Catalytic Pyrolysis of Biomass and Polymer Wastes


1
Cain Department of Chemical Engineering, Louisiana State University, Baton Rouge, LA 70803, USA
2
Chemical Sciences Division and Center for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA
3
Department of Bioproducts and Biosystems Engineering, University of Minnesota, St Paul 55108, MN 55108, USA
4
National Engineering Laboratory for Biomass Power Generation Equipment, North China Electric Power University, Beijing 102206, China
5
School of Chemical, Biological and Materials Engineering, The University of Oklahoma, Norman, OK 73019, USA
*
Author to whom correspondence should be addressed.
Catalysts 2018, 8(12), 659; https://doi.org/10.3390/catal8120659
Submission received: 1 November 2018
/
Revised: 30 November 2018
/
Accepted: 30 November 2018
/
Published: 13 December 2018
(This article belongs to the Special Issue Catalytic Pyrolysis)
Abstract
:Oil produced by the pyrolysis of biomass and co-pyrolysis of biomass with waste synthetic polymers has significant potential as a substitute for fossil fuels. However, the relatively poor properties found in pyrolysis oil—such as high oxygen content, low caloric value, and physicochemical instability—hampers its practical utilization as a commercial petroleum fuel replacement or additive. This review focuses on pyrolysis catalyst design, impact of using real waste feedstocks, catalyst deactivation and regeneration, and optimization of product distributions to support the production of high value-added products. Co-pyrolysis of two or more feedstock materials is shown to increase oil yield, caloric value, and aromatic hydrocarbon content. In addition, the co-pyrolysis of biomass and polymer waste can contribute to a reduction in production costs, expand waste disposal options, and reduce environmental impacts. Several promising options for catalytic pyrolysis to become industrially viable are also discussed.
Keywords:
pyrolysis; catalyst; biomass; waste plastics; waste tires; bio-oil; zeolites; coke; co-pyrolysis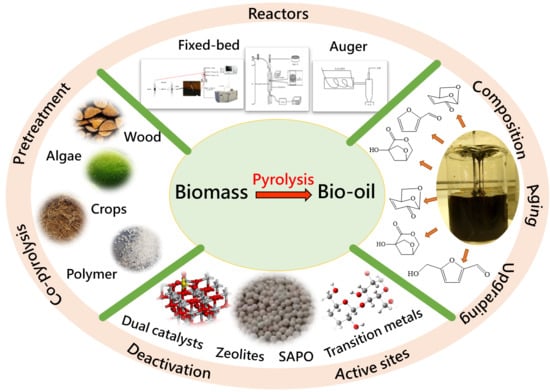
1. Introduction
1.1. Drive for Sustainable Energy
World energy is, and continues to be, dominated by fossil fuels. With the progress of science and technology, especially after the industrial revolution, wealth growth globally has largely depended on utilizing fossil fuels to power internal combustion engines. World-wide, approximately 95% of energy used comes from fossil fuels [1], and even with an increase in drilling capabilities, world oil demand and utilization have increased more than its discoveries and what enhanced oil recovery methods can provide. In 1999, the American Petroleum Institute estimated that oil supplies would be depleted between 2062 and 2094 [2], and a more recent estimation lowered the depletion year to 2057 [3].
Environmental concerns related to fossil, nuclear, and hydroelectric energy also increases the attraction for alternative and diversified sources. For example, CO2 emissions resulting from fossil fuels combustion has increased by ~11% between 2005 and 2009 [4]. Continued high rates of greenhouse emissions in the transportation and industrial sectors will negatively impact not only the environment, but eventually the economy. In addition to environmental and industry driven changes to the energy sector, government regulations on standards, and taxes for fuels and emissions, as well renewable energy production subsidies, increasingly mandate a diversification of energy options. In the U.S. and Europe, policies have been successfully implemented resulting in reduced air pollution and acid rain levels [5]. As determined by the U.S. Energy Information Administration (EIA), the fossil fuel component of total energy usage will decrease 5% by 2040, while renewable energy will grow from 9% to 12% in the same time frame [6]. Rapid consumption and greater environmental awareness over the past few decades have highlighted challenges in continued use of fossil fuels as energy sources, including their non-renewability, limits to access and distribution, and environmental impacts from recovery and use. As a result, the development of renewable energy and green chemistry options have increasingly become a focus of research and industrial efforts.
