Geddie, H.V.; Olivier, L.; Cairns, M.; Truter, N.; Beselaar, L.; Essop, M.F. Chronic stress and endothelial dysfunction: Mechanisms, experimental challenges and way ahead. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H488–H506. [Google Scholar] [CrossRef]
- Barbiero, S.; Aimo, A.; Castiglione, V.; Giannoni, A.; Vergaro, G.; Passino, C.; Emdin, M. Healthy hearts at hectic pace: From daily life stress to abnormal cardiomyocyte function and arrhythmias. Eur. J. Prev. Cardiol. 2018, 25, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, F.A.; McDonnell, B.M.; Wolf-Johnston, A.S.; Kanai, A.J.; Shiva, S.; Chelimsky, T.; Rodriguez, L.; Birder, L.A. Stress-induced autonomic dysregulation of mitochondrial function in the rat urothelium. Neurourol. Urodyn. 2019, 38, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Matsuhisa, F.; Kitamura, N.; Satoh, E. Effects of acute and chronic psychological stress on platelet aggregation in mice. Stress 2014, 17, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Solanki, N.; Salvi, A.; Patki, G.; Salim, S. Modulating oxidative stress relieves stress-induced behavioral and cognitive impairments in rats. Int. J. Neuropsychopharmacol. 2017, 20, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Turdi, S.; Yuan, M.; Leedy, G.M.; Wu, Z.; Ren, J. Chronic social stress induces cardiomyocyte contractile dysfunction and intracellular Ca2+ derangement in rats. Physiol. Behav. 2012, 105, 498–509. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S.; Mehta, S.L.; Li, P.A. Glutamate induces mitochondrial dynamic imbalance and autophagy activation: Preventive effects of selenium. PLoS ONE 2012, 7, e39382. [Google Scholar] [CrossRef]
- Napiwotzki, J.; Shinzawa-Itoh, K.; Yoshikawa, S.; Kadenbach, B. ATP and ADP bind to cytochrome c oxidase and regulate its activity. Biol. Chem. 1997, 378, 1013–1021. [Google Scholar] [CrossRef]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar]
- O’Shea, P.S.; Petrone, G.; Casey, R.P.; Azzi, A. The current– voltage relationships of liposomes and mitochondria. Biochem. J. 1984, 219, 719–726. [Google Scholar] [CrossRef] [Green Version]
- Steverding, D.; Kadenbach, B. Influence of N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline modification on proton translocation and membrane potential of reconstituted cytochrome c oxidase support “proton slippage”. J. Biol. Chem. 1991, 266, 8097–8101. [Google Scholar] [CrossRef]
- Babcock, G.T.; Wikström, M. Oxygen activation and the conservation of energy in cell respiration. Nature 1992, 356, 301–309. [Google Scholar] [CrossRef]
- Frank, V.; Kadenbach, B. Regulation of the H+/e--stoichiometry of cytochrome c oxidase from bovine heart by intraliposomal ATP/ADP ratios. FEBS Lett. 1996, 382, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, S. A cytochrome c oxidase proton pumping mechanism that excludes the O2 reduction site. FEBS Lett. 2003, 555, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, S.; Muramoto, K.; Sinzawa-Itoh, K.; Aoyama, H.; Tsukihara, T.; Shimokata, K.; Katayama, Y.; Shimada, H.H. Proton pumping mechanism of bovine heart cytochrome c oxidase. Biochim. Biophys. Acta 2006, 1757, 1110–1116. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, S.; Muramoto, K.; Shinzawa-Itoh, K. The O(2) reduction and proton pumping gate mechanism of bovine heart cytochrome c oxidase. Biochim. Biophys. Acta 2011, 1807, 1279–1286. [Google Scholar] [CrossRef] [Green Version]
