
Gut Microbiome Profiles and Associated Metabolic Pathways in HIV-Infected Treatment-Naïve Patients

 , , , ,
, , , ,  , and
, and
Abstract
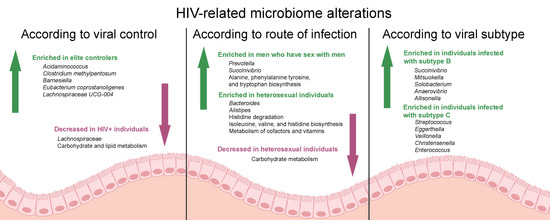
:
1. Introduction
2. Methods
2.1. Study Population
2.2. Sample Collection and Processing
2.3. Viral Subtype Analysis
2.4. Sequencing and Bioinformatics Analysis
2.5. Statistical Analysis
3. Results
3.1. Epidemiological Features
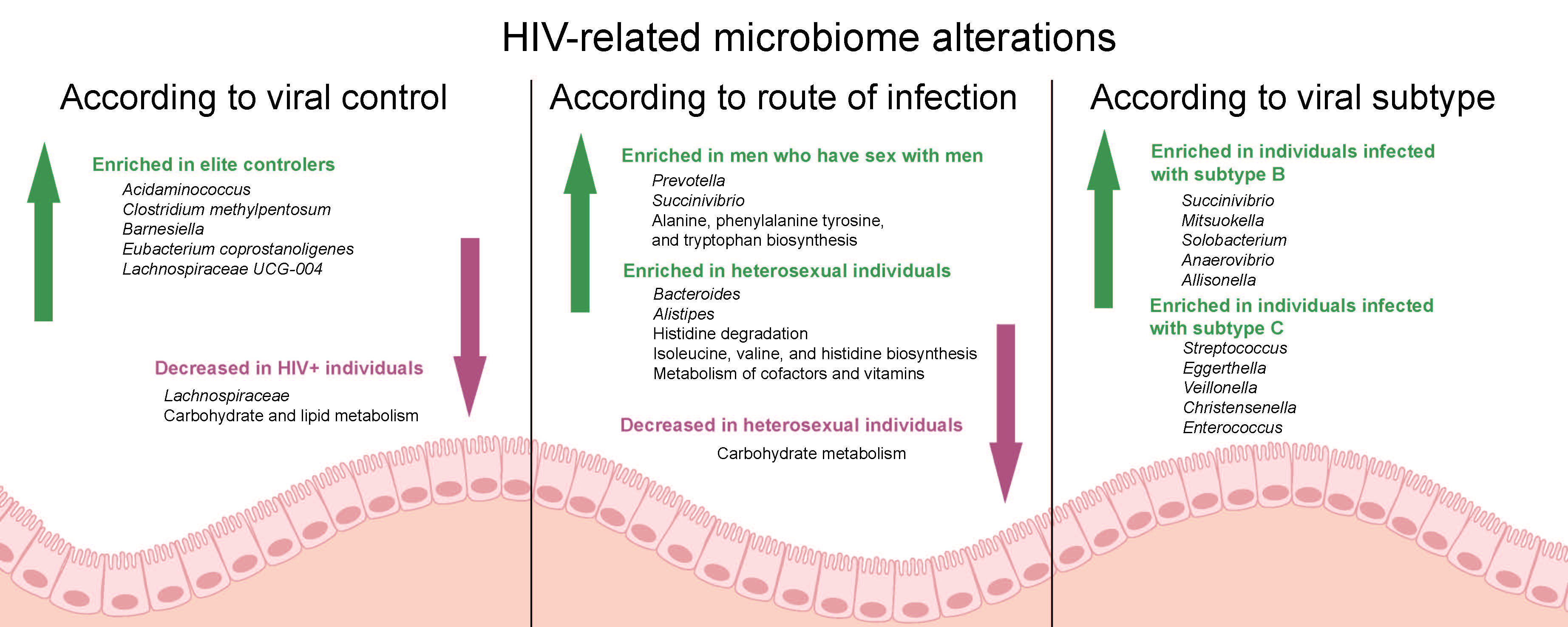
3.2. Comparison of Microbiota Profile between HIV+ and HD
3.3. Differences in Microbiota Composition between EC, NC, and HD
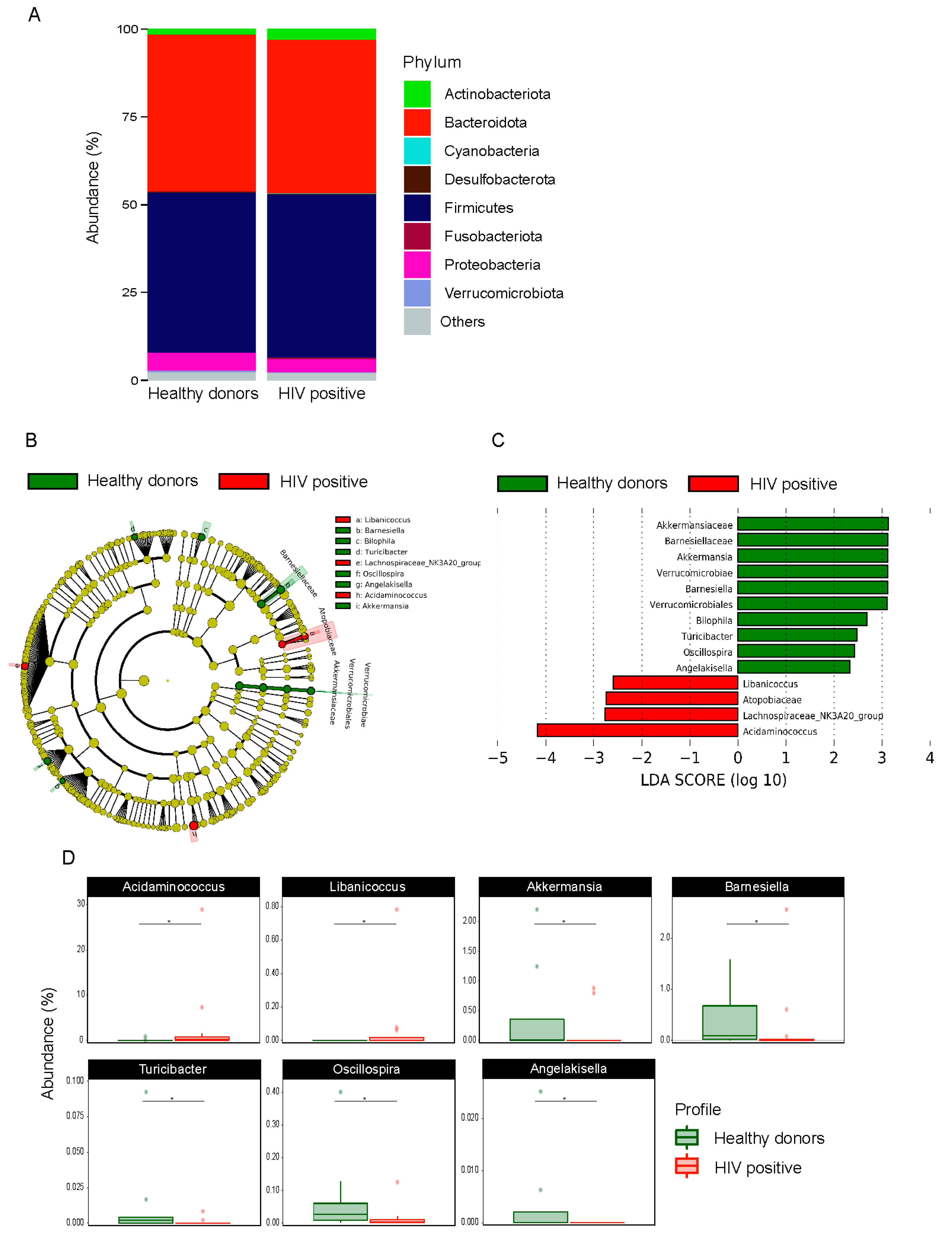
3.4. HIV Transmission Route Influences Composition of Gut Microbiome
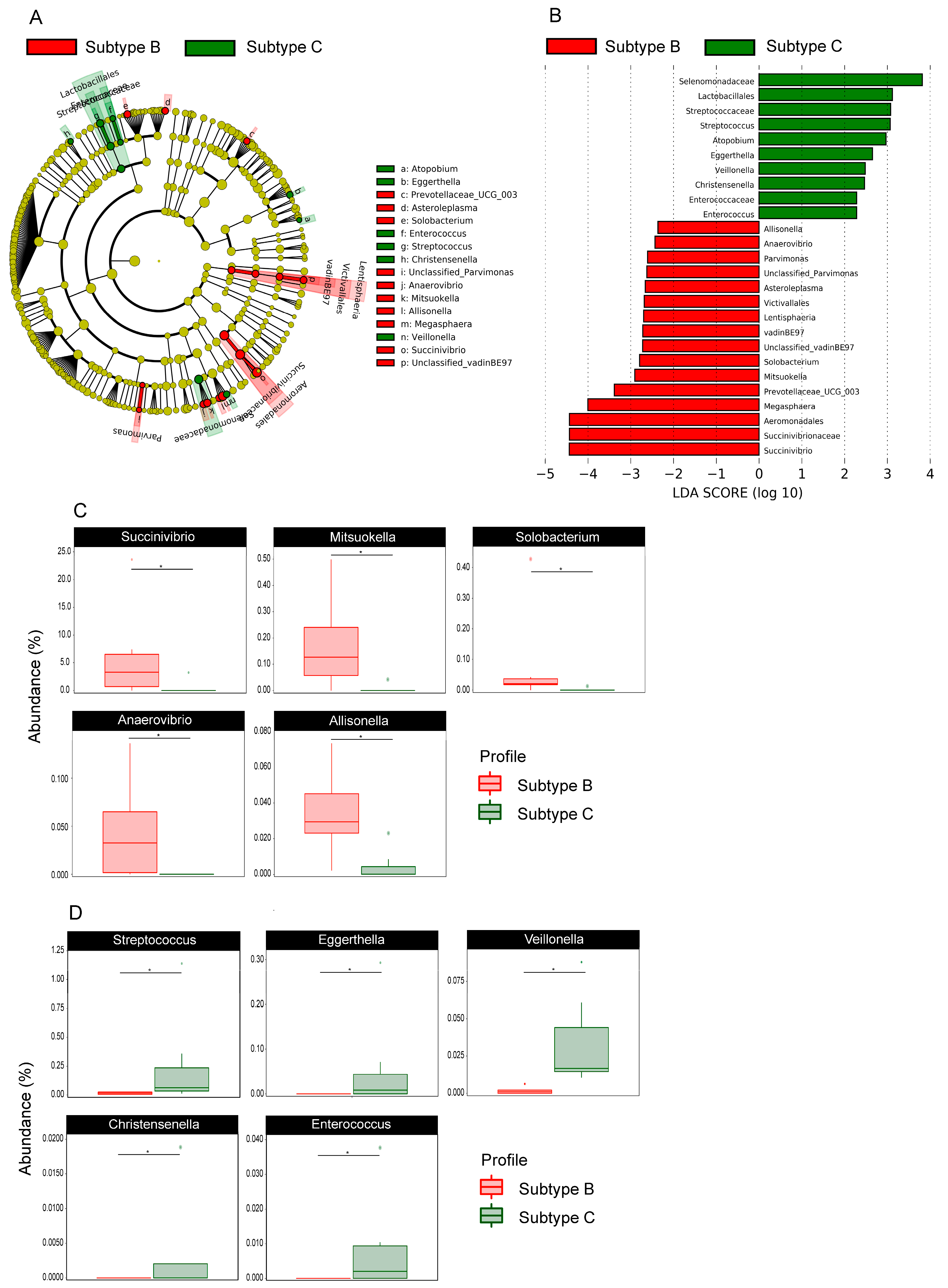
3.5. Gut Bacterial Microbiota Profile Differs Between Patients InfeCTED with HIV-1 Subtype B and C
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNAIDS. UNAIDS Data 2020; UNAIDS: Geneva, Switzerland, 2020; Available online: https://www.unaids.org/sites/default/files/media_asset/2020_aids-data-book_en.pdf (accessed on 26 September 2020).