Biomass accounts for over 50% of the total renewable energy produced. Biomass utilization for energy production has no negative impact on the atmospheric CO2 content if the CO2 consumed during plant growth is equal to or greater than that produced during combustion—or if CO2 differences can be offset by replanting. EIA predicted that biomass to liquid production (excluding ethanol and biodiesel) will increase by 32,200 barrels of oil per day (bbl/d) from 2012 to 2040 [6]. Figure 1 shows three main strategies for the production of fuels from biomass [7]. Enzymatic hydrolysis where lignocellulosic biomass is converted to fermentable sugars is a complex process and only limited success has been realized using low(er) cost techniques [8]. Conversion of biomass to fuels using catalysis can be accomplished via gasification and pyrolysis [9]. Catalytic steam-reforming of biomass produces synthesis gas that can further be transformed to biofuels. The more economical and environmentally-friendly method is to produce biofuels and fuel additives by subsequent catalytic upgrading of bio-oil that was produced by either catalytic or non-catalytic pyrolysis of biomass. Many upgrading techniques using catalysts that have been utilized to produce renewable fuels with quality comparable to conventional fossil fuels. These upgrading techniques include low temperature esterification with alcohols, catalytic cracking, and hydrotreating processes [10,11]. Interestingly, the side-products from biomass valorization—e.g., methyl levulinate, furfural, and humins—have also gained focus because of a significant potential in conversion into useful derivatives [12]. Recently a few efforts have employed dielectric heating from microwave radiation for the pyrolysis of biomass, which has advantages in uniform volumetric heating, energy savings and efficiency, and process control flexibility in the conversion of lignin to valuable chemicals and fuels [13].
In addition to quickly depleting fossil fuel resources, the large quantity of polymers produced, used, and disposed annually is causing severe environmental problems, that will accumulate as the polymer waste accumulates. One of the largest segments in the polymer products market is single-use plastic packaging materials that are discarded after a very short-term period of use. Another critical issue in polymeric material recycling and disposal is vehicle tires and other materials that have limitations on the ability to reform and reuse them. The vast quantity of polymeric material produced and inappropriate management of these materials as wastes has resulted in significant harm to the environment. Chemical recycling to convert polymers to smaller chain hydrocarbons or monomers is a promising method to help solve the problem of waste plastic and tire accumulation.
Pyrolysis of lignocellulosic biomass has been widely investigated, and it is considered to be the most promising production method for sustainable biofuels. However, the lower energy density, poor quality and stability of resulting liquid, and a high formation of coke in pyrolysis biofuels hampers the scale-up of production to commercial levels. The addition of a hydrogen-rich co-reactant, such as waste plastics and tires, can significantly improve the yield of aromatics and reduce coke formation [14,15]. The process of biomass co-pyrolysis with plastic waste allows for increased energy security and also reduced environmental pollution [15].
1.2. Catalytic Fast Pyrolysis of Biomass and Waste Polymers
Pyrolysis, an efficient and eco-friendly process, is a feasible alternative for the production of fuels, chemicals, and hydrogen from biomasses and waste polymers. Pyrolysis methods can be divided into two major categories, i.e., slow and fast pyrolysis [16]. Slow pyrolysis involves a slow heating rates of 0.1–1 °C/s, a residence time varying from minutes to hours and a temperature range of 400–600 °C. Slow pyrolysis has been used to produce methanol, but also results in the formation of large amounts of side products—such as char, gas, and low-quality liquid [17]. In contrast, fast pyrolysis rapidly heats samples to 400–600 °C with a heating rate of 10–1000 °C/s in a non-oxidizing environment during a short residence time (<2 s) and produced pyrolysis vapors are rapidly quenched [18]. Fast pyrolysis is promising because of the high yield of liquid product (~70 wt %), known as pyrolysis oil or bio-oil, obtained. In the late 1970s, Garrett and Mallan [19] disclosed a fast pyrolysis process of waste solids, including trash, rubber, and natural raw materials, which were intermixed with char and carrier gas and passed through a pyrolysis zone under turbulent conditions at temperatures ranging from 300 to 2000 °F with residence times under 10 s. Char was used as the heat source and was superior than using hot carrier gas alone due to its higher heat capacity. In addition, the char could be reused after degasification and desulfurization. This early effort focused on using fast pyrolysis for the recovery of chemicals and fuel values from waste materials. Significant research in fast pyrolysis have continued over the past several decades.