- Salje, J.; Ludwig, B.; Richter, O.M. Is a third proton-conducting pathway operative in bacterial cytochrome c oxidase? Biochem. Soc. Trans. 2005, 33, 829–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, L.E.; Reynafarje, B.; Lehninger, A.L. Stoichiometry of mitochondrial H+ translocation coupled to succinate oxidation at level flow. J. Biol. Chem. 1984, 259, 4802–4811. [Google Scholar] [CrossRef]
- Reynafarje, B.; Alexandre, A.; Davies, P.; Lehninger, A.L. Proton translocation stoichiometry of cytochrome oxidase: Use of a fast-responding oxygen electrode. Proc. Natl. Acad. Sci. USA 1982, 79, 7218–7222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynafarje, B.; Costa, L.E.; Lehninger, A.L. Upper and lower limits of the proton stoichiometry of cytochrome c oxidation in rat liver mitoplasts. J. Biol. Chem. 1986, 261, 8254–8262. [Google Scholar] [CrossRef]
- Setty, O.H.; Shrager, R.I.; Bunow, B.; Reynafarje, B.; Lehninger, A.L.; Hendler, R.W. Direct measurement of the initial proton extrusion to oxygen uptake ratio accompanying succinate oxidation by rat liver mitochondria. Biophys. J. 1986, 50, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef] [Green Version]
- Weishaupt, A.; Kadenbach, B. Selective removal of subunit VIb increases the activity of cytochrome c oxidase. Biochemistry 1992, 31, 11477–11481. [Google Scholar] [CrossRef]
- Nesterov, S.; Chesnokov, Y.; Kamyshinsky, R.; Panteleeva, A.; Lyamzaev, K.; Vasilov, R.; Yaguzhinsky, L. Ordered clusters of the complete oxidative phosphorylation system in cardiac mitochondria. Int. J. Mol. Sci. 2021, 22, 1462. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of oxidative damage in human disease. Clin. Chem. 2006, 52, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncola, J.; Cronin, M.T.D.; Mazura, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhang, H.; Zhang, W.; Feng, L.; Du, M.; Zhou, Y.; Chen, Z.; Pelicano, H.; Plunkett, W.; Wierda, W.G.; et al. Redox regulation of cell survival. Antioxi. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Sreedhar, A.; Aguilera-Aguirre, L.; Singh, K.K. Mitochondria in skin health, aging, and disease. Cell. Death Dis. 2020, 11, 444. [Google Scholar] [CrossRef]
- Go, Y.M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–245. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxidative Med. Cell. Longev. 2016, 4350965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.S. Generating, partitioning, targeting and functioning of superoxide in mitochondria. Biosci. Rep. 1997, 17, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Afonso, V.; Champy, R.; Mitrovic, D.; Collin, P.; Lomri, A. Reactive oxygen species and superoxide dismutases: Role in joint diseases. Jt. Bone Spine 2007, 74, 324–329. [Google Scholar] [CrossRef]
- Kaim, G.; Dimroth, P. ATP synthesis by F-type ATP synthase is obligatorily dependent on the transmembrane voltage. EMBO J. 1999, 18, 4118–4127. [Google Scholar] [CrossRef] [Green Version]
- Hüttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef]
