
Intracellular Sources of ROS/H2O2 in Health and Neurodegeneration: Spotlight on Endoplasmic Reticulum



1
Department of Clinical Neurosciences, UK Dementia Research Institute, University of Cambridge, Cambridge CB2 0AH, UK
2
CCMAR—Centro de Ciências do Mar, Campus de Gambelas, Universidade do Algarve, 8005-139 Faro, Portugal
3
Cambridge Institute for Medical Research, University of Cambridge, Cambridge CB2 0XY, UK
*
Authors to whom correspondence should be addressed.
Cells 2021, 10(2), 233; https://doi.org/10.3390/cells10020233
Submission received: 17 November 2020
/
Revised: 15 January 2021
/
Accepted: 18 January 2021
/
Published: 25 January 2021
(This article belongs to the Section Intracellular and Plasma Membranes)
Abstract
:Reactive oxygen species (ROS) are produced continuously throughout the cell as products of various redox reactions. Yet these products function as important signal messengers, acting through oxidation of specific target factors. Whilst excess ROS production has the potential to induce oxidative stress, physiological roles of ROS are supported by a spatiotemporal equilibrium between ROS producers and scavengers such as antioxidative enzymes. In the endoplasmic reticulum (ER), hydrogen peroxide (H2O2), a non-radical ROS, is produced through the process of oxidative folding. Utilisation and dysregulation of H2O2, in particular that generated in the ER, affects not only cellular homeostasis but also the longevity of organisms. ROS dysregulation has been implicated in various pathologies including dementia and other neurodegenerative diseases, sanctioning a field of research that strives to better understand cell-intrinsic ROS production. Here we review the organelle-specific ROS-generating and consuming pathways, providing evidence that the ER is a major contributing source of potentially pathologic ROS.
1. Introduction
Substantial quantities of reactive oxygen species (ROS), H2O2 in particular, are produced intracellularly. H2O2 is generated endogenously as a functional entity and as a by-product of reducing/oxidising (redox) chemistry: H2O2 can be generated by regulated enzymatic processes to serve as a signal molecule, fuel protein disulfide bonding or as a controlled toxin in immunological-activity. Imperfections in electrochemical reactions can result in unintended H2O2/ROS (e.g., mitochondrial respiration chain with oxygen as terminal electron acceptor). Some ROS, specifically free radicals, are short-lived as a product of their high reactivity with biological macromolecules, including nucleic acids and lipids, which in turn can render them hazardous. By contrast, H2O2, a non-radical ROS, is less reactive with biomolecules and as such has a relatively long biological lifespan (cellular half-life of ~1 ms) [1,2]. Yet H2O2 is also capable of producing harmful radicals, through fenton chemistry [3]. Therefore, ROS generating pathways have co-evolved with mechanisms to control their spatio–temporal distribution. Thus, cellular compartments handling the protein-encoding nucleic acids maintain a strictly reducing environment (redox potential estimated at −300 mV and −280 mV in the nucleus and cytoplasm, respectively [4]) (see glossary). These compartments are equipped with ROS quenching enzymes, e.g., catalase and glutathione peroxidases. The reducing nature of such environments allows transient and localised usage of H2O2 as a second messenger. As such, H2O2 complements the cell’s repertoire of second messengers as it can induce rapid and reversible functional changes to proteins through intra/inter molecular disulfide bonding and other cysteine modifications such as sulfenylation and sulfonation. This principle is exemplified by a bacterial H2O2 sensing system, where the key protein, OxyR contains a peroxidatic cysteine residue, surrounded by positive charges, that is preferentially reactive with H2O2. This reaction results in a sulfenic intermediate that is highly reactive with the thiolate of a second “resolving” cysteine, producing an intramolecular disulfide that converts the protein into a functional transcription factor that activates the production of ROS-antagonising enzymes [5]. The kinetic advantage of H2O2, in such a system, over the reducing forces of the cell allows this reaction to prevail even when its concentration is 2–3 orders of magnitudes lower than the antagonising reductants [6]. As such, OxyR has been utilised as a template in engineering a genetically encoded fluorescent H2O2 probe, HyPer, sensitive to physiological H2O2 concentrations [7].
The short-lived nature of H2O2 relative to the biomolecules it reacts with and the overwhelming cellular reductive force ensure the locality and transience of H2O2-mediated signalling events. Several signalling pathways of mammalian cells take advantage of this chemistry, enzymatically producing local H2O2 that can activate factors such as kinases. For instance, oxidation of cysteine residues in SRC, a proto-oncogene non-receptor tyrosine kinase, activates this enzyme, regulating cell proliferation and survival by activating downstream pathways such as ERK and AKT signalling [8]. Several transcription factors, such as NF-κB and KEAP1-NRF2, also sense H2O2 to activate transcriptional programs [9,10].
The reducing, ROS disfavouring environment of the nucleus, cytoplasm and mitochondria contrasts with a relatively oxidising milieu in the organelles of the secretory pathway. The endoplasmic reticulum (ER) lumen, in particular, maintains an oxidising environment similar to that of the extracellular conditions. This is favoured by the relatively oxidising reduction potential of glutathione (EGSH/GSSG), a measure of redox conditions, estimated at −208 mV in the ER [11,12]. Moreover, the ER maintains a relatively high turn-over of H2O2 as its oxidative protein folding machinery both produces and utilises H2O2 continuously. H2O2 apparent confinement to the ER (discussed herein) can be compromised when redox homeostasis is perturbed [13,14].
This notion places the ER in the spotlight as a potential source of ROS associated with pathologies, including those associated with neurodegenerative diseases. Excessive ROS load entails damage of structural and functional macromolecules including lipids, protein, RNA, and perhaps with the most severe repercussions to DNA, where a single unrepaired damage event can impact on all its downstream products. As such, compromised insulation of organelles that maintain naturally high ROS (such as ER) may contribute to pathological elevations in ROS.
With an emphasis on the ER, here we review the findings contributing to the current understanding of organelles’ ROS producing/consuming pathways and discuss how an imbalance in the activity of ROS generating and antagonising pathways as well as organellar ROS permeability dysregulation can lead to an increase in their steady-state load. The intracellular sources of H2O2 are discussed in the context of their association with neurodegenerative diseases.
2. ER Sources of ROS/H2O2
The ER is devoid of ROS-vulnerable genetic material and therefore, predictably, can tolerate a high abundance of ROS. In addition, H2O2 reactivity with most protein functional groups is energetically unfavourable, suggesting it is unlikely to significantly contribute to protein misfolding directly [15]. ER oxidative protein folding both generates, and is at least partially fuelled by, H2O2 (described in detail below) and basal ER ROS content is reported to be relatively high [13]. This is unsurprising, as approximately 30% of cellular proteins enter the ER, many of them en route to the cell membrane or secretion. A 1970s ex vivo study using rat liver homogenate reports that H2O2 in the microsomal fraction, which contains the ER predominantly, accounts for 45% of the cell total, three times that derived from mitochondrial extracts of the same samples [16,17]. Thus, the ER appears uniquely capable of producing and utilising H2O2 as fuel for enzymatic oxidative folding process on a massive scale.
The core of the ER oxidative folding machinery is a group of oxidoreductase-chaperone enzymes of the protein disulphide isomerase (PDI) family, which functionally interact with their substrates via redox-active CXXC motifs of their catalytic thioredoxin domains (Figure 1) [18]. PDI itself catalyses disulphide bond formation in substrate proteins, leading to reduction of PDI CXXC motifs that are in turn reoxidised by ER oxidoreductin 1 (ERO1). The later produces H2O2 as two electrons per thiol pair flow from PDIs to molecular oxygen [19]. In this process, ERO1 is estimated to consume 25% of the oxygen available in the cell [20,21,22]. Yet the by-product of this reaction, H2O2, can be consumed by another process that drives PDI reoxidation, catalysed by the ER-localised peroxiredoxin PRDX4 (pathway described below). Notably, even though ERO1 gene expression is up-regulated in response to stimuli such as hypoxia and the unfolded proteins response (UPR) [23,24,25,26], knockdown of ERO1 gene leads to a resistance to tunicamycin-induced ER stress in worms and ER stress-induced apoptosis in mouse macrophages [27,28]. It seems that up-regulation of ERO1 results in excessive ROS production rather than improving oxidative folding capacity. Indeed, excess of ERO1 activity caused by hyperactive mutants or overexpression has been reported to increase ROS production and cell death [29,30]. Noteworthily, knockdown of ERO1 in worms extends their lifespan by 32% and is accompanied by a reduction in peroxide levels [31]. Thus, through its role in oxidative protein folding, excessive ERO1 activity contributes to an ER-emanating ROS hazard.
Although ERO1 deficit is lethal in yeasts [32,33], this gene is at least partially dispensable in mammals (knockout of all ERO1 isoforms, Ero1a and Ero1b, gives rise to only a mild phenotypic defect in mice [34,35]). In the absence of ERO1, the oxidative folding process can be sustained by peroxiredoxin 4 (PRDX4), an ER-localised antioxidative enzyme that catalyses PDI reoxidation [36]. PRDX4 scavenges H2O2, forming water with the concomitant relay of oxidative equivalent to PDIs (Figure 1). In normal conditions, PRDX4 works presumably in parallel to ERO1 in oxidative recycling of PDI. Fuelled by H2O2 generated as a by-product of ERO1 activity, PRDX4 seems to work alongside ERO1 as a wasteless system of transferring electrons from cysteine thiols to oxygen generating water in the process. The robustness of ER oxidative protein folding in higher organisms may be in part attributed to the evolutionary acquisition of these parallel pathways (the PDI-PRDX4 pathway is absent in yeast [36]). As such, ER-accumulation of H2O2 that could have detrimental effects in other cellular compartments is dealt with “in house” by the ER, through a mechanism that further promotes oxidative protein folding capacity.
The triple knockout mouse model that lacks Prdx4, Ero1a and Ero1b is viable, whilst showing a compromised oxidative folding kinetics [6] and a scurvy-like phenotype, presumably due to impaired collagen maturation [37]. This provides evidence to support that additional proteins are capable of driving oxidative protein folding in the ER (Figure 1).
The importance of PRDX4-mediated H2O2 quenching in the ER is highlighted by phenotypic consequences of its genetic manipulation: Its overexpression led to protection against glutamate-induced neuronal cell death, increased insulin production, and inhibition of adipogenesis [38,39,40]. Prdx4 knockout mice showed phenotypes such as abnormal spermatogenesis and susceptibility to drug-induced colitis, indicating a systemic requirement for this protein [41,42]. Moreover, the expression level of PRDX4 positively correlates with the prognosis of patients with lung adenocarcinoma [43]. The apparent ability of cells to maintain ER oxidative protein folding in the face of PRDX4 loss suggests that ERO1 activity is likely sufficient to perform this function, albeit driving further H2O2 production.
Besides PRDX4, ER H2O2 is also utilised by ER-resident glutathione peroxidases, GPX7 and GPX8 (ER luminal and lumen-facing membrane anchored, respectively [44]). Both can oxidise PDI in the presence of H2O2 and interact with ERO1, catalysing oxidative protein folding [29,44,45]. They can also protect insulin-secreting cells from ER stress induced by saturated fatty acids (FFAs) [45]. Metformin, a therapy for diabetes, is reported to provide antiaging and cognitive decline-diminishing benefits that are thought to be mediated by the upregulation of GPX7 [46]. Maintaining GPX7 levels seems to be important to protect from hyperactivation of ERO1 and ER stress triggered by homocysteine, which causes hyperhomocysteinemia-related vascular diseases [47]. Gpx7 knockout mice show systemic oxidative stress, carcinogenesis and shortened lifespan [48]. Given that GPX7 can protect both the ER and nuclear DNA from oxidative damage, the effects of ER ROS appear to reach beyond the lumen of the organelle [49]. Supporting this view, removal of GPX8 increases H2O2 levels in the cytosol, indicating that GPX8 may play a role in preventing leakage of H2O2 from the ER [14].
The membrane localised enzyme NADPH oxidase 4 (NOX4) provides another source of ER H2O2. This enzyme preferentially produces H2O2 rather than superoxide through a highly conserved histidine residue in its extracytosolic loop (E-loop), not present in other NOX family proteins [50]. NOX4-derived H2O2 drives multiple biological events inside and outside the ER (e.g., Ras signalling activation through ER Ca2+ efflux by sarco/endoplasmic reticulum calcium-ATPase (SERCA) oxidation [51,52], H2O2 traversal of ER membrane in this extra-ER activity is likely mediated by aquaporins, see discussion in the next section). Although NOX4 is found in other organelles, this protein has been shown to be predominantly expressed on the ER membrane in human neuroblastoma SH-SY5Y cells, human hepatocyte Hep G2 cells, and human umbilical vein endothelial (HUVEC) cells [51,53]. NOX4 interacts with calnexin, an ER membrane lectin-chaperone, with a degree of dependence on this factor: elimination of calnexin reduces NOX4 expression level as well as ROS production [54]. Moreover, NOX4 also interacts with PDI [55], and its interaction controls uncoupling of endothelial nitric oxide synthase (eNOS) and expression of β-galactosidase, which are involved in ageing of vascular endothelial cells [56].
Protective roles have been suggested for ER-localised NOX4 activity. NOX4 has been suggested to drive prolonged integrated stress response signalling by oxidatively inhibiting protein phosphatase 1 (PP1) and protecting against acute kidney injury in vivo [57].
The lumen of the ER appears to be endowed with an additional crucial layer of controlling H2O2 load. Measurements using an ER-tuned genetically encoded H2O2 sensor pointed to the role of reduced glutathione in buffering the load of ER-intrinsically generated H2O2 [13]. Relatively high ER-glutathione (approximately 10–15 mM total, mostly reduced [58,59]) may compensate for poor kinetics of GSH–H2O2 reactivity. Regulation of ER redox by glutathione seems to be mediated by its flux and Ca2+ mobilisation, at least in part [60]. However, H2O2-degrading enzymes, such as the ER-resident PRDX4 and glutathione peroxidases GPX7 and GPX8, are significantly more efficient hydrogen peroxide-degrading catalysts than GSH alone. However, above a threshold concentration, calculated to be within the range of concentrations at which H2O2 acts as a second messenger, chemical reduction of H2O2 by GSH becomes kinetically competitive with enzymatic activity, aiding the maintenance of H2O2 levels. Accordingly, when glutathione is depleted, levels of H2O2 in the ER augment, and the viability of cells with reduced cytosolic antioxidant capacity (e.g., a pancreatic β-cell model, with natural catalase deficiency) becomes strongly compromised, particularly when H2O2 production is further stimulated through the biosynthesis of disulphide-containing substrates (e.g., pro-insulin) [13]. This highlights the physiological relevance of the chemical reduction of H2O2 by GSH and further suggests that the ER constitutes a significant source of pathological ROS.