Catalytic fast pyrolysis (CFP) has been widely recognized as a promising platform for the thermochemical conversion of lignocellulosic biomass and waste polymers to useful chemicals and fuel additives. Catalytic upgrading of pyrolysis oils increases the H/C ratio and energy density of the final products by elimination and substitution of oxygen and oxygen-containing functional groups. Various catalyst types, metal oxide, molecular sieve, mineral, transition metals, etc., have been examined to optimize the resulting bio-oil yields and properties, such as composition and stability. A notable review by Bridgewater et al. [18] comprehensively reviewed feedstock processing, reactor design and maintenance, catalyst design and regeneration, vapor condensation, optimization of product distribution, resulting oil filtration, and catalytic upgrading.
Biochar is a side product of CFP. The application of biochar for pollution removal during water treatment and agriculture soil amendment has attracted wide attention [20]. Without further manufacture and simply burying the biochar in soil, the biochar is considered as an ideal adsorbent for the removal diverse pollutants including heavy metals such as Zn, Cd, Cu, Cr [21], organic compounds such as polycyclic aromatic hydrocarbons [22], polybrominated diphenyl ethers [23], and greenhouse gas [24]. Regarding the interaction between the pollutants and biochar, physical adsorption, ion exchange, electrostatic attraction, surface complexation and precipitation, hydrogen bond, hydrophobic effect, and pore filling are proposed [25]. Besides pollution removal, the benefits of biochar also include depressing soil disease microbial growth and increasing soil enzymes activities [26]. Biochar is also considered as a precursor of activated carbon with further dehydrating treatment that can be used as catalyst support [27].
Post-pyrolysis catalytic upgrading [28,29] of pyrolysis oils has gained much attention in the last decade. Metal catalysts mainly work via two pathways, i.e., hydrodeoxygenation and hydrogenation, while zeolites are primarily used to produce aromatics from bio-oil. However, a broad generalization of the effect of catalyst type is not possible due to the wide variety of catalyst types and properties [30]. Moreover, the usage of two different CFP processes, in-bed and in-situ catalytic vapor upgrading, makes discerning the impact of specific catalyst properties on the pyrolysis oil even more difficult. In this review, we strictly separate catalysts used for in-situ catalytic upgrading and post-pyrolysis catalytic upgrading.
Generally, the use of catalysts in biomass (co)pyrolysis has the potential to decrease the pyrolysis temperature—reducing equipment and energy costs, increase biomass conversion, and change the distribution of products. We will examine the research and development of CFP in recent years by providing a review of studies published in the technical literature. The discussion will be focused on developing multi-functional catalysts that would provide an advantage of combining in-situ both metal catalyst hydrodeoxygenation and zeolite cracking. Successful development of catalysts for fast pyrolysis with high selectivity, stability, and activity will help to achieve a globally impactful goal of sustainable fuels and chemicals production from biomass and plastics wastes.
2. Fundamentals of Catalytic Fast Pyrolysis (CFP)
2.1. Feedstock
A consistent and lower-cost feed supply are critical to the economic feasibility of a given catalytic fast pyrolysis (CFP) process. The formulation of feedstocks or feedstock blending technique can impact the properties and yield of bio-oil, in addition to the cost of the final product. Likewise, in considering options for thermal and/or chemical pretreatment the ability to maintain a stable feedstock supply, in terms of continuous availability and low cost, should weigh heavily in the decision.
2.1.1. Plant Biomass and Its Pretreatment
It has been reported more than 100 different biomass types [31] have been tested as pyrolysis feedstocks. The biomass sources range from forest/agricultural residues to energy crops to solid wastes such as swine mature and sewage sludge. Any form of biomass could theoretically be utilized as a feedstock for CFP. Woody biomass is by far the most widely-investigated because of its low ash formation and higher-quality bio-oil. However, the availability and cost of biomass resources vary greatly by locale. While not covered here, detailed information about the impact of feedstock type and pretreatments on bio-oil yield, product distribution, and upgradability can be found in some recent reviews [32,33].