- Kadenbach, B.; Arnold, S.; Lee, I.; Hüttemann, M. The possible role of cytochrome c oxidase in stress-induced apoptosis and degenerative diseases. Biochim. Biophys. Acta 2004, 1655, 400–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skulachev, V.P. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 2006, 11, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, R.; Staniek, K.; Kadenbach, B.; Vogt, S. Mitochondrial respiration and membrane potential are regulated by the allosteric ATP-inhibition of cytochrome c oxidase. Biochim. Biophys. Acta 2010, 1797, 1672–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Zurek, M.P.; Turner, A.A.; Khobeir, A.; Yazdi, N.; Hakim, L.; Liu, J.; Vaishnav, A.; et al. Cytochrome c phosphorylation: Control of mitochondrial electron transport chain flux and apoptosis. Intern. J. Biochem. Cell Biol. 2020, 121, 105704. [Google Scholar] [CrossRef] [PubMed]
- Yegorov, Y.E.; Poznyak, A.; Nikiforov, N.G.; Sobenin, I.A.; Orekhov, A.N. The link between chronic stress and accelerated aging. Biomedicines 2020, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Musatov, A.; Ortega-Lopez, J.; Robinson, N.C. Detergent-solubilized bovine cytochrome c oxidase: Dimerization depends on the amphiphilic environment. Biochemistry 2000, 39, 12996–13004. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H.; Pfeiffer, K. The ratio of oxidative phosphorylation complexes I–V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J. Biol. Chem. 2001, 276, 37861–37867. [Google Scholar] [CrossRef]
- Musatov, A.; Robinson, N.C. Cholate-induced dimerization of detergent- or phospholipid-solubilized bovine cytochrome c oxidase. Biochemistry 2002, 41, 4371–4376. [Google Scholar] [CrossRef]
- Lee, I.; Bender, E.; Arnold, S.; Kadenbach, B. Minireview-Hypothesis. New control of mitochondrial membrane potential and ROS-formation. Biol. Chem. 2001, 382, 1629–1633. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Grossman, L.I.; Doan, J.W.; Sanderson, T.H. Phosphorylation of mammalian cytochrome c and cytochrome c oxidase in the regulation of cell destiny: Rion, apoptosis, and human disease. Adv. Exp. Med. Biol. Chapter X 2012, 748, 237–264. [Google Scholar]
- Kadenbach, B. Complex IV—The regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion 2021, 56, S1567–S7249. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Bender, E.; Kadenbach, B. Control of mitochondrial membrane potential and ROS formation by reversible phosphorylation of cytochrome c oxidase. Mol. Cell. Biochem. 2002, 234–235, 63–70. [Google Scholar] [CrossRef]
- Covian, R.; Balaban, R.S. Cardiac mitochondrial matrix and respiratory complex protein phosphorylation. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H940–H966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helling, S.; Hüttemann, M.; Ramzan, R.; Kim, S.H.; Lee, I.; Müller, T.; Langenfeld, E.; Meyer, H.E.; Kadenbach, B.; Vogt, S.; et al. Multiple phosphorylations of cytochrome c oxidase and their functions. Proteomics 2012, 12, 950–959. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.K.; Prabu, S.K.; Sepuri, N.B.; Raza, H.; Anandatheerthavarada, H.K.; Galati, D.; Spear, J.; Avadhani, N.G. Site specific phosphorylation of cytochrome c oxidase subunits I, IVi1 and Vb in rabbit hearts subjected to ischemia/reperfusion. FEBS Lett. 2007, 581, 1302–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadenbach, B.; Hartmann, R.; Glanville, R.; Buse, G. Tissue-specific genes code for polypeptide VIa of beef liver and heart cytochrome c oxidase. FEBS Lett. 1982, 138, 236–238. [Google Scholar] [CrossRef] [Green Version]