H2O2 Transport Across the ER Membrane
The abundance of H2O2 in the ER suggests that the ER membrane likely limits its conductance towards other, potentially more ROS-vulnerable cellular compartments. The H2O2 quenching mechanisms on the cytosolic side of the ER membrane likely contribute to the insulation. However, mechanistic details of what promotes or limits H2O2 permeability over membranes remains only partially understood. A view point in favour of unlimited H2O2 transit across the ER membrane was expressed [61].
Since artificially imposed H2O2 removal in other organelles does not affect H2O2 content of the ER, the influx of H2O2 into the ER from other organelles seems to be tightly restricted at steady state [6]. However, in light of measurements showing that ER-derived H2O2 can leak to the cytosol under conditions of luminal H2O2 overload, potentially causing oxidative damage in other organelles [13,14], transport of ER H2O2 across the membrane is a plausible toxicity-determining parameter. The general principles of controlled material exchange between organelles through passive channels or active transporters seem to apply in the case of H2O2: aquaporins (AQPs, described initially as water channels) have been suggested to conduct H2O2 across membranes [62,63,64,65].
In mammals, the AQP family of proteins consists of thirteen isoforms. They share a common topology of six transmembrane helices, five loops (three extracellular and two intracellular), and two highly conserved asparagine–proline–alanine (NPA) motifs. Assembled as tetramers, these proteins form an hourglass-like functional pore [66]. They are classified by three sub-groups; classical AQPs (AQP0, AQP1, AQP2, AQP4, AQP5, AQP6, AQP8), aquaglyceroporins (AQP3, AQP7, AQP9, AQP10), and unorthodox or superaquaporins (AQP11, AQP12), based on their substrates. In addition, recently a new category—peroxiporins has been identified (AQP1, AQP3, AQP5, AQP8, AQP9, AQP11), whose common feature is the ability to transport H2O2 with a degree of preference [63,67,68]. It is worth noting that all AQPs can potentially enable transport of H2O2 to some extent [69].
As all AQPs start their journey through the secretory pathway at the ER, each can potentially contribute to H2O2 transport across its membrane. Notably, AQP8 was explicitly shown to mediate H2O2 transport across the ER membrane [67]. Partial ER-localisation of AQP4 and AQP9 have also been observed [68]. Further, human AQP11 showed ER localisation and the capacity to transport H2O2 across the ER membrane [70,71].
The precise function of peroxiporins on the ER membrane currently remains obscure. One can speculate that AQPs may maintain redox homeostasis or even support cellular signalling by modulating H2O2 efflux from the ER, as well as the influx to the ER. This is indirectly supported by the finding that the expression level of NOX2 and renal oxidative damage are augmented in Aqp11 knockout mice [72]. Recent findings on hydrogen sulphide (H2S)-mediated regulation of an ER aquaporin’s conductivity support such a possibility [73].
It is conceivable that regulated and localised H2O2 conductivity of the ER membrane may constitute a mechanism for time-space specific releases of H2O2 from its “ER-store”. Such a mechanism, hitherto uncharacterized, could be considered analogous to regulated calcium releases from ER stores. In the case of the short-lived H2O2 the “storage” is afforded by its constant high productions rate in the confined space of the ER.
3. Non-ER Sources of ROS/H2O2
3.1. Mitochondrial ROS/H2O2
Though the mitochondrial matrix maintains a relatively reducing environment (redox potential of −300 mV [4]), it is thought to constitute a major source of intracellular ROS, as a by-product of its multicomponent electrochemistry machinery. However, only 15% of total H2O2 was accounted for in a mitochondrial fraction of cell extracts [16,17]. This is consistent with the abundant and diverse antioxidant enzymology of the organelle, affording protection to other organelles and its precious DNA content from oxidative damage.
Mitochondria synthesise ATP through oxidative phosphorylation, coupled to the electron transport chain (ETC). Leakage of electrons from the ETC, as well as other related enzymes, leads to a partial reduction of oxygen to form superoxide, and it is subsequently converted to H2O2 (a less reactive oxidant) and oxygen by superoxide dismutase 1 (SOD1) in mitochondrial intermembrane space, and SOD2 in mitochondrial matrix. Although most of the oxygen in the cell is consumed by mitochondria, only 1–2% seems to be converted to ROS [17]. Systemic homozygous knockout of SOD2 in mice led to pre-weaning lethality accompanied by multiple tissue dysfunctions such as cardiomyopathy, fatty liver, metabolic acidosis, and neurodegeneration [74,75]. In contrast, overexpression of both SOD2 and catalase protects the cell from antiretroviral-induced oxidative stress and cardiomyopathy [76].
To date, eleven ETC-related enzymes have been identified as the source of ROS in mitochondria [77,78]. Four of them are present in distinct 2-oxoacid dehydrogenase complexes, which catalyse the oxidative decarboxylation of different 2-oxoacids to the corresponding acyl-CoA and NADH. These are 2-oxoglutarate dehydrogenase complex (OGDHC), pyruvate dehydrogenase complex (PDHC), branched-chain 2-oxoacids dehydrogenase complex (BCOADHC), and 2-oxoadipate dehydrogenase complex (OADHC). ROS production from these complexes seems to be much higher than that from complex I, which has been previously considered as one of the major mitochondrial ROS sources [79]. Complex I (NADH ubiquinone oxidoreductase) generates H2O2 at a flavin-containing site and a quinone-binding site. Notably, an improved genetically encoded H2O2-specific sensor (HyPer7), detected complex1-derived H2O2, which is driven by its selective inhibitor rotenone, appears to be released toward the mitochondrial matrix side [80]. The remaining four of the ROS sources are located in dehydrogenases, linked to ubiquinone: dihydroorotate dehydrogenase (DHODH), the electron-transferring flavoprotein (ETF)–ubiquinone oxidoreductase system (ETF-QO), mitochondrial glycerol-3-phosphate dehydrogenase (mGPDH) as well as complex II (succinate dehydrogenase, SDH). ROS can also emanate from the outer ubiquinone-binding site of complex III [81].
Furthermore, mitochondria-localising NADPH oxidase can provide a source of H2O2. NOX4 is predominantly localised to mitochondria in certain cells such as mouse podocytes, mesangial cells and kidney cortex [82,83]. Mitochondrial NOX4-derived H2O2 was implicated in ageing-dependent aortic stiffening and drug resistance [84,85].
Of interest, cardiomyopathy triggered by the ablation of desmin, a major muscle-specific intermediate filament protein, is ameliorated by catalase overexpression but deteriorated by SOD2 overexpression [86]. This can be explained by either augmenting mitochondrial H2O2 or diminished superoxide and implies that balancing mitochondrial ROS maintains cellular homeostasis. Indeed, mitochondrial ROS seems to regulate hypoxic signalling [87]. In addition, mitochondrial ROS are reported to be required for expressions of hypoxia-inducible genes such as erythropoietin and Hypoxia-inducible factor 1 (HIF-1) [88], as well as enhancing cytosolic calcium levels in pulmonary arterial smooth muscle cells (PASMCs) during hypoxic pulmonary vasoconstriction (HPV) [89,90].
Notably, the majority of proteins in mitochondrial intermembrane space are known to contain disulfide bonds as a post-translational modification [91], and an electron relay mediated by ERV1, a mitochondrial FAD-linked sulfhydryl oxidase which mediates disulfide bond formation, results in H2O2 production analogous to the ERO1-pathway. H2O2 produced by ERV1 is scavenged by cytochrome C, likely forming another mode by which mitochondria are protected from the toxicity of H2O2 [92]. Uncoupling proteins (UCPs), the modulators of thermogenesis by uncoupling mitochondrial proton gradient and ATP synthesis, also seem to be important in mitochondrial H2O2 production regulation [93,94,95].
3.2. Peroxisomal ROS/H2O2
Peroxisomes are single-membrane-bounded organelles present in most eukaryotes. A variety of oxidation reactions, such as β-oxidation of fatty acids, biosynthesis of cholesterol, and metabolism of amino acids and purines are carried out in this organelle. Accordingly, the production of peroxisomal ROS is reportedly mediated by: xanthine oxidase, D-amino oxidase, urate oxidase, and Acyl-CoA oxidase [96]. Despite its relatively small size, the peroxisome is estimated to generate about 35% of total intracellular H2O2 and therefore is considered as a major source of intracellular ROS [97,98,99].
Peroxisomal ROS can function as a signal molecule and seem to be functionally associated with autophagy in particular. For instance, the negative regulators of mammalian target of rapamycin complex 1 (mTORC1), tuberous sclerosis complex 1 (TSC1) and 2 are localised to peroxisomes, where they suppress mTORC1 signalling and thus induce autophagy in response to ROS [100]. Moreover, peroxisomal ROS induce selective autophagy of peroxisomes themselves (pexophagy) [101]. Ataxia-telangiectasia mutated (ATM), a serine/threonine kinase responsive to DNA damage, is also activated at peroxisomes in response to peroxisomal ROS to induce autophagy and pexophagy [102]. It also has been suggested that peroxisomal ROS serve as a measure for quality control of the organelle’s function [103].
Use of a peroxisome-targeted H2O2 sensor revealed that peroxisomal redox milieu is kept reductive at steady-state despite abundant ROS productions [104]. This can be attributed to abundant peroxisome-resident antioxidative enzymes such as catalase, SOD1, SOD2, PRDX1, PRDX5, epoxide hydrolase, a peroxisomal membrane protein 20 (PMP 20), and potentially a GPX [105]. Although no subtype of GPX has been identified in mammals as of yet, Gpx1 has been found in the peroxisomal matrix in yeast [106]. Indeed, the neuronal-specific ablation of peroxin 5 (Pex5, a peroxisomal biogenesis factor) causes peroxisome dysfunction and axonal myelin loss without causing oxidative stress in the central nervous system [107]. Furthermore, deficiency of PEX5 and PEX12 shifts the localisation of catalase from peroxisome to cytoplasm, and protects cells from oxidative damage by preferentially removing cytosolic ROS [108]. Thus, although peroxisomes are a significant source of ROS, they do not seem to be a critical source of oxidative stress, unless their multiple ROS quenching enzymes fail collectively.
3.3. Nuclear ROS/H2O2
The nucleus is the most reducing organelle in the cell [109], a fact not incongruous with its function of storing DNA, a most precious and oxidation sensitive cellular component. The reductive nuclear milieu is thought to be fuelled by thiol reductants such as thioredoxin-1 (TRX1), GPXs, PRDXs, glutathione (GSH) and specific isoforms of glutathione S-transferases. These reductants seem to scavenge nuclear ROS and function in the nucleus as well as other subcellular localisations. For instance, TRX1 localises to both nucleus and cytosol at steady state. However, in the presence of excess ROS, TRX1 preferentially localises in the nucleus [110]. Redox potential shift of nuclear TRX1 (about 60 mV oxidation from −300 mV at steady state) indicates that TRX1 attends to the oxidative stress in the nucleus [110]. Moreover, cytosolic TRX1 can interact with apoptosis-inducing factor (AIF) by forming a disulphide bond that facilitates delivery of AIF to the nucleus [111]. After this delocalisation, the reduction of TRX1–AIF complex releases AIF in the nucleus where it functions to promote apoptosis.
GSH accumulates in the nucleus during cell proliferation and spreads throughout the cell when cells are confluent. [112,113]. It can be speculated that this serves as a defensive measure to protect the nucleus from oxidative stress, as intracellular ROS increase during mitosis [114]. Indeed, depletion of GSH impairs cell proliferation [115]. GSH not only directly removes nuclear ROS but also introduces glutathionylation to nuclear proteins, such as histone H3, to regulate nucleosome stability by blocking the irreversible oxidation of cysteine residues [116]. The level of glutathionylation of H3 is high in rapidly proliferating cells such as cancer cells and low in aged cells, suggesting this redox event may be relevant to ageing and diseases.
Despite the abundance of nuclear reductants, ROS can unavoidably oxidise DNA in the nucleus. 8-Oxo-2’-deoxyguanosine (8-oxo-dG) is the most common product of DNA oxidation and has long been considered a useful, if somewhat biologically uninteresting, marker of oxidative damage. However, studies using genetic ablation of 8-oxoguanine DNA glycosylase 1 (OGG1), an enzyme that binds and removes 8-oxo-dG, have demonstrated a direct relevance of 8-oxo-dG to physical dysfunctions such as age-associated neuronal loss and susceptibility to a dopaminergic toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [117,118]. Such phenotypes are likely mediated by DNA methylation status because 8-oxo-dG recruits a DNA demethylase ten-eleven translocation 1 (TET1) via OGG1 [119]. Indeed, a negative correlation between 8-oxo-dG and 5-methyl cytosine (5mC), a methylated cytosine involved in gene transcriptions and various disorders, has been reported in gliomas [120]. Moreover, some gene expressions are upregulated by 8-oxo-dG on their loci promoter [121,122,123,124,125,126,127].
NOX4 appears as a significant nucleus-intrinsic source of ROS. Nuclear ROS emanating from NOX4 have been demonstrated to cause DNA damage, relevant to diseases such as myelodysplastic syndromes and viral infection [128,129], with a conceivable extension to neurodegeneration-associated DNA damage. Knockdown of NOX4 in human endothelial cells diminishes DNA damage and expanded cellular lifespan, displaying a strong relevance to senescence [130]. Intriguingly, NOX4 activity is negatively regulated by FYN, a tyrosine kinase belonging to SRC family kinases, implying programmed regulation of nuclear ROS by NOX4 as a result of cellular signalling, which seems to be relevant to cardiac remodelling [131].
3.4. Golgi ROS/H2O2
There are fewer reports related to ROS production and oxidative stress in the Golgi apparatus than perhaps any other organelle. Since the main functions of the Golgi are secretory proteins’ glycosylation and sorting, studies exploring redox reactions in this compartment are relatively infrequent. However, the Golgi’s oxidative environment, which is equivalent to that of the ER, supports the plausibility of ROS production in this compartment [132].