- Dillon, S.M.; Frank, D.N.; Wilson, C.C. The gut microbiome and HIV-1 pathogenesis: A two-way street. Aids 2016, 30, 2737–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okulicz, J.F.; Lambotte, O. Epidemiology and clinical characteristics of elite controllers. Curr. Opin. HIV AIDS 2011, 6, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Walker, B.D. Human Immunodeficiency Virus Controllers: Mechanisms of Durable Virus Control in the Absence of Antiretroviral Therapy. Immunity 2007, 27, 406–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gori, A.; Tincati, C.; Rizzardini, G.; Torti, C.; Quirino, T.; Haarman, M.; Amor, K.B.; Van Schaik, J.; Vriesema, A.; Knol, J.; et al. Early impairment of gut function and gut flora supporting a role for alteration of gastrointestinal mucosa in human immunodeficiency virus pathogenesis. J. Clin. Microbiol. 2008, 46, 757–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubourg, G.; Surenaud, M.; Lévy, Y.; Hüe, S.; Raoult, D. Microbiome of HIV-infected people. Microb. Pathog. 2017, 106, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Usyk, M.; Sollecito, C.C.; Qiu, Y.; Williams-Nguyen, J.; Hua, S.; Gradissimo, A.; Wang, T.; Xue, X.; Kurland, I.J.; et al. Altered Gut Microbiota and Host Metabolite Profiles in Women With Human Immunodeficiency Virus. Clin. Infect. Dis. 2019, 10461, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dinh, D.M.; Volpe, G.E.; Duffalo, C.; Bhalchandra, S.; Tai, A.K.; Kane, A.V.; Wanke, C.A.; Ward, H.D. Intestinal Microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J. Infect. Dis. 2015, 211, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesterbacka, J.; Rivera, J.; Noyan, K.; Parera, M.; Neogi, U.; Calle, M.; Paredes, R.; Sönnerborg, A.; Noguera-Julian, M.; Nowak, P. Richer gut microbiota with distinct metabolic profile in HIV infected Elite Controllers. Sci. Rep. 2017, 7, 6269. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, E.A.; Keshavarzian, A.; Losurdo, J.; Swanson, G.; Siewe, B.; Forsyth, C.; French, A.; DeMarais, P.; Sun, Y.; Koenig, L.; et al. A Compositional Look at the Human Gastrointestinal Microbiome and Immune Activation Parameters in HIV Infected Subjects. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguera-Julian, M.; Rocafort, M.; Guillén, Y.; Rivera, J.; Casadellà, M.; Nowak, P.; Hildebrand, F.; Zeller, G.; Parera, M.; Bellido, R.; et al. Gut Microbiota Linked to Sexual Preference and HIV Infection. EBioMedicine 2016, 5, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Ministério da Saúde. Protocolo clínico e diretrizes para manejo da infecção pelo HIV em adultos, 1st ed.; Ministério da Saúde: Brasília, DF, Brazil, 2018. Available online: http://www.aids.gov.br/pt-br/pub/2013/protocolo-clinico-e-diretrizes-terapeuticas-para-manejo-da-infeccao-pelo-hiv-em-adultos (accessed on 12 October 2018).
- Azevedo, S.S.D.; Caetano, D.G.; Côrtes, F.H.; Teixeira, S.L.M.; Santos Silva, K.; Hoagland, B.; Grinsztejn, B.; Veloso, V.G.; Morgado, M.G.; Bello, G. Highly divergent patterns of genetic diversity and evolution in proviral quasispecies from HIV controllers. Retrovirology 2017, 14, 1–13. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, 1–11. [Google Scholar] [CrossRef]