- Anthony, G.; Stroh, A.; Lottspeich, F.; Kadenbach, B. Different isozymes of cytochrome c oxidase are expressed in bovine smooth muscle and skeletal or heart muscle. FEBS Lett. 1990, 277, 97–100. [Google Scholar] [CrossRef] [Green Version]
- Schlerf, A.; Droste, M.; Winter, M.; Kadenbach, B. Characterization of two different genes (cDNA) for cytochrome c oxidase subunit VIa from heart and liver of the rat. EMBO J. 1988, 7, 2387–2391. [Google Scholar] [CrossRef] [PubMed]
- Sinkler, C.A.; Kalpage, H.; Shay, J.; Lee, I.; Malek, M.H.; Grossman, L.I.; Hüttemann, M. Tissue- and condition-specific isoforms of mammalian cytochrome c oxidase subunits: From function to human disease. Oxid Med. Cell Longev. 2017, 2017, 1534056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hüttemann, M.; Kadenbach, B.; Grossman, L.I. Mammalian subunit IV isoforms of cytochrome c oxidase. Gene 2001, 267, 111–123. [Google Scholar] [CrossRef]
- Endou, M.; Yoshida, K.; Hirota, M.; Nakajima, C.; Sakaguchi, A.; Kurihara, Y. Coxfa4l3, a novel mitochondrial electron transport chain Complex 4 subunit protein, switches from Coxfa4 during spermatogenesis. Mitochondrion 2020, 52, 1–7. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvat, S.; Beyer, C.; Arnold, S. Effect of hypoxia on the transcription pattern of subunit isoforms and the kinetics of cytochrome c oxidase in cortical astrocytes and cerebellar neurons. J. Neurochem. 2006, 99, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Jaradat, S.; Grossman, L.I. Cytochrome c oxidase of mammals contains a testes-specific isoform of subunit VIb—the counterpart to testes-specific Cytochrome c? Mol. Reprod. Dev. 2003, 66, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Segade, F.; Hurle, B.; Claudio, E.; Ramos, S.; Lazo, P.S. Identification of an additional member of the cytochrome c oxidase subunit VIIa family of proteins. J. Biol. Chem. 1996, 271, 12343–12349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Wang, G.; Zhang, X.; Hüttemann, P.P.; Qiu, Y.; Liu, J.; Mitchell, A.; Lee, I.; Zhang, C.; Lee, J.S.; et al. COX7AR is a Stress-inducible Mitochondrial COX Subunit that Promotes Breast Cancer Malignancy. Sci. Rep. 2016, 6, 31742. [Google Scholar] [CrossRef] [Green Version]
- Indrieri, A.; van Rahden, V.A.; Tiranti, V.; Morleo, M.; Iaconis, D.; Tammaro, R.; D’Amato, I.; Conte, I.; Maystadt, I.; Demuth, S.; et al. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am. J. Hum. Genet. 2012, 91, 942–949. [Google Scholar] [CrossRef] [Green Version]
- Hüttemann, M.; Schmidt, T.R.; Grossman, L.I. A third isoform of cytochrome c oxidase subunit VIII is present in mammals. Gene 2003, 312, 95–102. [Google Scholar] [CrossRef]
- Bonne, G.; Seibel, P.; Possekel, S.; Marsac, C.; Kadenbach, B. Expression of human cytochrome c oxidase subunits during fetal development. Eur. J. Biochem. 1993, 217, 1099–1107. [Google Scholar] [CrossRef]