Quiescin-sulfhydryl oxidases (QSOXs), flavin-linked sulfhydryl oxidases, are the most likely candidate for Golgi ROS production. All QSOX isoforms possess an ER signal peptide and are transported from the ER to the Golgi like generic secretory proteins lacking ER retention signals. Immunohistochemical analysis has also indicated a significant Golgi localised pool of this protein [133,134,135,136]. QSOX isoforms consist of two thioredoxin domains containing CxxC motifs and FAD-binding domain at the N-terminus. As such, they can oxidise thiol groups to form disulphide bonds with the reduction of oxygen and production of H2O2, much like PDI and ERO1 in the ER [137]. Indeed, a recent report demonstrates QSOX1-dependent H2O2 production in human placenta-derived trophoblast and involvement of this protein in the pathogenesis of preeclampsia [138]. On the other hand, QSOX1 overexpression protects rat pheochromocytoma PC12 cells from oxidative stress-induced cell death, although it has also been reported to slow down cellular proliferation [139], hence potential benefits of QSOXs (and Golgi ROS in general) may depend on tissue and cell type. Moreover, upregulation of QSOXs expression is found in several types of cancers such as breast, pancreas, and prostate cancers, suggesting the relevance of this gene to cancer development [140,141]. Interestingly, higher expression of QSOX1 seems to reduce tumorigenesis and is associated with better outcomes for breast cancer patients (relations to known functions of QSOXs remains unclear) [142]. Exogenously expressed QSOX1 restores impaired viability and disulphide bond formation capacity in Ero1p deficient yeast, suggesting that QSOX can rectify oxidative folding defects in proteins that, notwithstanding the ER quality control system, make it to the Golgi [136]. On the other hand, QSOX1 knocked-down fibroblasts fail to incorporate an extracellular matrix (ECM) protein—laminin, but not collagen IV and fibronectin [133]. Markedly, this defect is rescued by the addition of recombinant QSOX1 to the cell culture media [133]. These suggest that QSOX1, and its single-chain arrangement of ERO1 and PDI equivalents may specialise on a subset of secretory proteins and act extracellularly.
The existence of other redox enzymes further implies the importance of redox regulation in the Golgi. In yeast, glutaredoxin 6 and 7 (Grx6, Grx7) are found to locate to the cis-Golgi preferentially [143]. Grx6 localises at both the ER and the Golgi, and Grx7 predominantly localises at the Golgi. The glutaredoxin family are thought to play roles in detoxification of oxidative damage together with GSH, GPXs, and NADPH, preventing H2O2 production [144]. The Golgi is apparently protected from damage by Grx6 and Grx7 upregulation in response to several stresses such as sodium and peroxides (as well as calcium, specifically for Grx6), but not in response to heat shock and osmotic stress [145]. Although mammalian orthologs of these genes have not yet been identified, similar enzymes may exist that determine the level of H2O2 in the Golgi.
In addition to the above-mentioned proteins, one isoform of NOS may be considered as another source of H2O2 in the Golgi. The NOS family are the enzymes that catalyse the production of NO from L-arginine and consists of three isozymes: neuronal NOS (nNOS), cytokine-inducible NOS (iNOS), and endothelial NOS (eNOS). Of these, eNOS is reported to locate to the Golgi membrane as well as the plasma membrane caveolae in endothelial cells [146,147,148]. All NOS proteins require dimerisation and several cofactors, such as nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), calmodulin (CaM), Haeme, and tetrahydrobiopterin (BH4). Although NOS-derived NO underlies productions of reactive nitrogen species (RNS), a subset of free radicals, uncoupled eNOS resulting from L-arginine and/or BH4 deficit, lead to the production of superoxide and subsequent H2O2 (and molecular oxygen). This reaction can occur spontaneously or as SOD-catalysed dismutation [149,150].
The biological consequences of H2O2 produced in the Golgi are still elusive. Recently, a novel Golgi-targeted H2O2 specific probe termed Np-Golgi has been developed and revealed augmented H2O2 in the Golgi in hypertensive mice [151].
4. ROS/H2O2 and Neurodegeneration
Oxidative stress is a hallmark of several diseases, including dementia and related neurodegeneration pathologies. Among ROS, H2O2 is often considered as the central player of redox-regulated events disrupted in neurodegenerative disorders [152,153,154]. A direct link between ROS and disease is provided by the pathology associated with SOD, an enzyme that plays a role in redox homeostasis by converting superoxide anion radicals into H2O2 and oxygen. Mutations in SOD1 have been shown to cause Amyotrophic Lateral Sclerosis (ALS) [155]. SOD1 aggregation in the motor neurons seems to be caused by oxidation of two of its free cysteines in the mitochondria where oxidative stress may occur [156]. Moreover, the excess activity of SOD1 due to a third copy of its encoding gene in Down syndrome results in the accumulation of H2O2 that may reach toxic levels sufficient to promote neuronal death [157]. An extensive body of evidence implicates oxidative stress in Alzheimer’s Disease (AD). In particular, peroxiredoxin levels are altered in AD, and lipid peroxidation is found in positive correlation with brain amyloid beta (Aβ)-plaque load (the hallmark and genetic determinant of AD), suggesting a contribution of ROS to AD pathogenesis [158,159]. Furthermore, in Huntington’s disease, oxidative damage compromises low-molecular-weight antioxidant metabolism (i.e., reduced cysteine, GSH levels [160,161,162,163], and the uptake of reduced ascorbate [164,165]) and elevates the production of free radicals. Abundant evidence also suggests redox imbalance in other neurodegenerative diseases such as Parkinson’s disease [160], and in prion pathology, where a burst of ROS accompanies neurodegeneration [166].
Thus, the substantial association of ROS with neurodegeneration suggested the use of antioxidants as a therapeutic strategy. Scavenging ROS by administration of antioxidants has been considered as a valid therapeutic strategy for several diseases, including neurodegeneration. For instance, trolox (a water-soluble vitamin E analogue) decreases aggregation of mutant SOD1 (SOD1G86R), which is a causing factor of familial ALS, in vitro [167]. Ascorbic acid (vitamin C) and a natural antioxidative compound berberine-derivative can suppress Aβ aggregation and thus AD pathology [168,169]. In addition, in vivo studies have demonstrated neuroprotective effects of some antioxidants, such as rosmarinic acid, coenzyme Q10, and α-tocopherol (vitamin E), in AD, PD, and Down syndrome animal models, respectively [170,171,172,173].
However, despite these encouraging results using antioxidants as neuroprotectants in cell systems [174], beneficial outcomes at the organism level are inconsistent and, in large part, disappointing [175,176,177]. The reasons for those failures are not immediately clear and may have to do with their effective bioavailability and/or antioxidants’ effects on physiological roles of ROS in cell signalling, (H2O2, in particular [178,179]). Significant antioxidant beneficial effects against neurodegeneration are predominantly seen at concentrations nearing their toxicity range when redox-dependent vital signalling pathways are also likely to be perturbed [176]. Developing organelle-selective antioxidants such as 2-[2-(triphenylphosphonio)ethyl]-3,4-dihydro-2, 5,7,8-tetramethyl-2H-1-benzopyran-6-ol bromide (TPPB) and MitoQ, mitochondria-targeting antioxidants, may half-circumvent some of these limitations [180,181].
Rescuing redox homeostasis poses a challenge ahead to effective treatments against oxidative stress, which requires targeted strategies that are often confounded by the low specificity of most antioxidants in general. Favourably, metabolic antioxidants acting on specific metabolic transformations seem to have exerted alleviating effects in several animal models of neurodegenerative diseases [176]. Specific examples in cultured cell models include the apparent benefit afforded through modulating ROS-detoxifying selenoenzymes such as glutathione peroxidases, thioredoxin reductases and selenoprotein P [182,183,184].
Another major hurdle in developing neuroprotective antioxidants seems to be their low blood–brain barrier permeability [177]. ROS are short-lived species due to their high reactivity. Some are able to oxidise cysteine and methionine residues, the DNA base guanine is prone to oxidative damage by ROS, lipids can undergo peroxidation, and even carbohydrates are prone to oxidative degradation and depolymerisation [160]. Quenching of ROS by antioxidants to prevent oxidative damage requires fast kinetics of reduction, which are not a given for all antioxidants. The problem may be further aggravated by limited bioavailability of antioxidants at the relevant location. For instance, quenching of H2O2 by reduced glutathione in the ER becomes kinetically competitive with enzyme catalysis only at 0.23 µM of H2O2 [13]. Thus, slow kinetics of antioxidant-ROS reactions may permit oxidative damage prior to ROS quenching events.
The challenge posed is to nullify disease-relevant ROS with spatiotemporal specificity whilst leaving physiological ROS intact, which requires the drawback of antioxidant’s low-specificity to be circumvented. In targeting disease-relevant enzymatic sources of ROS, ROS-toxifiers (that convert relatively non-toxic ROS to more reactive species) or proteins that repair other oxidatively damaged proteins has been proposed as a more rational strategy to interfere with ROS than the use of antioxidants [185]. Understanding intracellular ROS–source pathways is a key to informing such strategies.
Reports establishing a connection between oxidative stress and neurodegenerative disorders are innumerous and very disparate regarding a rational and causative link between them. Whether oxidative stress elicits neurodegeneration or is a consequence of neuronal cell death triggered by a combination of genetic and epigenetic factors and environmental interactions is still a matter of debate. The table below compiles a selection of findings on these relationships (Table 1).
ER H2O2 and Neurodegeneration
The extent of UPR activation strongly correlates with the pathology of human patients carrying neurodegenerative diseases [231], and neurodegeneration-causing factors, such as Aβ1–42 oligomer and tau, can also augment UPR [232,233], suggesting that excess UPR (ER stress) synergically contributes to the development of neurodegeneration. Given that UPR is originally an adaptive signalling pathway that governs quality control of proteins in the ER, target genes of UPR should be expected to be neuroprotective. PDI in particular, is considered as a potential target UPR-gene. Indeed, PDIA3 (ERp57) protects from ischemia-induced brain damage [234]. Further, upregulation of PDI expression in the central nervous system (CNS) seems to be a common feature observed in the patients and animal model of several neurodegenerative diseases such as AD, PD, ALS, and HD [235,236,237,238,239]. Interestingly, reversible/irreversible inhibitors of PDI show neuroprotective effects in vitro [240,241]. In addition, inhibitions of PDI by CCF642 or LOC14 appear neuroprotective in a mouse model of Experimental autoimmune encephalomyelitis (EAE) and HD [242,243]. ERO1 inhibitor EN460 also antagonises neurotoxicity [244]. Seemingly paradoxical observations that both inhibitors of these proteins and their upregulation show neuroprotective effects [169,245,246] can be reconciled under the assumption that their activity is crucial, but their by-product-ROS is hazardous. Consistently, small molecule antioxidants relieve not only oxidative load but also ER stress [247], probably at least in part by removing PDI and ERO1-derived ROS.
Further, ER-antioxidative enzymes benefit neuronal health: PRDX4 can protect from Aβ oligomer- or glutamate-mediated cell death presumably by suppressing ER stress [38,248]. While GPX7 deficient mice show an accelerated, aging-related neurodegeneration, accompanied by elevated ROS, in spinal motor neuron [245]. A brain transcriptome analysis reveals that this gene is significantly upregulated in response to neurotoxin MPTP [246,249].
Together these observations suggest that if excess ER-ROS can be eliminated from the cell, the beneficial outcomes of UPR should be expected to antagonise the neuropathophysiology more effectively (Figure 2).
5. Conclusions and Perspectives
In this review, we have outlined the sources and known functionalities of ROS from the ER and other organelles. Organelles harbour pathways that produce ROS as a by-product of redox reactions and/or to be utilised as a signalling factor. Uniquely, the ER utilises a ROS, H2O2 to directly fuel is anabolic activity—the oxidative protein folding. This organelle satisfies its own demand of ROS by internally producing abundant H2O2, on a scale that may well exceed that of any other organelle. As the ER constitutes a major source of ROS in the cells, the organelle’s homeostasis often correlates with cellular redox, explaining the simultaneous presence of ER stress and oxidative stress in the process of ageing and diseases.
It is plausible that antagonising ROS with antioxidants may have utility in alleviating the oxidative damage in certain disease states. However, the use of antioxidants as a therapeutic strategy is still controversial due to the uncertainty of their usefulness in humans. This may be because the comprehensive scavenging of systemic ROS not only relieves oxidative stress but also deprives the physiological advantages of ROS, which drive biological reactions by physiological oxidation of target molecules. Although distinguishing ROS benefit and toxicity in vivo is a significant challenge, this dilemma must be resolved to some extent by source-selective suppression of excessive ROS. As mentioned in this review, some molecules have indeed been developed such as TPPB and mitoQ (mitochondria-targeting antioxidants) and CCF642, LOC14, and EN460 (PDI and ERO1 inhibitor, respectively) to decrease local ROS productions. Therefore, controlling spatial ROS productions will be a future focus for providing antioxidants a utility as therapeutics. Finally, visualisation of ROS in live-cells has become clearer by the development of organelle-targeted genetically encoded redox sensors, such as roGFP and HyPer, as well as chemical probes. Most of these probes can detect ROS reversibly, allowing the monitoring of homeostatic ROS production and consumption. Indeed, these techniques also capture the redox state of tissues in vivo [250,251]. In addition to these tools, further development of biosensor implementations of image acquisition technology such as two-photon microscopy, super-resolution microscopy, and single-molecule analysis will likely enable mapping of physiological ROS with higher spatiotemporal resolution. The elucidation of local ROS functionality will be expected to provide new clues to aid our understanding of the pathophysiology not only of neurodegenerative diseases, but also ageing, cancer, and other related diseases.
Author Contributions
Conceptualisation, E.A.; original draft preparation, T.K., E.P.M., J.E.C. and E.A.; All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant from the UK Dementia Research Institute, which receives its funding from the Medical Research Council, Alzheimer’s Research and Alzheimer’s Society UK. EPM acknowledges Portuguese national funds, FCT—Foundation for Science and Technology through project UID/Multi/04326/2019, from the operational programs CRESC Algarve 2020 and COMPETE 2020 through project EMBRC.PT ALG-01-0145-FEDER-022121. JEC acknowledges MRC-MRC Ref MCMB MR/R009120/1, Alpha-1 foundation, and a Grifols ALTA training award.