- Machiavelli, A.; Duarte, R.T.D.; de Souza Pires, M.M.; Zarate-Bladés, C.R.; Pinto, A.R. The impact of in utero HIV exposure on gut microbiota, inflammation, and microbial translocation. Gut Microbes 2019, 10, 599–614. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murali, A.; Bhargava, A.; Wright, E.S. IDTAXA: A novel approach for accurate taxonomic classification of microbiome sequences. Microbiome 2018, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Castellanos, J.F.; Serrano-Villar, S.; Latorre, A.; Artacho, A.; Ferrús, M.L.; Madrid, N.; Vallejo, A.; Sainz, T.; Martínez-Botas, J.; Ferrando-Martínez, S.; et al. Altered metabolism of gut microbiota contributes to chronic immune activation in HIV-infected individuals. Mucosal Immunol. 2015, 8, 760–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, M.; Collado, M.C.; Ben-Amor, K.; Salminen, S.; De Vos, W.M. The mucin degrader Akkermansia muciniphila is an abundant resident of the human intestinal tract. Appl. Environ. Microbiol. 2008, 74, 1646–1648. [Google Scholar] [CrossRef] [Green Version]
- Png, C.W.; Lindén, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H.J. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol. 2010, 105, 2420–2428. [Google Scholar] [CrossRef]
- Kang, C.S.; Ban, M.; Choi, E.J.; Moon, H.G.; Jeon, J.S.; Kim, D.K.; Park, S.K.; Jeon, S.G.; Roh, T.Y.; Myung, S.J.; et al. Extracellular Vesicles Derived from Gut Microbiota, Especially Akkermansia muciniphila, Protect the Progression of Dextran Sulfate Sodium-Induced Colitis. PLoS ONE 2013, 8, e76520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, M.; Van Baarlen, P.; Hooiveld, G.; Norin, E.; Müller, M.; de Vos, W.M. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucin-degrader Akkermansia muciniphila. Front. Microbiol. 2011, 2, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottman, N.; Reunanen, J.; Meijerink, M.; Pietila, T.E.; Kainulainen, V.; Klievink, J.; Huuskonen, L.; Aalvink, S.; Skurnik, M.; Boeren, S.; et al. Pili-like proteins of Akkermansia muciniphila modulate host immune responses and gut barrier function. PLoS ONE 2017, 12, e0173004. [Google Scholar] [CrossRef]
- Meehan, C.J.; Beiko, R.G. A phylogenomic view of ecological specialization in the lachnospiraceae, a family of digestive tract-associated bacteria. Genome Biol. Evol. 2014, 6, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, A.; Bennett, N.; Ahmed, B.; Whelan, J.; Donohoe, D.R. Butyrate decreases its own oxidation in colorectal cancer cells through inhibition of histone deacetylases. Oncotarget 2018, 9, 27280–27292. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Mancabelli, L.; Milani, C.; Lugli, G.A.; Turroni, F.; Cocconi, D.; van Sinderen, D.; Ventura, M. Identification of universal gut microbial biomarkers of common human intestinal diseases by meta-analysis. FEMS Microbiol. Ecol. 2017, 93, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Villar, S.; Rojo, D.; Martínez-Martínez, M.; Deusch, S.; Vázquez-Castellanos, J.F.; Bargiela, R.; Sainz, T.; Vera, M.; Moreno, S.; Estrada, V.; et al. Gut Bacteria Metabolism Impacts Immune Recovery in HIV-infected Individuals. EBioMedicine 2016, 8, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wang, Q.; Zhao, J.; Gong, L.; Zhang, Y.; Wang, X.; Yuan, Z. Altered diversity and composition of the gut microbiome in patients with cervical cancer. AMB Express 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shkoporov, A.N.; Chaplin, A.V.; Khokhlova, E.V.; Shcherbakova, V.A.; Motuzova, O.V.; Bozhenko, V.K.; Kafarskaia, L.I.; Efimov, B.A. Alistipes inops sp. Nov. And coprobacter secundus sp. Nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2015, 65, 4580–4588. [Google Scholar] [CrossRef]
- Gartner, M.J.; Roche, M.; Churchill, M.J.; Gorry, P.R.; Flynn, J.K. Understanding the mechanisms driving the spread of subtype C HIV-1. EBioMedicine 2020, 53, 102682. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, E.; Engelbrecht, S.; De Oliveira, T. History and origin of the HIV-1 subtype C epidemic in South Africa and the greater southern African region. Sci. Rep. 2015, 5, 16897. [Google Scholar] [CrossRef] [Green Version]