- Ewart, G.D.; Zhang, Y.Z.; Capaldi, R.A. Switching of bovine cytochrome c oxidase subunit VIa isoforms in skeletal muscle during development. FEBS Lett. 1991, 292, 79–84. [Google Scholar]
- Ogórek, M.; Gąsior, Ł.; Pierzchała, O.; Daszkiewicz, R.; Lenartowicz, M. Role of copper in the process of spermatogenesis. Postepy Hig Med. Dosw (Online) 2017, 71, 663–683. [Google Scholar] [CrossRef] [PubMed]
- Cottin, S.C.; Roussel, G.; Gambling, L.; Hayes, H.E.; Currie, V.J.; McArdle, H.J. The effect of maternal iron deficiency on zinc and copper levels and on genes of zinc and copper metabolism during pregnancy in the rat. Br. J. Nutr. 2019, 121, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, M.A.; Krzyaniak, M.D.; Xu, S.; McCracken, J.; Ferguson-Miller, S. EPR evidence of cyanide binding to the Mn(Mg) center of cytochrome c oxidase: Support for Cu(A)-Mg involvement in proton pumping. Biochemistry 2009, 48, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Florens, L.; Schmidt, B.; McCracken, J.; Ferguson-Miller, S. Fast deuterium access to the buried magnesium/manganese site in cytochrome c oxidase. Biochemistry 2001, 40, 7491–7497. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Hydrogen sulfide, an endogenous stimulator of mitochondrial function in cancer cells. Cells 2021, 10, 220. [Google Scholar] [CrossRef]
- Buhrow, L.; Hiser, C.; Van Voorst, J.R.; Ferguson-Miller, S.; Kuhn, L.A. Computational prediction and in vitro analysis of potential physiological ligands of the bile acid binding site in cytochrome c oxidase. Biochemistry 2013, 52, 6995–7006. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Mills, D.A.; Buhrow, L.; Hiser, C.; Ferguson-Miller, S. A conserved steroid binding site in cytochrome C oxidase. Biochemistry 2008, 47, 9931–9933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoji, K.; Giuffro, A.; D’Itri, E.; Hagiwara, K.; Yamanaka, T.; Brunori, M.; Sarti, P. The ratio between the fast and slow forms of bovine cytochrome c oxidase is changed by cholate or nucleotides bound to the cholate-binding site close to the cytochrome a3/CuB binuclear centre. Cell Mol. Life Sci. 2000, 57, 1482–1487. [Google Scholar] [CrossRef]
- Arnold, S.; Goglia, F.; Kadenbach, B. 3,5-Diiodothyronine binds to subunit Va of cytochrome c oxidase and abolishes the allosteric inhibition of respiration by ATP. Eur. J. Biochem. 1998, 252, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kadenbach, B. Palmitate decreases proton pumping of liver-type cytochrome c oxidase. Eur. J. Biochem. 2001, 268, 6329–6334. [Google Scholar] [CrossRef]
- Tu, L.F.; Cao, L.F.; Zhang, Y.H.; Guo, Y.L.; Zhou, Y.F.; Lu, W.Q.; Zhang, T.Z.; Zhang, T.; Zhang, G.X.; Kurihara, H.; et al. Sirt3-dependent deacetylation of COX-1 counteracts oxidative stress-induced cell apoptosis. FASEB J. 2019, 33, 14118–14128. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, Y.; Tang, R.; Jiang, X.; Wang, Y.; Gu, T. Effect of TFAM on ATP content in tachypacing primary cultured cardiomyocytes and atrial fibrillation patients. Mol. Med. Rep. 2020, 22, 5105–5112. [Google Scholar] [CrossRef] [PubMed]
- Xiyang, Y.B.; Liu, R.; Wang, X.Y.; Li, S.; Zhao, Y.; Lu, B.T.; Xiao, Z.C.; Zhang, L.F.; Wang, T.H.; Zhang, J. COX5A Plays a vital role in memory impairment associated with brain aging via the BDNF/ERK1/2 signaling pathway. Front. Aging Neurosci. 2020, 12, 215. [Google Scholar] [CrossRef]