Acknowledgments
We profoundly thank Ilir Mehmeti (Hannover Medical School) for critical reading this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Glossary
| Superoxide (O2−) | A radical ROS produced by the one-electron reduction of molecular oxygen. It is converted to hydrogen peroxide and molecular oxygen by spontaneous and/or SOD-catalysing dismutation. |
| Hydrogen peroxide (H2O2) | A non-radical, relatively stable and functional ROS, hazardous in excess. |
| Hydroxyl radical (·OH) | Highly reactive and short-lived radical. Its production is driven by Fenton reaction, between ferrous ion (Fe2+) and hydrogen peroxide. |
| Redox potential | Expressed in millivolts (mV), a measure of redox state that reflects the tendency of a chemical to receive/donate electrons, and concentrations of its reduced and oxidised species, given by the Nernst equation |
| Superoxide dismutase (SOD) | An antioxidant enzyme catalysing dismutation of superoxide to hydrogen peroxide and molecular oxygen. SOD family consists of Cu/Zn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3). Mutations in the SOD1 gene affect approximately 20% of familial amyotrophic lateral sclerosis (ALS). |
| Peroxiredoxin (PRDX) | An antioxidant enzyme scavenging peroxides including H2O2. PRDX family consists of six isoforms (PRDX1-6). PRDX4 is localised in the ER. |
| Glutathione peroxidase (GPX) | An antioxidant enzyme scavenging H2O2 as well as lipid peroxides by utilising reduced glutathione. GPX family consists of eight isoforms (GPX1-8). GPX1-4 and GPX6 contain an active site selenocysteine (selenoproteins). GPX7 and GPX8 are localised in the ER. GSH peroxidase activity of GPX7 and GPX8 is low. |
| Glutathione | An abundant antioxidant tripeptide consisting of alanine–cysteine–glutamate (with cysteine and glutamate linked by gamma–glutamyl bond). Reduced glutathione (GSH) drives GPXs reactions and as a result GSH is converted to the oxidised form (GSSG), in which two molecules are bound by disulfide bond. GSSG is recycled by reducing by glutathione reductase (GR). |
| Catalase | An antioxidant enzyme that scavenges H2O2. The iron-bound heme group in the active site catalyses the conversion of H2O2 to water (H2O) and molecular oxygen (O2). It mainly localises in the peroxisomes. |
| Protein disulfide isomerase (PDI) | An ER chaperone possessing oxidoreductase activity via CXXC catalytic motif-containing thioredoxin domains. Catalyses protein disulphide bond formation and isomerisation during folding of ER client proteins. |
| ER oxidoreductin 1 (ERO1) | A sulfhydryl oxidase that supplies oxidising equivalents to recycle PDI, in the process ERO1 produces H2O2 by reducing O2 with its active site (CXXCXXC motif/flavin adenine dinucleotide (FAD) prosthetic group). |
| NADPH (nicotinamide adenine dinucleotide phosphate) oxidase (NOX) | An enzyme catalysing the production of a superoxide by transferring one electron to oxygen from NADPH. The NOX family consists of NOX1~NOX5, Dual oxidase 1 (DUOX1) and DUOX2. Among NOXs, NOX4 preferentially produces H2O2 rather than superoxide. |
References
- Paravicini, T.M.; Touyz, R.M. NADPH oxidases, reactive oxygen species, and hypertension: Clinical implications and therapeutic possibilities. Diabetes Care 2008, 31 (Suppl. 2), S170–S180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Jones, D.P.; Go, Y.M. Redox compartmentalization and cellular stress. Diabetes Obes. Metab. 2010, 12, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Åslund, F.; Storz, G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 1998, 279, 1718–1721. [Google Scholar] [CrossRef]
- Konno, T.; Melo, E.P.; Lopes, C.; Mehmeti, I.; Lenzen, S.; Ron, D.; Avezov, E. ERO1-independent production of H2O2 within the endoplasmic reticulum fuels Prdx4-mediated oxidative protein folding. J. Cell Biol. 2015, 211, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 3, 281–286. [Google Scholar] [CrossRef]
- Giannoni, E.; Buricchi, F.; Grimaldi, G.; Parri, M.; Cialdai, F.; Taddei, M.L.; Raugei, G.; Ramponi, G.; Chiarugi, P. Redox regulation of anoikis: Reactive oxygen species as essential mediators of cell survival. Cell Death Differ. 2008, 15, 867–878. [Google Scholar] [CrossRef]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen peroxide activates NF-κB through tyrosine phosphorylation of IκBα and serine phosphorylation of p65. Evidence for the involvement of IκBα kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef] [Green Version]
- Fourquet, S.; Guerois, R.; Biard, D.; Toledano, M.B. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J. Biol. Chem. 2010, 285, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Birk, J.; Meyer, M.; Aller, I.; Hansen, H.G.; Odermatt, A.; Dick, T.P.; Meyer, A.J.; Appenzeller-Herzog, C. Endoplasmic reticulum: Reduced and oxidized glutathione revisited. J. Cell Sci. 2013, 126, 1604–1617. [Google Scholar] [CrossRef] [Green Version]
- Kojer, K.; Bien, M.; Gangel, H.; Morgan, B.; Dick, T.P.; Riemer, J. Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J. 2012, 31, 3169–3182. [Google Scholar] [CrossRef] [Green Version]
- Melo, E.P.; Lopes, C.; Gollwitzer, P.; Lortz, S.; Lenzen, S.; Mehmeti, I.; Kaminski, C.F.; Ron, D.; Avezov, E. TriPer, an optical probe tuned to the endoplasmic reticulum tracks changes in luminal H2O2. BMC Biol. 2017, 15. [Google Scholar] [CrossRef] [Green Version]
- Ramming, T.; Hansen, H.G.; Nagata, K.; Ellgaard, L.; Appenzeller-Herzog, C. GPx8 peroxidase prevents leakage of H2O2 from the endoplasmic reticulum. Free Radic. Biol. Med. 2014, 70, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. In Methods in Enzymology; Academic Press Inc.: Cambridge, MA, USA, 2013; Volume 528, pp. 3–25. ISBN 9780124058811. [Google Scholar]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Ellgaard, L. The human PDI family: Versatility packed into a single fold. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shergalis, A.G.; Hu, S.; Bankhead, A.; Neamati, N. Role of the ERO1-PDI interaction in oxidative protein folding and disease. Pharmacol. Ther. 2020, 210, 107525. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Princiotta, M.F.; Finzi, D.; Qian, S.B.; Gibbs, J.; Schuchmann, S.; Buttgereit, F.; Bennink, J.R.; Yewdell, J.W. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 2003, 18, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Gess, B.; Hofbauer, K.H.; Wenger, R.H.; Lohaus, C.; Meyer, H.E.; Kurtz, A. The cellular oxygen tension regulates expression of the endoplasmic oxidoreductase ERO1-Lα. Eur. J. Biochem. 2003, 270, 2228–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemori, Y.; Sakaguchi, A.; Matsuda, S.; Mizukami, Y.; Sakurai, H. Stress-induced transcription of the endoplasmic reticulum oxidoreductin gene ERO1 in the yeast Saccharomyces cerevisiae. Mol. Genet. Genomics 2006, 275, 89–96. [Google Scholar] [CrossRef]
- Pagani, M.; Fabbri, M.; Benedetti, C.; Fassio, A.; Pilati, S.; Bulleid, N.J.; Cabibbo, A.; Sitia, R. Endoplasmic reticulum oxidoreductin 1-Lβ (ERO1-Lβ), a human gene induced in the course of the unfolded protein response. J. Biol. Chem. 2000, 275, 23685–23692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Ramming, T.; Okumura, M.; Kanemura, S.; Baday, S.; Birk, J.; Moes, S.; Spiess, M.; Jenö, P.; Bernèche, S.; Inaba, K.; et al. A PDI-catalyzed thiol-disulfide switch regulates the production of hydrogen peroxide by human Ero1. Free Radic. Biol. Med. 2015, 83, 361–372. [Google Scholar] [CrossRef]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Curran, S.P.; Ruvkun, G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007, 3, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Frand, A.R.; Kaiser, C.A. The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol. Cell 1998, 1, 161–170. [Google Scholar] [CrossRef]
- Pollard, M.G.; Travers, K.J.; Weissman, J.S. Ero1p: A novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol. Cell 1998, 1, 171–182. [Google Scholar] [CrossRef]
- Zito, E.; Chin, K.T.; Blais, J.; Harding, H.P.; Ron, D. ERO1-β, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J. Cell Biol. 2010, 188, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, Å.; Neubert, T.A.; Ron, D. Oxidative Protein Folding by an Endoplasmic Reticulum-Localized Peroxiredoxin. Mol. Cell 2010, 40, 787–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zito, E.; Hansen, H.G.; Yeo, G.S.H.; Fujii, J.; Ron, D. Endoplasmic Reticulum Thiol Oxidase Deficiency Leads to Ascorbic Acid Depletion and Noncanonical Scurvy in Mice. Mol. Cell 2012, 48, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.H.; Kim, M.H.; Lee, H.J.; Huh, J.W.; Lee, H.S.; Lee, D.S. Peroxiredoxin 4 attenuates glutamate-induced neuronal cell death through inhibition of endoplasmic reticulum stress. Free Radic. Res. 2020, 54, 207–220. [Google Scholar] [CrossRef]
- Mehmeti, I.; Lortz, S.; Elsner, M.; Lenzen, S. Peroxiredoxin 4 improves insulin biosynthesis and glucoseinduced insulin secretion in insulin-secreting INS-1E cells. J. Biol. Chem. 2014, 289, 26904–26913. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kim, M.H.; Lee, H.J.; Huh, J.W.; Lee, S.R.; Lee, H.S.; Lee, D.S. Peroxiredoxin 4 inhibits insulin-induced adipogenesis through regulation of ER stress in 3T3-L1 cells. Mol. Cell. Biochem. 2020, 468, 97–109. [Google Scholar] [CrossRef]
- Iuchi, Y.; Okada, F.; Tsunoda, S.; Kibe, N.; Shirasawa, N.; Ikawa, M.; Okabe, M.; Ikeda, Y.; Fujii, J. Peroxiredoxin 4 knockout results in elevated spermatogenic cell death via oxidative stress. Biochem. J. 2009, 419, 149–158. [Google Scholar] [CrossRef]
- Takagi, T.; Homma, T.; Fujii, J.; Shirasawa, N.; Yoriki, H.; Hotta, Y.; Higashimura, Y.; Mizushima, K.; Hirai, Y.; Katada, K.; et al. Elevated ER stress exacerbates dextran sulfate sodium-induced colitis in PRDX4-knockout mice. Free Radic. Biol. Med. 2019, 134, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, K.; Guo, X.; Shioya, A.; Zhang, J.; Zheng, J.; Kurose, N.; Ishibashi, H.; Motono, N.; Uramoto, H.; Yamada, S. The impact of PRDX4 and the EGFR mutation status on cellular proliferation in lung adenocarcinoma. Int. J. Med. Sci. 2019, 16, 1199–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.R.; Lappi, A.K.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.H.; Wang, C.C.; Ruddock, L.W. Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Mehmeti, I.; Lortz, S.; Avezov, E.; Jörns, A.; Lenzen, S. ER-resident antioxidative GPx7 and GPx8 enzyme isoforms protect insulin-secreting INS-1E β-cells against lipotoxicity by improving the ER antioxidative capacity. Free Radic. Biol. Med. 2017, 112, 121–130. [Google Scholar] [CrossRef]
- Fang, J.; Yang, J.; Wu, X.; Zhang, G.; Li, T.; Wang, X.; Zhang, H.; Wang, C.C.; Liu, G.H.; Wang, L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 2018, 17. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C.C.; Feng, J.; Wang, L. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019, 20, 46–59. [Google Scholar] [CrossRef]
- Wei, P.C.; Hsieh, Y.H.; Su, M.I.; Jiang, X.; Hsu, P.H.; Lo, W.T.; Weng, J.Y.; Jeng, Y.M.; Wang, J.M.; Chen, P.L.; et al. Loss of the Oxidative Stress Sensor NPGPx Compromises GRP78 Chaperone Activity and Induces Systemic Disease. Mol. Cell 2012, 48, 747–759. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.F.; Belkhiri, A.; Hu, T.L.; Chaturvedi, R.; Asim, M.; Wilson, K.T.; Zaika, A.; El-Rifai, W. Glutathione peroxidase 7 protects against oxidative DNA damage in oesophageal cells. Gut 2012, 61, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.-F.; Ma, Z.; Liu, Z.; Terada, L.S. Nox4-Derived H2O2 Mediates Endoplasmic Reticulum Signaling through Local Ras Activation. Mol. Cell. Biol. 2010, 30, 3553–3568. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.F.; Liao, C.; Hatoum, H.; Fu, G.; Ochoa, C.D.; Terada, L.S. RasGRF couples Nox4-dependent endoplasmic reticulum signaling to Ras. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auer, S.; Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Breitenbach-Koller, H.; Geisberger, R.; Aigner, E.; Cadamuro, J.; Richter, K.; Sopjani, M.; et al. The human NADPH oxidase, Nox4, regulates cytoskeletal organization in two cancer cell lines, HepG2 and SH-SY5Y. Front. Oncol. 2017, 7, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, K.K.; Wittig, I.; Leisegang, M.S.; Groenendyk, J.; Weissmann, N.; Michalak, M.; Jansen-Dürr, P.; Shah, A.M.; Brandes, R.P. The endoplasmic reticulum chaperone calnexin is a NADPH oxidase NOX4 interacting protein. J. Biol. Chem. 2016, 291, 7045–7059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janiszewski, M.; Lopes, L.R.; Carmo, A.O.; Pedro, M.A.; Brandes, R.P.; Santos, C.X.C.; Laurindo, F.R.M. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J. Biol. Chem. 2005, 280, 40813–40819. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Zeeshan, H.M.A.; Kim, H.R.; Chae, H.J. Nox4 regulates the eNOS uncoupling process in aging endothelial cells. Free Radic. Biol. Med. 2017, 113, 26–35. [Google Scholar] [CrossRef]
- Santos, C.X.; Hafstad, A.D.; Beretta, M.; Zhang, M.; Molenaar, C.; Kopec, J.; Fotinou, D.; Murray, T.V.; Cobb, A.M.; Martin, D.; et al. Targeted redox inhibition of protein phosphatase 1 by Nox4 regulates eIF 2α-mediated stress signaling. EMBO J. 2016, 35, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Bass, R.; Ruddock, L.W.; Klappa, P.; Freedman, R.B. A Major Fraction of Endoplasmic Reticulum-located Glutathione Is Present as Mixed Disulfides with Protein. J. Biol. Chem. 2004, 279, 5257–5262. [Google Scholar] [CrossRef] [Green Version]