- Gräf, T.; Machado Fritsch, H.; de Medeiros, R.M.; Maletich Junqueira, D.; Esteves de Matos Almeida, S.; Pinto, A.R. Comprehensive Characterization of HIV-1 Molecular Epidemiology and Demographic History in the Brazilian Region Most Heavily Affected by AIDS. J. Virol. 2016, 90, 8160–8168. [Google Scholar] [CrossRef] [Green Version]
- Ariën, K.K.; Abraha, A.; Quiñones-mateu, M.E.; Vanham, G.; Arts, E.J.; Arie, K.K.; Quin, M.E.; Kestens, L. The replicative fitness of primary human immunodeficiency virus type 1 ( HIV-1 ) group M, HIV-1 group O, and HIV-2 Isolates. J. Virol. 2005, 79, 8979–8990. [Google Scholar] [CrossRef] [Green Version]
- Venner, C.M.; Nankya, I.; Kyeyune, F.; Demers, K.; Kwok, C.; Chen, P.L.; Rwambuya, S.; Munjoma, M.; Chipato, T.; Byamugisha, J.; et al. Infecting HIV-1 Subtype Predicts Disease Progression in Women of Sub-Saharan Africa. EBioMedicine 2016, 13, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vujkovic-Cvijin, I.; Sortino, O.; Verheij, E.; Sklar, J.; Wit, F.W.; Kootstra, N.A.; Sellers, B.; Brenchley, J.M.; Ananworanich, J.; van der Loeff, M.S.; et al. HIV-associated gut dysbiosis is independent of sexual practice and correlates with noncommunicable diseases. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Dang, A.T.; Cotton, S.; Sankaran-Walters, S.; Li, C.S.; Lee, C.Y.M.; Dandekar, S.; Paster, B.J.; George, M.D. Evidence of an increased pathogenic footprint in the lingual microbiome of untreated HIV infected patients. BMC Microbiol. 2012, 12, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Elite Controllers | Non-Controllers | Healthy Donors | |
|---|---|---|---|
| Number of individuals | 06 | 07 | 09 |
| Age (years, median (IQR)) | 42 (37–52) | 37 (32–55) | 29 (27–39) |
| Body mass index BMI | 26.98 (25.4–28.4) | 23.32 (20.7–25.4) | 26.03 (22.01–30.6) |
| Gender, n (%) | |||
| Female | 4 (67%) | 2 (29%) | 6 (67%) |
| Male | 2 (33%) | 5 (71%) | 3 (33%) |
| Ethnicity, n (%) | |||
| Euro-descendant | 8 (100%) | 6 (86%) | 8 (89%) |
| Afro-descendant | 0 | 1 (14%) | 1 (11%) |
| Category Exposure, n (%) | |||
| HET | 4 (67%) | 2 (28.5%) | - |
| MSM | 2 (33%) | 3 (43%) | - |
| IDU | 0 | 2 (28.5%) | - |
| Time since HIV-1 diagnosis years (median (IQR)) | 5.6 (5.0–9.3) | 6.4 (5.8–6.9) | - |
| CD4+ T-cell count (median (IQR)) | 1190 (605.5–1370) | 726 (521–866) | - |
| CD8+ T-cell count (median (IQR)) | 1057 (810–1356) | 1295 (798–1527) | - |
| CD4/CD8+ T-cell ratio (median (IQR) | 0.99 (0.59–1.49) | 0.56 (0.4–0.72) | - |
| Viral load (copies/mL) (median (IQR)) | Undetectable | 4203 (2316–5510) | - |
| HIV-1 Subtype (%) | |||
| B | 3 (50%) | 3 (43%) | - |
| C | 3 (50%) | 4 (57%) | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
do Nascimento, W.M.; Machiavelli, A.; Ferreira, L.G.E.; Cruz Silveira, L.; de Azevedo, S.S.D.; Bello, G.; Smith, D.P.; Mezzari, M.P.; Petrosino, J.F.; Delgado Duarte, R.T.; et al. Gut Microbiome Profiles and Associated Metabolic Pathways in HIV-Infected Treatment-Naïve Patients. Cells 2021, 10, 385. https://doi.org/10.3390/cells10020385
do Nascimento WM, Machiavelli A, Ferreira LGE, Cruz Silveira L, de Azevedo SSD, Bello G, Smith DP, Mezzari MP, Petrosino JF, Delgado Duarte RT, et al. Gut Microbiome Profiles and Associated Metabolic Pathways in HIV-Infected Treatment-Naïve Patients. Cells. 2021; 10(2):385. https://doi.org/10.3390/cells10020385
Chicago/Turabian Styledo Nascimento, Wellinton M., Aline Machiavelli, Luiz G. E. Ferreira, Luisa Cruz Silveira, Suwellen S. D. de Azevedo, Gonzalo Bello, Daniel P. Smith, Melissa P. Mezzari, Joseph F. Petrosino, Rubens Tadeu Delgado Duarte, and et al. 2021. "Gut Microbiome Profiles and Associated Metabolic Pathways in HIV-Infected Treatment-Naïve Patients" Cells 10, no. 2: 385. https://doi.org/10.3390/cells10020385