- Jiang, Y.; Bai, X.; Li, T.T.; Al-Hawwas, M.; Jin, Y.; Zou, Y.; Hu, Y.; Liu, L.Y.; Zhang, Y.; Liu, Q.; et al. COX5A over-expression protects cortical neurons from hypoxic ischemic injury in neonatal rats associated with TPI up-regulation. BMC Neurosci. 2020, 21, 18. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Shu, L.; Zhang, W.; Wang, Z. Cca-miR398 increases copper sulfate stress sensitivity via the regulation of CSD mRNA transcription levels in transgenic Arabidopsis thaliana. PeerJ 2020, 8, e9105. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Chen, Y.; Wu, Z.; Xu, Q.; Chen, M.; Shao, M.; Cao, X.; Zhou, Y.; Xie, M.; Shi, Y.; et al. Mitochondrial miR-181a-5p promotes glucose metabolism reprogramming in liver cancer by regulating the electron transport chain. Carcinogenesis 2020, 41, 972–983. [Google Scholar] [CrossRef]
- Singh, R.K.; Saini, S.K.; Prakasam, G.; Kalairasan, P.; Bamezai, R.N.K. Role of ectopically expressed mtDNA encoded cytochrome c oxidase subunit I (MT-COI) in tumorigenesis. Mitochondrion 2019, 49, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wu, P.; Xiao, J.; Jiang, L. Overexpression of COX6B1 protects against I/R-induced neuronal injury in rat hippocampal neurons. Mol. Med. Rep. 2019, 19, 4852–4862. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, Y.; Wan, J.; Zhang, P.; Pei, F. COX6B1 relieves hypoxia/reoxygenation injury of neonatal rat cardiomyocytes by regulating mitochondrial function. Biotechnol. Lett. 2019, 41, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Chen, X.; Feng, Y.; Wang, G.; Nawaz, I.; Hu, L.; Liu, P. COX7A1 suppresses the viability of human non-small cell lung cancer cells via regulating autophagy. Cancer Med. 2019, 8, 7762–7773. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.X.; Pervaiz, S. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death Differ. 2010, 17, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.P.; Sun, H.F.; Jiang, H.L.; Li, L.D.; Hu, X.; Xu, X.E.; Jin, W. Loss of COX5B inhibits, and promotes senescence via mitochondrial dysfunction in breast cancer. Oncotarget 2015, 6, 43363–43374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Xi, J. Identification of COX5B as a novel biomarker in high-grade glioma patients. Onco Targets Ther. 2017, 10, 5463–5470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.P.; Sun, H.F.; Fu, W.Y.; Li, L.D.; Zhao, Y.; Chen, M.T.; Jin, W. High expression of COX5B is associated with poor prognosis in breast cancer. Future Oncol. 2017, 13, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Krupar, R.; Hautmann, M.G.; Pathak, R.R.; Varier, I.; McLaren, C.; Gaag, D.; Hellerbrand, C.; Evert, M.; Laban, S.; Idel, C.; et al. Immunometabolic determinants of chemoradiotherapy response and survival in head and neck squamous cell carcinoma. Am. J. Pathol. 2018, 188, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, J.; Tenbrock, J.; Kristiansen, G.; Müller, S.C.; Ellinger, J. Systematic expression analysis of the mitochondrial respiratory chain protein subunits identifies COX5B as a prognostic marker in clear cell renal cell carcinoma. J. Urol. 2019, 26, 910–916. [Google Scholar] [CrossRef]