- Montero, D.; Tachibana, C.; Rahr Winther, J.; Appenzeller-Herzog, C. Intracellular glutathione pools are heterogeneously concentrated. Redox Biol. 2013, 1, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Lizák, B.; Birk, J.; Zana, M.; Kosztyi, G.; Kratschmar, D.V.; Odermatt, A.; Zimmermann, R.; Geiszt, M.; Appenzeller-Herzog, C.; Bánhegyi, G. Ca2+ mobilization-dependent reduction of the endoplasmic reticulum lumen is due to influx of cytosolic glutathione. BMC Biol. 2020, 18. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Bánhegyi, G.; Bogeski, I.; Davies, K.J.A.; Delaunay-Moisan, A.; Forman, H.J.; Görlach, A.; Kietzmann, T.; Laurindo, F.; Margittai, E.; et al. Transit of H2O2 across the endoplasmic reticulum membrane is not sluggish. Free Radic. Biol. Med. 2016, 94, 157–160. [Google Scholar] [CrossRef] [Green Version]
- Bienert, G.P.; Møller, A.L.B.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dynowski, M.; Schaaf, G.; Loque, D.; Moran, O.; Ludewig, U. Plant plasma membrane water channels conduct the signalling molecule H2O2. Biochem. J. 2008, 414, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta Biomembr. 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Hooijmaijers, C.; Rhee, J.Y.; Kwak, K.J.; Chung, G.C.; Horie, T.; Katsuhara, M.; Kang, H. Hydrogen peroxide permeability of plasma membrane aquaporins of Arabidopsis thaliana. J. Plant Res. 2012, 125, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.S.; Preston, G.M.; Smith, B.L.; Guggino, W.B.; Agre, P. Molecular structure of the water channel through aquaporin CHIP. The hourglass model—PubMed. J. Biol. Chem. 1994, 269, 14648–14654. [Google Scholar] [CrossRef]
- Bertolotti, M.; Bestetti, S.; García-Manteiga, J.M.; Medraño-Fernandez, I.; Dal Mas, A.; Malosio, M.L.; Sitia, R. Tyrosine Kinase signal modulation: A matter of H2O2 membrane permeability? Antioxid. Redox Signal. 2013, 19, 1447–1451. [Google Scholar] [CrossRef] [Green Version]
- Cavazzin, C.; Ferrari, D.; Facchetti, F.; Russignan, A.; Vescovi, A.L.; La Porta, C.A.M.; Gritti, A. Unique expression and localization of aquaporin- 4 and aquaporin-9 in murine and human neural stem cells and in their glial progeny. Glia 2006, 53, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Almasalmeh, A.; Krenc, D.; Wu, B.; Beitz, E. Structural determinants of the hydrogen peroxide permeability of aquaporins. FEBS J. 2014, 281, 647–656. [Google Scholar] [CrossRef]
- Bestetti, S.; Galli, M.; Sorrentino, I.; Pinton, P.; Rimessi, A.; Sitia, R.; Medraño-Fernandez, I. Human aquaporin-11 guarantees efficient transport of H2O2 across the endoplasmic reticulum membrane. Redox Biol. 2020, 28, 101326. [Google Scholar] [CrossRef]
- Morishita, Y.; Matsuzaki, T.; Hara-chikuma, M.; Andoo, A.; Shimono, M.; Matsuki, A.; Kobayashi, K.; Ikeda, M.; Yamamoto, T.; Verkman, A.; et al. Disruption of Aquaporin-11 Produces Polycystic Kidneys following Vacuolization of the Proximal Tubule. Mol. Cell. Biol. 2005, 25, 7770–7779. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, Y.; Sonoda, H.; Nishimura, R.; Mori, K.; Ishibashi, K.; Ikeda, M. Involvement of the NADPH oxidase 2 pathway in renal oxidative stress in Aqp11-/- mice. Biochem. Biophys. Rep. 2019, 17, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Bestetti, S.; Medraño-Fernandez, I.; Galli, M.; Ghitti, M.; Bienert, G.P.; Musco, G.; Orsi, A.; Rubartelli, A.; Sitia, R. A persulfidation-based mechanism controls aquaporin-8 conductance. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, J.J.; Cucoranu, I.; Fields, E.; Green, E.; He, S.; Hoying, A.; Russ, R.; Abuin, A.; Johnson, D.; Hosseini, S.H.; et al. Transgenic mitochondrial superoxide dismutase and mitochondrially targeted catalase prevent antiretroviral-induced oxidative stress and cardiomyopathy. Lab. Investig. 2009, 89, 782–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.L.S.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 2015, 290, 209–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Goncalves, R.L.S.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef] [Green Version]
- Pak, V.V.; Ezeriņa, D.; Lyublinskaya, O.G.; Pedre, B.; Tyurin-Kuzmin, P.A.; Mishina, N.M.; Thauvin, M.; Young, D.; Wahni, K.; Martínez Gache, S.A.; et al. Ultrasensitive Genetically Encoded Indicator for Hydrogen Peroxide Identifies Roles for the Oxidant in Cell Migration and Mitochondrial Function. Cell Metab. 2020, 31, 642–653.e6. [Google Scholar] [CrossRef]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Das, R.; Xu, S.; Quan, X.; Nguyen, T.T.; Kong, I.D.; Chung, C.H.; Lee, E.Y.; Cha, S.K.; Park, K.S. Upregulation of mitochondrial Nox4 mediates TGF-β-induced apoptosis in cultured mouse podocytes. Am. J. Physiol. Ren. Physiol. 2014, 306, F155–F167. [Google Scholar] [CrossRef] [PubMed]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. 2019, 26. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapti, K.; Diokmetzidou, A.; Kloukina, I.; Milner, D.J.; Varela, A.; Davos, C.H.; Capetanaki, Y. Opposite effects of catalase and MnSOD ectopic expression on stress induced defects and mortality in the desmin deficient cardiomyopathy model. Free Radic. Biol. Med. 2017, 110, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [Green Version]
- Waypa, G.B.; Guzy, R.; Mungai, P.T.; Mack, M.M.; Marks, J.D.; Roe, M.W.; Schumacker, P.T. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ. Res. 2006, 99, 970–978. [Google Scholar] [CrossRef] [Green Version]
- Waypa, G.B.; Chandel, N.S.; Schumacker, P.T. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ. Res. 2001, 88, 1259–1266. [Google Scholar] [CrossRef] [Green Version]
- Backes, S.; Herrmann, J.M. Protein translocation into the intermembrane space and matrix of mitochondria: Mechanisms and driving forces. Front. Mol. Biosci. 2017, 4, 83. [Google Scholar] [CrossRef]
- Bihlmaier, K.; Mesecke, N.; Terziyska, N.; Bien, M.; Hell, K.; Herrmann, J.M. The disulfide relay system of mitochondria is connected to the respiratory chain. J. Cell Biol. 2007, 179, 389–395. [Google Scholar] [CrossRef] [Green Version]
- Dlasková, A.; Clarke, K.J.; Porter, R.K. The role of UCP 1 in production of reactive oxygen species by mitochondria isolated from brown adipose tissue. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1470–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Yu, M.S.; Huang, Y.; Xiang, Z.; Chen, Y.P. MiR-30e-UCP2 pathway regulates alcoholic hepatitis progress by influencing ATP and hydrogen peroxide expression. Oncotarget 2017, 8, 64294–64302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nègre-Salvayre, A.; Hirtz, C.; Carrera, G.; Cazenave, R.; Troly, M.; Salvayre, R.; Pénicaud, L.; Casteilla, L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation—PubMed. FASEB J. 1997, 11, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Angermüller, S.; Islinger, M.; Völkl, A. Peroxisomes and reactive oxygen species, a lasting challenge. Histochem. Cell Biol. 2009, 131, 459–463. [Google Scholar] [CrossRef]
- Tripathi, D.N.; Walker, C.L. The peroxisome as a cell signaling organelle. Curr. Opin. Cell Biol. 2016, 39, 109–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 1755–1766. [Google Scholar] [CrossRef] [Green Version]
- De Duve, C.; Baudhuin, P. Peroxisomes (microbodies and related particles). Physiol. Rev. 1966, 46, 323–357. [Google Scholar] [CrossRef]
- Zhang, J.; Kim, J.; Alexander, A.; Cai, S.; Tripathi, D.N.; Dere, R.; Tee, A.R.; Tait-Mulder, J.; Di Nardo, A.; Han, J.M.; et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 2013, 15, 1186–1196. [Google Scholar] [CrossRef]
- Lee, J.N.; Dutta, R.K.; Maharjan, Y.; Liu, Z.Q.; Lim, J.Y.; Kim, S.J.; Cho, D.H.; So, H.S.; Choe, S.K.; Park, R. Catalase inhibition induces pexophagy through ROS accumulation. Biochem. Biophys. Res. Commun. 2018, 501, 696–702. [Google Scholar] [CrossRef]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Kawałek, A.; van der Klei, I.J. Peroxisomal quality control mechanisms. Curr. Opin. Microbiol. 2014, 22, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Mehmeti, I.; Lortz, S.; Lenzen, S. The H2O2-sensitive HyPer protein targeted to the endoplasmic reticulum as a mirror of the oxidizing thiol-disulfide milieu. Free Radic. Biol. Med. 2012, 53, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Dhaunsi, G.S.; Gupta, M.P.; Orak, J.K.; Asayama, K.; Singh, I. Demonstration of glutathione peroxidase in rat liver peroxisomes and its intraorganellar distribution. Arch. Biochem. Biophys. 1994, 315, 331–338. [Google Scholar] [CrossRef]
- Ohdate, T.; Inoue, Y. Involvement of glutathione peroxidase 1 in growth and peroxisome formation in Saccharomyces cerevisiae in oleic acid medium. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 1295–1305. [Google Scholar] [CrossRef] [Green Version]
- Bottelbergs, A.; Verheijden, S.; Van Veldhoven, P.P.; Just, W.; Devos, R.; Baes, M. Peroxisome deficiency but not the defect in ether lipid synthesis causes activation of the innate immune system and axonal loss in the central nervous system. J. Neuroinflamm. 2012, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Dubreuil, M.M.; Morgens, D.W.; Okumoto, K.; Honsho, M.; Contrepois, K.; Lee-McMullen, B.; Traber, G.M.A.; Sood, R.S.; Dixon, S.J.; Snyder, M.P.; et al. Systematic Identification of Regulators of Oxidative Stress Reveals Non-canonical Roles for Peroxisomal Import and the Pentose Phosphate Pathway. Cell Rep. 2020, 30, 1417–1433. [Google Scholar] [CrossRef]
- Malinouski, M.; Zhou, Y.; Belousov, V.V.; Hatfield, D.L.; Gladyshev, V.N. Hydrogen peroxide probes directed to different cellular compartments. PLoS ONE 2011, 6, e14564. [Google Scholar] [CrossRef] [Green Version]
- Watson, W.H.; Jones, D.P. Oxidation of nuclear thioredoxin during oxidative stress. FEBS Lett. 2003, 543, 144–147. [Google Scholar] [CrossRef] [Green Version]
- Shelar, S.B.; Kaminska, K.K.; Reddy, S.A.; Kumar, D.; Tan, C.T.; Yu, V.C.; Lu, J.; Holmgren, A.; Hagen, T.; Chew, E.H. Thioredoxin-dependent regulation of AIF-mediated DNA damage. Free Radic. Biol. Med. 2015, 87, 125–136. [Google Scholar] [CrossRef]
- Pallardó, F.V.; Markovic, J.; García, J.L.; Viña, J. Role of nuclear glutathione as a key regulator of cell proliferation. Mol. Asp. Med. 2009, 30, 77–85. [Google Scholar] [CrossRef]
- Söderdahl, T.; Enoksson, M.; Lundberg, M.; Holmgren, A.; Ottersen, O.P.; Orrenius, S.; Bolcsfoldi, G.; Cotgreave, I.A. Visualization of the compartmentalization of glutathione and protein-glutathione mixed disulfides in cultured cells. FASEB J. 2003, 17, 124–126. [Google Scholar] [CrossRef] [Green Version]
- Patterson, J.C.; Joughin, B.A.; van de Kooij, B.; Lim, D.C.; Lauffenburger, D.A.; Yaffe, M.B. ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst. 2019, 8, 163–167.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovic, J.; Mora, N.J.; Broseta, A.M.; Gimeno, A.; de-la-Concepción, N.; Viña, J.; Pallardó, F.V. The depletion of nuclear glutathione impairs cell proliferation in 3t3 fibroblasts. PLoS ONE 2009, 4, e6413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Giménez, J.L.; Olaso, G.; Hake, S.B.; Bönisch, C.; Wiedemann, S.M.; Markovic, J.; Dasí, F.; Gimeno, A.; Pérez-Quilis, C.; Palacios, Ò.; et al. Histone H3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Antioxid. Redox Signal. 2013, 19, 1305–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minowa, O.; Arai, T.; Hirano, M.; Monden, Y.; Nakai, S.; Fukuda, M.; Itoh, M.; Takano, H.; Hippou, Y.; Aburatani, H.; et al. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4156–4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardozo-Pelaez, F.; Sanchez-Contreras, M.; Nevin, A.B.C. Ogg1 null mice exhibit age-associated loss of the nigrostriatal pathway and increased sensitivity to MPTP. Neurochem. Int. 2012, 61, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zhuang, Z.; Wang, W.; He, L.; Wu, H.; Cao, Y.; Pan, F.; Zhao, J.; Hu, Z.; Sekhar, C.; et al. OGG1 is essential in oxidative stress induced DNA demethylation. Cell. Signal. 2016, 28, 1163–1171. [Google Scholar] [CrossRef]
- Barciszewska, A.M.; Giel-Pietraszuk, M.; Perrigue, P.M.; Naskręt-Barciszewska, M. Total DNA Methylation Changes Reflect Random Oxidative DNA Damage in Gliomas. Cells 2019, 8, 1065. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, M.; Dellino, I.G.; Gambino, V.; Roda, N.; Pelicci, P.G. On the epigenetic role of guanosine oxidation. Redox Biol. 2020, 29, 101398. [Google Scholar] [CrossRef]