Effect of Biomass Composition on Bio-Oil Properties
Plant biomass are mainly composed of cellulose, hemicelluloses, lignin, as well as organic extractives and inorganic minerals. Weight percentages of each compound varies significantly for each different plant species. Due to differences in chemical structure, cellulose, hemicelluloses, and lignin each differ in their thermal decomposition and chemical transformation during the CFP process [33,34]. In addition to difference in products formed, the bio-oil yields vary with the cellulose, hemicellulose, and lignin content in the feedstock. CFP of cellulose results in the maximum aromatic yield (38.4%) and the minimum non-condensable gas yield (18.4%). The three-dimensional network structure of lignin affects the breakdown products, and pyrolysis of lignin results in high levels of char and tar. Lignin CFP gives the highest coke yield at 68.6% and the lowest aromatic yield at 10.2%. Hemicellulose pyrolysis shows the lowest coke yield (29.4%) and the highest yield of non-condensable gas (39.1%) [35].
A set of commercial zeolites, laboratory-synthesized ZSM-5 and X and Y catalysts, and alumni and silica materials were evaluated for hydrocarbon production performance in batch and semi-continuous pyrolysis/catalytic cracking processing. Three types of biomass feedstocks—cellulose, lignin, and wood—were pyrolyzed in physical contact with different catalysts at five to 10 catalyst/feedstock ratios at temperatures from 400 to 600 °C [36]. Hydrocarbon yields were highest for nickel, cobalt, iron, and gallium substituted ZSM-5 catalysts. In the semi-continuous reactions, it was shown that coke formation on the catalyst decreased the deoxygenation activity. In addition to the impact of catalysts type and substitution of aluminum or hydrogen with specific metals, the ash content in the feedstock can also affect the yields of incondensable gases and carbonaceous solids. A feedstock with a higher ash content generates more gases (mainly CO2). A positive correlation between the cellulose content and CO yield and a negative correlation between the hemicellulose content and CO yield has been demonstrated [34].
Interactions between biomass components, i.e., cellulose, hemicellulose, and lignin, affect the CFP behavior and the resulting product distribution. The impacts of cellulose–hemicellulose and cellulose–lignin interactions on fast pyrolysis were shown by comparing the pyrolysis products from the native mixture of lignin-cellulose-hemicellulose, physical mixtures, and superpositions of the individual components [37]. Negligible or weak interactions were found for both binary physical mixtures and the native cellulose–hemicellulose mixture, but strong interactions were noted for other feedstock compositions, including that which may occur in biomass-waste co-pyrolysis. For herbaceous biomass, the native cellulose–lignin interaction significantly decreased yields of levoglucosan and increased yields of furans and low molecular weight compounds. Interestingly, this same effect was not observed for woody biomass, and may be due to differing covalent linkage contents. Cellulose-lignin co-pyrolysis could promote the formation of low molecular weight products (e.g., esters, aldehydes, ketones, phenols, and guaiacols), while inhibiting the formation of anhydro sugars [38]. The native cellulose-lignin feedstock mixture showed the most significant impact on the product distribution. The mixing method was the most influential parameter on interaction effects, followed by temperature, and mixing ratio were the least significant [38].
Lignin is the main component of plant cell walls and accounts for 15–30 wt % of biomass [39]. It is a crosslinked polymer with three different phenolic connected by ether, ester, and carbon-carbon bonds [40]. The lignin polymer has a hyperbranched topology with no regular repeating structure, but the structure of a given type/source of lignin has been found to change the pyrolysis product distribution. A CFP study comparing milled wood lignin (MWL) to organosolv lignin—isolated from red oak (hardwood), loblolly pine (softwood), or corn stover (herbaceous biomass)—found that corn stover lignin exhibited a significant potential for volatilization [41]. Corn stover lignins produced more aromatic hydrocarbons than lignins derived from red oak or loblolly pine. This is due to corn stover lignin having a highly branched polymer structure enriched in tricin, ferulate, and coumarate groups. The organosolv lignins produced fewer volatiles, more char and less phenolic oil—in comparison to corresponding MWL. Comparing MWL feedstocks, corn stover MWL exhibited higher selectivity toward phenolic monomers than either red oak MWL or loblolly pine MWL.