- Chu, Y.D.; Lin, W.R.; Lin, Y.H.; Kuo, W.H.; Tseng, C.J.; Lim, S.N.; Huang, Y.L.; Huang, S.C.; Wu, T.J.; Lin, K.H.; et al. COX5B-mediated bioenergetic alteration regulates tumor growth and migration by modulating AMPK-UHMK1-ERK cascade in hepatoma. Cancers 2020, 12, 1646. [Google Scholar] [CrossRef]
- Signes, A.; Fernandez-Vizarra, E. Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem. 2018, 62, 255–270. [Google Scholar] [PubMed]
- Letts, J.A.; Sazanov, L.A. Clarifying the supercomplex: The higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef]
- Baker, N.; Patel, J.; Khacho, M. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: How mitochondrial structure can regulate bioenergetics. Mitochondrion 2019, 49, 259–268. [Google Scholar] [CrossRef]
- Genova, M.L.; Lenaz, G. A critical appraisal of the role of respiratory supercomplexes in mitochondria. Biol. Chem. 2013, 394, 631–639. [Google Scholar] [CrossRef] [PubMed]
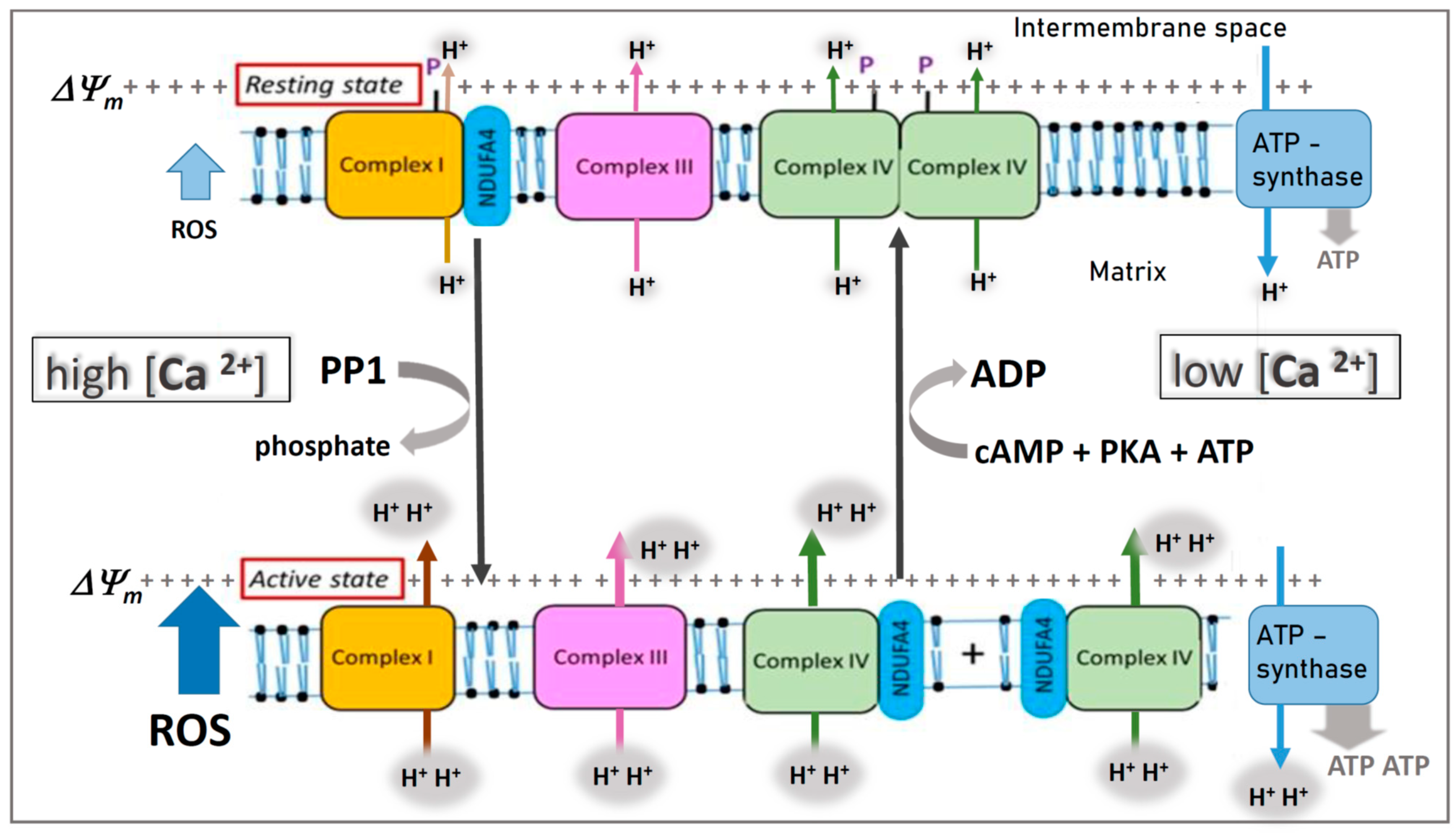
Figure 1.
Hypothesis: Variable binding of NDUFA4 to complex I or cytochrome c oxidase (COX) in the mitochondrial respiratory chain. It is assumed that, in the resting state, phosphorylation (P) of complex I and COX (complex IV) by a cAMP-dependent protein kinase A (PKA) at low cytosolic calcium (<1 µM) stabilizes binding of NDUFA4 to complex I and induces “allosteric ATP inhibition” of dimeric COX. In the active state, stress-induced increase of cytosolic calcium (> 1 µM) dephosphorylates complex I and COX via a calcium-activated protein phosphatase (PP1), accompanied by monomerization of COX, changed binding of NDUFA4 from complex I to monomeric COX, and switching off its allosteric ATP inhibition. Note, in the “active state”, there is higher proton pumping (H+) and ATP synthesis, but ROS production is also increased (blue arrow). Modified from Figure 1 in [92].
Figure 1.
Hypothesis: Variable binding of NDUFA4 to complex I or cytochrome c oxidase (COX) in the mitochondrial respiratory chain. It is assumed that, in the resting state, phosphorylation (P) of complex I and COX (complex IV) by a cAMP-dependent protein kinase A (PKA) at low cytosolic calcium (<1 µM) stabilizes binding of NDUFA4 to complex I and induces “allosteric ATP inhibition” of dimeric COX. In the active state, stress-induced increase of cytosolic calcium (> 1 µM) dephosphorylates complex I and COX via a calcium-activated protein phosphatase (PP1), accompanied by monomerization of COX, changed binding of NDUFA4 from complex I to monomeric COX, and switching off its allosteric ATP inhibition. Note, in the “active state”, there is higher proton pumping (H+) and ATP synthesis, but ROS production is also increased (blue arrow). Modified from Figure 1 in [92].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ramzan, R.; Kadenbach, B.; Vogt, S. Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase. Cells 2021, 10, 514. https://doi.org/10.3390/cells10030514
AMA Style
Ramzan R, Kadenbach B, Vogt S. Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase. Cells. 2021; 10(3):514. https://doi.org/10.3390/cells10030514
Chicago/Turabian StyleRamzan, Rabia, Bernhard Kadenbach, and Sebastian Vogt. 2021. "Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase" Cells 10, no. 3: 514. https://doi.org/10.3390/cells10030514
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.