- Redstone, S.C.J.; Fleming, A.M.; Burrows, C.J. Oxidative Modification of the Potential G-Quadruplex Sequence in the PCNA Gene Promoter Can Turn on Transcription. Chem. Res. Toxicol. 2019, 32, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Larsen, E.; Kwon, K.; Coin, F.; Egly, J.M.; Klungland, A. Transcription activities at 8-oxoG lesions in DNA. DNA Repair 2004, 3, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.; Zhang, Y.; Wan, X.; Zhang, C.; Huang, X.; Huang, W.; Pu, H.; Pei, C.; Wu, H.; et al. KDM1A triggers androgen-induced miRNA transcription via H3K4me2 demethylation and DNA oxidation. Prostate 2015, 75, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Amente, S.; Bertoni, A.; Morano, A.; Lania, L.; Avvedimento, E.V.; Majello, B. LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene 2010, 29, 3691–3702. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Hao, W.; Zheng, X.; Zeng, X.; Ahmed Abbasi, A.; Boldogh, I.; Ba, X. OGG1-DNA interactions facilitate NF-k B binding to DNA targets. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Guida, M.; Maraldi, T.; Beretti, F.; Follo, M.Y.; Manzoli, L.; De Pol, A. Nuclear Nox4-derived reactive oxygen species in myelodysplastic syndromes. Biomed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- De Mochel, N.S.R.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2010, 52, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Lener, B.; Kozieł, R.; Pircher, H.; Hütter, E.; Greussing, R.; Herndler-Brandstetter, D.; Hermann, M.; Unterluggauer, H.; Jansen-Dürr, P. The NADPH oxidase Nox4 restricts the replicative lifespan of human endothelial cells. Biochem. J. 2009, 423, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, S.; Kuroda, J.; Zhai, P.; Liu, T.; Ikeda, S.; Nagarajan, N.; Oka, S.I.; Yokota, T.; Kinugawa, S.; Hsu, C.P.; et al. Tyrosine kinase FYN negatively regulates NOX4 in cardiac remodeling. J. Clin. Investig. 2016, 126, 3403–3416. [Google Scholar] [CrossRef]
- Samoylenko, A.; Al Hossain, J.; Mennerich, D.; Kellokumpu, S.; Hiltunen, J.K.; Kietzmann, T. Nutritional countermeasures targeting reactive oxygen species in cancer: From mechanisms to biomarkers and clinical evidence. Antioxid. Redox Signal. 2013, 19, 2157–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilani, T.; Alon, A.; Grossman, I.; Horowitz, B.; Kartvelishvily, E.; Cohen, S.R.; Fass, D. A secreted disulfide catalyst controls extracellular matrix composition and function. Science 2013, 341, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Tury, A.; Mairet-Coello, G.; Poncet, F.; Jacquemard, C.; Risold, P.Y.; Fellmann, D.; Griffond, B. QSOX sulfhydryl oxidase in rat adenohypophysis: Localization and regulation by estrogens. J. Endocrinol. 2004, 183, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mairet-Coello, G.; Tury, A.; Esnard-Feve, A.; Fellmann, D.; Risold, P.Y.; Griffond, B. FAD-Linked Sulfhydryl Oxidase QSOX: Topographic, Cellular, and Subcellular Immunolocalization in Adult Rat Central Nervous System. J. Comp. Neurol. 2004, 473, 334–363. [Google Scholar] [CrossRef]
- Chakravarthi, S.; Jessop, C.E.; Willer, M.; Stirling, C.J.; Bulleid, N.J. Intracellular catalysis of disulfide bond formation by the human sulfhydryl oxidase, QSOX1. Biochem. J. 2007, 404, 403–411. [Google Scholar] [CrossRef]
- Raje, S.; Thorpe, C. Inter-domain redox communication in flavoenzymes of the quiescin/sulfhydryl oxidase family: Role of a thioredoxin domain in disulfide bond formation. Biochemistry 2003, 42, 4560–4568. [Google Scholar] [CrossRef]
- Li, J.; Tong, C.; Xu, P.; Wang, L.; Han, T.L.; Wen, L.; Luo, X.; Tan, B.; Zhu, F.; Gui, S.; et al. QSOX1 regulates trophoblastic apoptosis in preeclampsia through hydrogen peroxide production. J. Matern. Neonatal Med. 2019, 32, 3708–3715. [Google Scholar] [CrossRef] [Green Version]
- Morel, C.; Adami, P.; Musard, J.F.; Duval, D.; Radom, J.; Jouvenot, M. Involvement of sulfhydryl oxidase QSOX1 in the protection of cells against oxidative stress-induced apoptosis. Exp. Cell Res. 2007, 313, 3971–3982. [Google Scholar] [CrossRef]
- Lake, D.F.; Faigel, D.O. The emerging role of qsox1 in cancer. Antioxid. Redox Signal. 2014, 21, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Katchman, B.A.; Ocal, I.T.; Cunliffe, H.E.; Chang, Y.H.; Hostetter, G.; Watanabe, A.; LoBello, J.; Lake, D.F. Expression of quiescin sulfhydryl oxidase 1 is associated with a highly invasive phenotype and correlates with a poor prognosis in Luminal B breast cancer. Breast Cancer Res. 2013, 15, R28. [Google Scholar] [CrossRef] [Green Version]
- Pernodet, N.; Hermetet, F.; Adami, P.; Vejux, A.; Descotes, F.; Borg, C.; Adams, M.; Pallandre, J.R.; Viennet, G.; Esnard, F.; et al. High expression of QSOX1 reduces tumorogenesis, and is associated with a better outcome for breast cancer patients. Breast Cancer Res. 2012, 14, R136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesecke, N.; Spang, A.; Deponte, M.; Herrmann, J.M. A novel group of glutaredoxins in the cis-Golgi critical for oxidative stress resistance. Mol. Biol. Cell 2008, 19, 2673–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; Craig Ayre, D.; Christian, S.L. Induction of mitochondrial reactive oxygen species production by GSH mediated S-glutathionylation of 2-oxoglutarate dehydrogenase. Redox Biol. 2016, 8, 285–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo, A.; Casas, C.; Mühlenhoff, U.; Lillig, C.H.; Herrero, E. Saccharomyces cerevisiae Grx6 and Grx7 are monothiol glutaredoxins associated with the early secretory pathway. Eukaryot. Cell 2008, 7, 1415–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessa, W.C.; Garcia-Cardena, G.; Liu, J.; Keh, A.; Pollock, J.S.; Bradley, J.; Thiru, S.; Braverman, I.M.; Desai, K.M. The Golgi association of endothelial nitric oxide synthase is necessary for the efficient synthesis of nitric oxide. J. Biol. Chem. 1995, 270, 17641–17644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangwung, P.; Greco, T.M.; Wang, Y.; Ischiropoulos, H.; Sessa, W.C.; Iwakiri, Y. Proteomic identification of S-nitrosylated Golgi proteins: New insights into endothelial cell regulation by eNOS-derived NO. PLoS ONE 2012, 7, e31564. [Google Scholar] [CrossRef] [PubMed]
- García-Cardeña, G.; Oh, P.; Liu, J.; Schnitzer, J.E.; Sessa, W.C. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: Implications for nitric oxide signaling. Proc. Natl. Acad. Sci. USA 1996, 93, 6448–6453. [Google Scholar] [CrossRef] [Green Version]
- Druhan, L.J.; Forbes, S.P.; Pope, A.J.; Chen, C.A.; Zweier, J.L.; Cardounel, A.J. Regulation of eNOS-derived superoxide by endogenous methylarginines. Biochemistry 2008, 47, 7256–7263. [Google Scholar] [CrossRef]
- Sindler, A.L.; Delp, M.D.; Reyes, R.; Wu, G.; Muller-Delp, J.M. Effects of ageing and exercise training on eNOS uncoupling in skeletal muscle resistance arterioles. J. Physiol. 2009, 587, 3885–3897. [Google Scholar] [CrossRef]
- Wang, H.; He, Z.; Yang, Y.; Zhang, J.; Zhang, W.; Zhang, W.; Li, P.; Tang, B. Ratiometric fluorescence imaging of Golgi H2O2 reveals a correlation between Golgi oxidative stress and hypertension. Chem. Sci. 2019, 10, 10876–10880. [Google Scholar] [CrossRef] [Green Version]
- Tabner, B.J.; El-Agnaf, O.M.A.; Turnbull, S.; German, M.J.; Paleologou, K.E.; Hayashi, Y.; Cooper, L.J.; Fullwood, N.J.; Allsop, D. Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J. Biol. Chem. 2005, 280, 35789–35792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behl, C.; Davis, J.B.; Lesley, R.; Schubert, D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell 1994, 77, 817–827. [Google Scholar] [CrossRef]
- Tabner, B.J.; Turnbull, S.; El-Agnaf, O.M.A.; Allsop, D. Formation of hydrogen peroxide and hydroxyl radicals from Aβ and α-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Radic. Biol. Med. 2002, 32, 1076–1083. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Boca, M.; Girotto, S.; Martinelli, M.; Valentine, J.S.; Vieru, M. SOD1 and amyotrophic lateral sclerosis: Mutations and oligomerization. PLoS ONE 2008, 3, e1677. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.X.; Shi, Y.; Furukawa, Y.; Zhai, H.; Fu, R.; Liu, E.; Gorrie, G.H.; Khan, M.S.; Hung, W.Y.; Bigio, E.H.; et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7142–7147. [Google Scholar] [CrossRef] [Green Version]
- Sinet, P.M. Metabolism of Oxygen Derivatives in Down’s Syndrome. Ann. N. Y. Acad. Sci. 1982, 396, 83–94. [Google Scholar] [CrossRef]
- Kim, S.H.; Fountoulakis, M.; Cairns, N.; Lubec, G. Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer’s disease and Down Syndrome. J. Neural Transm. Suppl. 2001, 223–235. [Google Scholar] [CrossRef]
- Massaad, C.A. Neuronal and Vascular Oxidative Stress in Alzheimers Disease. Curr. Neuropharmacol. 2011, 9, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef]
- Paul, B.D.; Sbodio, J.I.; Xu, R.; Vandiver, M.S.; Cha, J.Y.; Snowman, A.M.; Snyder, S.H. Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 2014, 508, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Snyder, S.H. Neurodegeneration in Huntington’s disease involves loss of cystathionine γ-lyase. Cell Cycle 2014, 13, 2491–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Transcriptional control of amino acid homeostasis is disrupted in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 8843–8848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuña, A.I.; Esparza, M.; Kramm, C.; Beltrán, F.A.; Parra, A.V.; Cepeda, C.; Toro, C.A.; Vidal, R.L.; Hetz, C.; Concha, I.I.; et al. A failure in energy metabolism and antioxidant uptake precede symptoms of Huntington’s disease in mice. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covarrubias-Pinto, A.; Moll, P.; Solís-Maldonado, M.; Acuña, A.I.; Riveros, A.; Miró, M.P.; Papic, E.; Beltrán, F.A.; Cepeda, C.; Concha, I.I.; et al. Beyond the redox imbalance: Oxidative stress contributes to an impaired GLUT3 modulation in Huntington’s disease. Free Radic. Biol. Med. 2015, 89, 1085–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonati, T.; Reimann, R.R.; Falsig, J.; Baral, P.K.; O’Connor, T.; Hornemann, S.; Yaganoglu, S.; Li, B.; Herrmann, U.S.; Wieland, B.; et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013, 501, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matus, S.; Lopez, E.; Valenzuela, V.; Nassif, M.; Hetz, C. Functional Contribution of the Transcription Factor ATF4 to the Pathogenesis of Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e66672. [Google Scholar] [CrossRef] [PubMed]
- Monacelli, F.; Acquarone, E.; Giannotti, C.; Borghi, R.; Nencioni, A. Vitamin C, aging and Alzheimer’s disease. Nutrients 2017, 9, 670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasekhar, K.; Samanta, S.; Bagoband, V.; Murugan, N.A.; Govindaraju, T. Antioxidant Berberine-Derivative Inhibits Multifaceted Amyloid Toxicity. iScience 2020, 23, 101005. [Google Scholar] [CrossRef]
- Amato, A.; Terzo, S.; Mulè, F. Natural compounds as beneficial antioxidant agents in neurodegenerative disorders: A focus on Alzheimer’s disease. Antioxidants 2019, 8, 608. [Google Scholar] [CrossRef] [Green Version]
- Hase, T.; Shishido, S.; Yamamoto, S.; Yamashita, R.; Nukima, H.; Taira, S.; Toyoda, T.; Abe, K.; Hamaguchi, T.; Ono, K.; et al. Rosmarinic acid suppresses Alzheimer’s disease development by reducing amyloid β aggregation by increasing monoamine secretion. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Park, H.W.; Park, C.G.; Park, M.; Lee, S.H.; Park, H.R.; Lim, J.; Paek, S.H.; Choy, Y. Bin Intrastriatal administration of coenzyme Q10 enhances neuroprotection in a Parkinson’s disease rat model. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Lockrow, J.; Prakasam, A.; Huang, P.; Bimonte-Nelson, H.; Sambamurti, K.; Granholm, A.C. Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Exp. Neurol. 2009, 216, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Danino, O.; Grossman, S.; Fischer, B. ATP-γ-S-(α,β-CH2) protects against oxidative stress and amyloid beta toxicity in neuronal culture. Biochem. Biophys. Res. Commun. 2015, 460, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, H.; Speckmann, B.; Sies, H. Toward understanding success and failures in the use of selenium for cancer prevention. Antioxid. Redox Signal. 2013, 19, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Moosmann, B.; Behl, C. Antioxidants as treatment for neurodegenerative disorders. Expert Opin. Investig. Drugs 2002, 11, 1407–1435. [Google Scholar] [CrossRef]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Sies, H. Findings in redox biology: From H2O2 to oxidative stress. J. Biol. Chem. 2020, 295, 13458–13473. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling functions of reactive oxygen species. Biochemistry 2010, 49, 835–842. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.A.J.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef]
- Tauskela, J.S. MitoQ—A mitochondria-targeted antioxidant—PubMed. IDrugs 2007, 10, 399–412. [Google Scholar]
- Dringen, R.; Kussmaul, L.; Gutterer, J.M.; Hirrlinger, J.; Hamprecht, B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem. 1999, 72, 2523–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, P.D.; Pérez-De La Cruz, V.; Torres-Ramos, M.; Silva-Islas, C.; Lecona-Vargas, R.; Lugo-Huitrón, R.; Blanco-Ayala, T.; Ugalde-Muñiz, P.; Vázquez-Cervantes, G.I.; Fortoul, T.I.; et al. Selenium-induced antioxidant protection recruits modulation of thioredoxin reductase during excitotoxic/pro-oxidant events in the rat striatum. Neurochem. Int. 2012, 61, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, H.; Alili, L.; Bilgic, E.; Sies, H.; Brenneisen, P. Involvement of selenoprotein P in protection of human astrocytes from oxidative damage. Free Radic. Biol. Med. 2006, 40, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Casas, A.I.; Dao, V.T.V.; Daiber, A.; Maghzal, G.J.; Di Lisa, F.; Kaludercic, N.; Leach, S.; Cuadrado, A.; Jaquet, V.; Seredenina, T.; et al. Reactive Oxygen-Related Diseases: Therapeutic Targets and Emerging Clinical Indications. Antioxid. Redox Signal. 2015, 23, 1171–1185. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Sullards, M.C.; Olzmann, J.A.; Rees, H.D.; Weintraub, S.T.; Bostwick, D.E.; Gearing, M.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J. Biol. Chem. 2006, 281, 10816–10824. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Ko, H.S.; Thomas, B.; Tsang, F.; Chew, K.C.M.; Tay, S.P.; Ho, M.W.L.; Lim, T.M.; Soong, T.W.; Pletnikova, O.; et al. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function. Hum. Mol. Genet. 2005, 14, 3885–3897. [Google Scholar] [CrossRef]
- Dryanovski, D.I.; Guzman, J.N.; Xie, Z.; Galteri, D.J.; Volpicelli-Daley, L.A.; Lee, V.M.Y.; Miller, R.J.; Schumacker, P.T.; James Surmeier, D. Calcium entry and α-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci. 2013, 33, 10154–10164. [Google Scholar] [CrossRef] [Green Version]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [Green Version]
- Cristóvão, A.C.; Guhathakurta, S.; Bok, E.; Je, G.; Yoo, S.D.; Choi, D.H.; Kim, Y.S. Nadph oxidase 1 mediates α-synucleinopathy in Parkinson’s disease. J. Neurosci. 2012, 32, 14465–14477. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Nakamura, T.; Yao, D.; Shi, Z.Q.; Gu, Z.; Ma, Y.; Masliah, E.; Nomura, Y.; Lipton, S.A. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006, 441, 513–517. [Google Scholar] [CrossRef]
- Pearce, R.K.B.; Owen, A.; Daniel, S.; Jenner, P.; Marsden, C.D. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J. Neural Transm. 1997, 104, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.L.; Yong, V.W. Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci. Lett. 1986, 67, 269–274. [Google Scholar] [CrossRef]
- Vural, G.; Gumusyayla, S.; Bektas, H.; Deniz, O.; Alisik, M.; Erel, O. Impairment of dynamic thiol–disulphide homeostasis in patients with idiopathic Parkinson’s disease and its relationship with clinical stage of disease. Clin. Neurol. Neurosurg. 2017, 153, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Hastings, T.G.; Lewis, D.A.; Zigmond, M.J. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc. Natl. Acad. Sci. USA 1996, 93, 1956–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, A.H.; Zeevalk, G.D. Characterization of reduced and oxidized dopamine and 3,4- dihydrophenylacetic acid, on brain mitochondrial electron transport chain activities. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yamada, N.; Maruyama, W.; Osawa, T. Formation of dopamine adducts derived from brain polyunsaturated fatty acids: Mechanism for Parkinson disease. J. Biol. Chem. 2008, 283, 34887–34895. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.R.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [Green Version]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein: A possible molecular link between parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [Green Version]
- Martin-Bastida, A.; Lao-Kaim, N.P.; Loane, C.; Politis, M.; Roussakis, A.A.; Valle-Guzman, N.; Kefalopoulou, Z.; Paul-Visse, G.; Widner, H.; Xing, Y.; et al. Motor associations of iron accumulation in deep grey matter nuclei in Parkinson’s disease: A cross-sectional study of iron-related magnetic resonance imaging susceptibility. Eur. J. Neurol. 2017, 24, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Zhang, F.; Zhou, H.; Kam, W.; Wilson, B.; Hong, J.S. Neuroinflammation and α-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ. Health Perspect. 2011, 119, 807–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated α-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Seok Ko, H.; Il Lee, Y.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunico, C.R.; Sultan, A.; Nakamura, T.; Dolatabadi, N.; Parker, J.; Shan, B.; Han, X.; Yates, J.R.; Masliah, E.; Ambasudhan, R.; et al. Role of sulfiredoxin as a peroxiredoxin-2 denitrosylase in human iPSC-derived dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2016, 113, E7564–E7571. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Hou, S.; Jiang, J.; Sekutowicz, M.; Kelly, J.; Bacskai, B.J. Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 7904–7909. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.W.; Lu, S.; Su, Y.J.; Xue, D.; Yu, X.L.; Wang, S.W.; Zhang, H.; Xu, P.X.; Xie, X.X.; Liu, R.T. Decreasing oxidative stress and neuroinflammation with a multifunctional peptide rescues memory deficits in mice with Alzheimer disease. Free Radic. Biol. Med. 2014, 74, 50–63. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [Green Version]
- Bradley-Whitman, M.A.; Timmons, M.D.; Beckett, T.L.; Murphy, M.P.; Lynn, B.C.; Lovell, M.A. Nucleic acid oxidation: An early feature of Alzheimer’s disease. J. Neurochem. 2014, 128, 294–304. [Google Scholar] [CrossRef] [Green Version]
- Borghi, R.; Patriarca, S.; Traverso, N.; Piccini, A.; Storace, D.; Garuti, A.; Cirmena, G.; Odetti, P.; Tabaton, M. The increased activity of BACE1 correlates with oxidative stress in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1009–1014. [Google Scholar] [CrossRef]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-Jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, N.; Trivedi, M.; Muratore, C.; Li, S.; Deth, R. Soluble oligomers of amyloid-β cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimer Dis. 2013, 36, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Sang, W.S.; Hamby, A.M.; Liu, J.; Wai, Y.C.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Lee, H.G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; McKee, A.C.; Beal, M.F.; Graham, B.H.; Wallace, D.C. Marked changes in mitochondrial dna deletion levels in alzheimer brains. Genomics 1994, 23, 471–476. [Google Scholar] [CrossRef]
- Lu, T.; Aron, L.; Zullo, J.; Pan, Y.; Kim, H.; Chen, Y.; Yang, T.H.; Kim, H.M.; Drake, D.; Liu, X.S.; et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 2014, 507, 448–454. [Google Scholar] [CrossRef] [Green Version]
- Molokanova, E.; Akhtar, M.W.; Sanz-Blasco, S.; Tu, S.; Piña-Crespo, J.C.; Mckercher, S.R.; Lipton, S.A. Differential effects of synaptic and extrasynaptic NMDA receptors on Aβ-induced nitric oxide production in cerebrocortical neurons. J. Neurosci. 2014, 34, 5023–5028. [Google Scholar] [CrossRef] [Green Version]
- Acevedo-Torres, K.; Berríos, L.; Rosario, N.; Dufault, V.; Skatchkov, S.; Eaton, M.J.; Torres-Ramos, C.A.; Ayala-Torres, S. Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington’s disease. DNA Repair 2009, 8, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.K.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in huntington’s disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Chen, C.M.; Wu, Y.R.; Cheng, M.L.; Liu, J.L.; Lee, Y.M.; Lee, P.W.; Soong, B.W.; Chiu, D.T.Y. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 2007, 359, 335–340. [Google Scholar] [CrossRef]
- Klepac, N.; Relja, M.; Klepac, R.; Hećimović, S.; Babić, T.; Trkulja, V. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: A cross-sectional study. J. Neurol. 2007, 254, 1676–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.Q.; Chen, Q.; Wang, X.; Wang, Q.C.; Wang, Y.; Cheng, H.P.; Guo, C.; Sun, Q.; Chen, Q.; Tang, T.S. Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J. Biol. Chem. 2013, 288, 3070–3084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartzokis, G.; Cummings, J.; Perlman, S.; Hance, D.B.; Mintz, J. Increased basal ganglia iron levels in Huntington disease. Arch. Neurol. 1999, 56, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.N.; Yu, Y.V.; Gundemir, S.; Jo, C.; Cui, M.; Tieu, K.; Johnson, G.V.W. Impaired Mitochondrial Dynamics and Nrf2 Signaling Contribute to Compromised Responses to Oxidative Stress in Striatal Cells Expressing Full-Length Mutant Huntingtin. PLoS ONE 2013, 8, e57932. [Google Scholar] [CrossRef] [Green Version]
- Johri, A.; Chandra, A.; Beal, M.F. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med. 2013, 62, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Weiduschat, N.; Mao, X.; Hupf, J.; Armstrong, N.; Kang, G.; Lange, D.J.; Mitsumoto, H.; Shungu, D.C. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci. Lett. 2014, 570, 102–107. [Google Scholar] [CrossRef]
- Beretta, S.; Sala, G.; Mattavelli, L.; Ceresa, C.; Casciati, A.; Ferri, A.; Carrì, M.T.; Ferrarese, C. Mitochondrial dysfunction due to mutant copper/zinc superoxide dismutase associated with amyotrophic lateral sclerosis is reversed by N-acetylcysteine. Neurobiol. Dis. 2003, 13, 213–221. [Google Scholar] [CrossRef]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Flint Beal, M.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [Green Version]
- Duran-Aniotz, C.; Cornejo, V.H.; Espinoza, S.; Ardiles, Á.O.; Medinas, D.B.; Salazar, C.; Foley, A.; Gajardo, I.; Thielen, P.; Iwawaki, T.; et al. IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 2017, 134, 489–506. [Google Scholar] [CrossRef]
- Baleriola, J.; Walker, C.A.; Jean, Y.Y.; Crary, J.F.; Troy, C.M.; Nagy, P.L.; Hengst, U. Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 2014, 158, 1159–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C.; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, D.Y.; Cho, S.B.; Jung, H.Y.; Kim, W.; Lee, K.Y.; Kim, J.W.; Moon, S.M.; Won, M.H.; Choi, J.H.; Yoon, Y.S.; et al. Protein disulfide-isomerase A3 significantly reduces ischemia-induced damage by reducing oxidative and endoplasmic reticulum stress. Neurochem. Int. 2019, 122, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Perri, E.R.; Thomas, C.J.; Parakh, S.; Spencer, D.M.; Atkin, J.D. The unfolded protein response and the role of protein disulfide isomerase in neurodegeneration. Front. Cell Dev. Biol. 2016, 3, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Won, S.M.; Suh, J.; Son, S.J.; Moon, G.J.; Park, U.J.; Gwag, B.J. Induction of the unfolded protein response and cell death pathway in alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 2010, 42, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Conn, K.J.; Gao, W.; McKee, A.; Lan, M.S.; Ullman, M.D.; Eisenhauer, P.B.; Fine, R.E.; Wells, J.M. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004, 1022, 164–172. [Google Scholar] [CrossRef]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef]
- Safren, N.; El Ayadi, A.; Chang, L.; Terrillion, C.E.; Gould, T.D.; Boehning, D.F.; Monteiro, M.J. Ubiquilin-1 overexpression increases the lifespan and delays accumulation of huntingtin aggregates in the R6/2 mouse model of huntington’s disease. PLoS ONE 2014, 9, e87513. [Google Scholar] [CrossRef] [Green Version]
- Hoffstrom, B.G.; Kaplan, A.; Letso, R.; Schmid, R.S.; Turmel, G.J.; Lo, D.C.; Stockwell, B.R. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 2010, 6, 900–906. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, A.; Gaschler, M.M.; Dunn, D.E.; Colligan, R.; Brown, L.M.; Iii, A.G.P.; Lo, D.C.; Stockwell, B.R. Small molecule-induced oxidation of protein disulfide isomerase is neuroprotective. Proc. Natl. Acad. Sci. USA 2015, 112, E2245–E2252. [Google Scholar] [CrossRef] [Green Version]
- Kamarehei, M.; Pejman, S.; Kaboudanian Ardestani, S.; Zahednasab, H.; Firouzi, M.; Harirchian, M.H. Inhibition of protein disulfide isomerase has neuroprotective effects in a mouse model of experimental autoimmune encephalomyelitis. Int. Immunopharmacol. 2020, 82, 106286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, G.; Kaplan, A.; Gaschler, M.M.; Zhang, X.; Hou, Z.; Jiang, M.; Zott, R.; Cremers, S.; Stockwell, B.R.; et al. Small molecule modulator of protein disulfide isomerase attenuates mutant huntingtin toxicity and inhibits endoplasmic reticulum stress in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2018, 27, 1545–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehtonen, Š.; Sonninen, T.M.; Wojciechowski, S.; Goldsteins, G.; Koistinaho, J. Dysfunction of cellular proteostasis in Parkinson’s disease. Front. Neurosci. 2019, 13, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, Y.L.; Su, F.Y.; Tsai, L.K.; Huang, C.C.; Ko, Y.L.; Su, L.W.; Chen, K.Y.; Shih, H.M.; Hu, C.M.; Lee, W.H. NPGPx-Mediated Adaptation to Oxidative Stress Protects Motor Neurons from Degeneration in Aging by Directly Modulating O-GlcNAcase. Cell Rep. 2019, 29, 2134–2143.e7. [Google Scholar] [CrossRef]
- Skladnev, N.V.; Ganeshan, V.; Kim, J.Y.; Burton, T.J.; Mitrofanis, J.; Stone, J.; Johnstone, D.M. Widespread brain transcriptome alterations underlie the neuroprotective actions of dietary saffron. J. Neurochem. 2016, 139, 858–871. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA. 2008, 105, 18525–18530. [Google Scholar] [CrossRef] [Green Version]
- Kam, M.K.; Lee, D.G.; Kim, B.; Lee, H.S.; Lee, S.R.; Bae, Y.C.; Lee, D.S. Peroxiredoxin 4 ameliorates amyloid beta oligomer-mediated apoptosis by inhibiting ER-stress in HT-22 hippocampal neuron cells. Cell Biol. Toxicol. 2019, 35, 573–588. [Google Scholar] [CrossRef]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Galvan, D.L.; Badal, S.S.; Long, J.; Chang, B.H.; Schumacker, P.T.; Overbeek, P.A.; Danesh, F.R. Real-time in vivo mitochondrial redox assessment confirms enhanced mitochondrial reactive oxygen species in diabetic nephropathy. Kidney Int. 2017, 92, 1282–1287. [Google Scholar] [CrossRef]
- Van Hameren, G.; Campbell, G.; Deck, M.; Berthelot, J.; Gautier, B.; Quintana, P.; Chrast, R.; Tricaud, N. In vivo real-time dynamics of ATP and ROS production in axonal mitochondria show decoupling in mouse models of peripheral neuropathies. Acta Neuropathol. Commun. 2019, 7, 86. [Google Scholar] [CrossRef]
Figure 1.
Schema of pathways producing and utilising endoplasmic reticulum (ER)-derived H2O2. Reduced thiols (-SH) of cysteine residues in nascent proteins are oxidised by catalytically competent protein disulphide isomerase (PDI)s to promote protein folding with native disulphide bonds (S-S). PDI can be re-oxidised by ERO1, with flavin adenine dinucleotide (FAD) embedded in its active site, which reduces molecular oxygen (O2) to hydrogen peroxide (H2O2). ER-resident antioxidants PRDX4, GPX7, and GPX8 oxidise their catalytic domain by utilising H2O2 (-SOH = sulfenic acid) and mediate “the disulphide relay” to PDI. NOX4 produces H2O2 over superoxide on the ER membrane, unlike other NOX family proteins.
Figure 1.
Schema of pathways producing and utilising endoplasmic reticulum (ER)-derived H2O2. Reduced thiols (-SH) of cysteine residues in nascent proteins are oxidised by catalytically competent protein disulphide isomerase (PDI)s to promote protein folding with native disulphide bonds (S-S). PDI can be re-oxidised by ERO1, with flavin adenine dinucleotide (FAD) embedded in its active site, which reduces molecular oxygen (O2) to hydrogen peroxide (H2O2). ER-resident antioxidants PRDX4, GPX7, and GPX8 oxidise their catalytic domain by utilising H2O2 (-SOH = sulfenic acid) and mediate “the disulphide relay” to PDI. NOX4 produces H2O2 over superoxide on the ER membrane, unlike other NOX family proteins.

Figure 2.
Schema of unfolded proteins response (UPR)-induced neurodegeneration and potential therapeutic strategies. UPR activated by a variety of factors, such as single nucleotide polymorphisms (SNPs) as genetic factors and aging or metabolic dysfunction as environmental factors, augments the expressions of target genes such as PDI and ERO1, and enhances the oxidative folding capacity of the ER. This can reduce the accumulation of unfolded proteins and results in neuroprotection, whereas excess H2O2, which is concomitantly produced by this process, exacerbates neurodegeneration as an oxidative stress agent. Inhibitors against PDI or ERO1 (such as CCF642, LOC14, and EN460) and antioxidants are considered promising in mitigating the development of neurodegenerative pathology by reducing ER-derived H2O2.
Figure 2.
Schema of unfolded proteins response (UPR)-induced neurodegeneration and potential therapeutic strategies. UPR activated by a variety of factors, such as single nucleotide polymorphisms (SNPs) as genetic factors and aging or metabolic dysfunction as environmental factors, augments the expressions of target genes such as PDI and ERO1, and enhances the oxidative folding capacity of the ER. This can reduce the accumulation of unfolded proteins and results in neuroprotection, whereas excess H2O2, which is concomitantly produced by this process, exacerbates neurodegeneration as an oxidative stress agent. Inhibitors against PDI or ERO1 (such as CCF642, LOC14, and EN460) and antioxidants are considered promising in mitigating the development of neurodegenerative pathology by reducing ER-derived H2O2.


{kind=link}
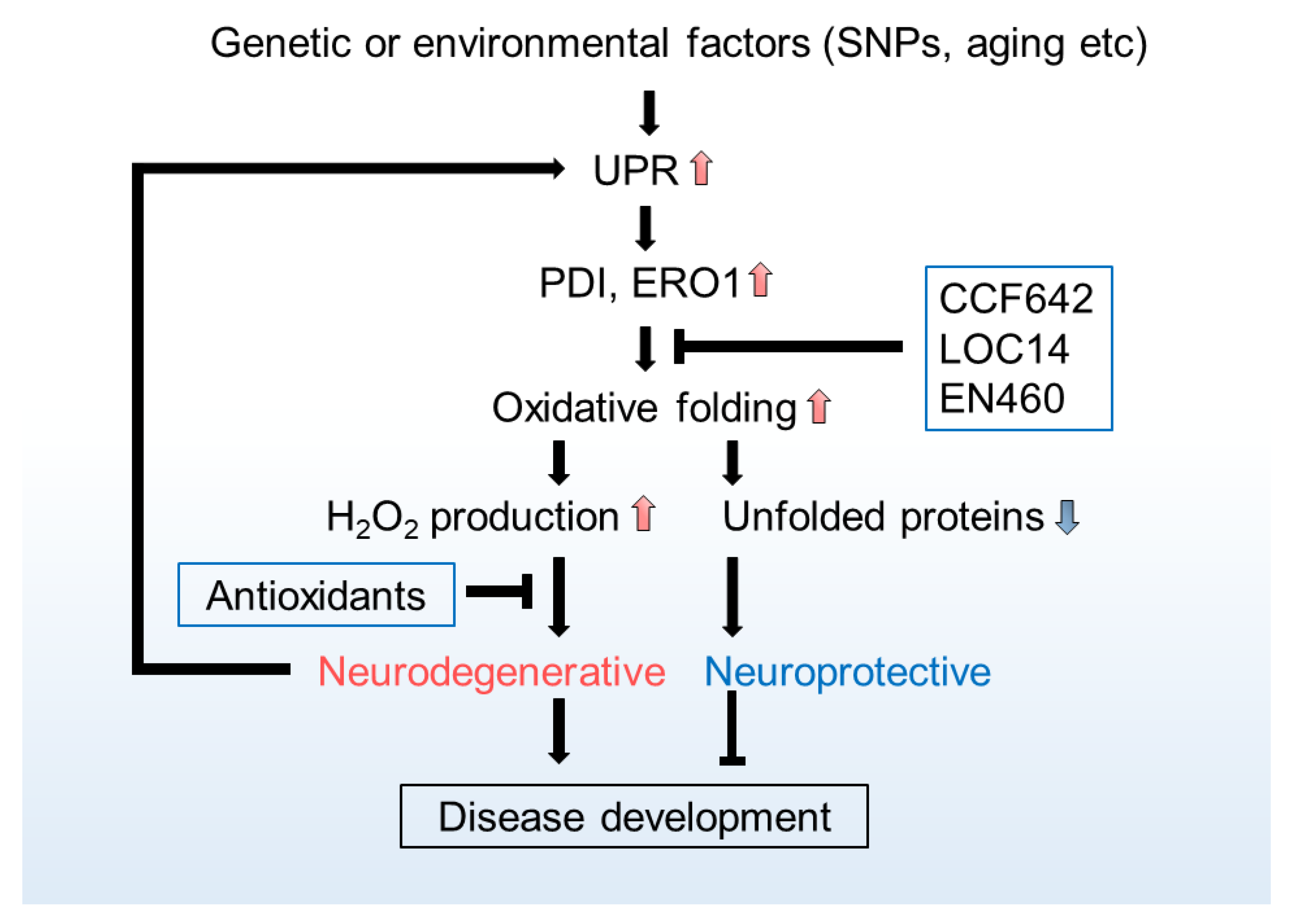{kind=link}
Table 1.
Potential relations between oxidative stress and neurodegenerative disorders.
| Neurodegenerative Disorder | Possible Source of Oxidative Stress | Cell Component Affected | Reference |
|---|---|---|---|
| Parkinson disease | Not determined | Oxidized DJ-1 1 protein | [186] |
| H2O2/rotenone/6-hydroxydopamine | Parkin | [187] | |
| Ca2+ influx through L-type channels/α-synuclein intracellular inclusions | Mitochondrial oxidant stress | [188,189] | |
| NADPH oxidase | α-synuclein | [190] | |
| NOS 2 | S-nitrosylation of PDI 3 | [191] | |
| Not determined | Depletion of GSH | [192,193,194] | |
| Intrastriatal dopamine injections | Protein-bound cysteinyl catechols/Loss of tyrosine hydroxylase | [195] | |
| Loss of dopamine homeostasis | Inhibition of NADH oxidase/mitochondrial dysfunction | [196] | |
| Formation of the hexanoyl dopamine adduct | Mitochondrial abnormality/apoptosis | [197] | |
| α-synuclein oligomer-induced ROS production | Depletion of GSH | [198] | |
| α-synuclein oligomers | Mitochondrial dysfunction | [199] | |
| Not determined | Mitochondrial respiratory-chain Complex I | [200] | |
| Iron deposition in deep brain nuclei | Fibrillation of α-synuclein/dopaminergenic neurodegeneration | [201,202] | |
| Inflammogen lipopolysaccharide/ NOS 2 and NADPH oxidase | Aggregated nitrated α-synuclein/upregulation of inflammatory markers | [203] | |
| Extracellular aggregated α-synuclein | Activation of NADPH oxidase/microglial activation | [204] | |
| Activation of NOS 2 | Decrease in parkin sulfhydration/increase in parkin nitrosylation | [205] | |
| Nitric oxide | S-nitrosylation of Prx2 4 | [206] | |
| Alzheimer’s disease | Not determined | Oxidized DJ-1 1 protein | [186] |
| Aβ plaques | Neurites | [207] | |
| Aβ plaques | Reduced GSH levels/Decreased SOD activity | [208] | |
| Aβ plaques | Mitochondrial protein import channels | [209] | |
| Not determined | Nuclear and mitochondrial DNA damage | [210] | |
| Increase in BACE1 5 activity | Lipid peroxidation | [211] | |
| Not determined | JNK/SAPK 6 dysregulation | [212] | |
| Aβ oligomers | Inhibition of EAAT3 7-mediated cysteine uptake/decreased GSH levels/decreased DNA methylation | [213,214] | |
| Inhibition of GSH synthesis | Increased levels of tau phosphorylated | [215] | |
| Age-dependent | Mitochondrial DNA deletion | [216] | |
| Not determined | Loss of REST 8 | [217] | |
| NOS 2 | S-nitrosylation of PDI 3 | [191] | |
| Aβ(1-42) oligomers | Activation of NMDARs 9/increase in Nitric oxide | [218] | |
| Huntington’s disease | Age-dependent | Mitochondrial DNA damage | [219] |
| Not determined | Cytosolic SOD 10 activity/nuclear DNA/oxidative phosphorylation enzymes | [220] | |
| Not determined | Cu/Zn-SOD 10 activity/glutathione peroxidase activity/mitochondrial DNA | [221] | |
| Not determined | Reduced GSH levels/plasma lipid peroxidation | [222] | |
| Mitochondrial Ca2+ signalling and superoxide generation | Mitochondrial DNA damage | [223] | |
| Depletion of cystathione γ-lyase 11 | Low levels of GSH/protein carbonylation/protein nitration/lipid peroxidation | [161] | |
| Impaired cysteine biosynthesis and transport | Failure of activating transcriptional factor 4 (ATF4) induction | [163] | |
| Uptake of vitamin C | Impaired trafficking of vitamin C transporters/impaired glucose transporter GLUT3 to the membrane | [164,165] | |
| Iron metabolism | Not determined | [224] | |
| Age-dependent | Oxidative DNA damage (CAG trinucleotide expansion) | [225] | |
| Reduced activity of Nrf2 12 | Impaired mitochondrial dynamics/decreased mitochondrial fusion | [226] | |
| Impaired function of PGC1α 13 | Mitochondrial dysfunction | [227] | |
| Not determined | Reduced levels of GSH | [228] | |
| Amyotrophic lateral sclerosis | Mutations in Cu/Zn SOD 10 (familiar cases) | Increased cytosolic and mitochondrial ROS/impaired mitochondrial respiration | [229,230] |
1 Ubiquitously expressed protein of the DJ-1/ThiJ/PfpI superfamily whose biochemical function is unknown; 2 Nitric oxide synthase; 3 Protein disulphide isomerase; 4 Peroxiredoxin-2; 5 β-secretase, β-site amyloid precursor protein cleaving enzyme 1; 6 c-Jun N-terminal kinase/Stress activated protein kinase; 7 Excitatory amino acid transporter 3; 8 Repressor element 1-silencing transcription factor, represses genes that promote cell death and induces expression of stress response genes; 9 N-methyl-D-aspartate-type glutamate receptor; 10 SOD—superoxide dismutase; 11 Biosynthetic enzyme for cysteine; 12 Erythroid 2-related factor 2, a master transcriptional regulator of the cellular antioxidant stress response; 13 Peroxisome proliferator-activated receptor γ coactivator 1α, a master transcriptional co-regulator of mitochondrial biogenesis and metabolism and antioxidant defences.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Konno, T.; Melo, E.P.; Chambers, J.E.; Avezov, E. Intracellular Sources of ROS/H2O2 in Health and Neurodegeneration: Spotlight on Endoplasmic Reticulum. Cells 2021, 10, 233. https://doi.org/10.3390/cells10020233
AMA Style
Konno T, Melo EP, Chambers JE, Avezov E. Intracellular Sources of ROS/H2O2 in Health and Neurodegeneration: Spotlight on Endoplasmic Reticulum. Cells. 2021; 10(2):233. https://doi.org/10.3390/cells10020233
Chicago/Turabian StyleKonno, Tasuku, Eduardo Pinho Melo, Joseph E. Chambers, and Edward Avezov. 2021. "Intracellular Sources of ROS/H2O2 in Health and Neurodegeneration: Spotlight on Endoplasmic Reticulum" Cells 10, no. 2: 233. https://doi.org/10.3390/cells10020233
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.