Cleavage of intermolecular linkages in lignin is crucial for the pyrolysis of lignin. Four typical β-O-4 lignin dimer [42] compounds with different substituents at the Cα and Cβ positions (Figure 2) were synthesized and used to investigate the effect of the substituent group on chemical reactions during lignin pyrolysis. The cleavage of intermolecular linkages dominated reactions during pyrolysis below 300 °C. The presence of the CH2OH group at the Cβ position greatly inhibited the formation of volatile products and promoted char formation. The compound with only α =O group substitution at Cα showed the highest volatility [43]. The formation of guaiacol indicated the break of Cβ-O. Oxidation of the Cα-OH group to Cα=O facilitated cleavage of the Cα-Cβ bond, which in turn led to the formation of 2-methoxy-benzaldehyde. In addition, new dimers were detected which could further undergo Cβ-O and Cα-Cβ bond cleavage.
The molecular weight of lignin also significantly impacts the pyrolysis process. Different molecular weights of alkali lignin, including pure alkali lignin (AL), a > 10 kDa lignin fraction, a 5–10 kDa lignin fraction, and a 1–5 kDa lignin fraction, were prepared by membrane ultrafiltration [43]. Pyrolysis of low molecular weight fractions of alkali lignin led to higher yields of CH4, CO, CO2, phenols and alkyl phenols, and pyrolysis of the high molecular weight fraction favored the generation of guaiacol and alkyl guaiacols.
Effect of Feedstock Elemental Composition
The elemental composition of feedstock, as measured by H/Ceff ratios, has been found significantly influence the ratio of olefins, aromatics, and coke production during pyrolytic reactions. CFP results using HZSM-5 catalyst with ten feedstocks, glucose, glycerol, sorbitol, tetrahydrofuran, methanol, and different hydrogenated bio-oil fractions, indicated that yields of olefins and aromatics, the olefins/aromatics ratio, and gas yields (mainly CO and CO2), increased with increasing H/Ceff feedstock ratios [44]. Thermogravimetric analysis (TGA) showed that coke formation decreased with increasing H/Ceff, which results in a prolonged catalyst lifespan due to reduced coke chocking of the catalyst pores.
While hydrogenation of the bio-oil feedstock to increase the H/Ceff ratio is not very practical, co-pyrolyzing biomass with a hydrogen donor such as methanol [45], tetralin [46], and olefin [47] has been investigated. Hydrocarbon products, including alkylated phenols and aromatics, increased during co-pyrolysis. Methanol was shown to help increase the amount of oxygen released via the vapor phase as water. Additionally, the content of alkylated aromatic hydrocarbon products increased significantly [45].
The catalytic pyrolysis of lignin and tetralin was investigated in a micropyrolyzer-GCMS system using both HY and HZSM-5 catalysts. The static pore size of HZSM-5 was smaller than the molecular size of tetralin. In contrast, HY zeolite has larger pores, which allow tetralin into its pore channels. Co-pyrolysis of lignin and tetralin over HY zeolite led to significant increases in aromatic hydrocarbon yield and decreases in coke yield. In comparison, the co-pyrolysis of lignin and tetralin over HZSM-5 was less effective, and nearly no synergistic effect was observed at low temperatures. The bigger pores and stable acid sites of the HY zeolite may promote the synergistic interactions between lignin and tetralin. Hydrogen transfer from tetralin to phenols may take place within the HY zeolite pores that facilitates hydrodeoxygenation and suppress decarboxylation or decarbonylation. Moreover, tetralin can also react with lignin to produce alkylated aromatics [46].
The H/Ceff ratio can also be adjusted by changing the relative amounts of alcohol and biomass as feedstock components. CFP of pine wood along with alcohols—methanol, 1-propanol, 1-butanol, 2-butanol, and their mixtures—using ZSM-5 as the catalyst was conducted in a bubbling fluidized bed reactor [48]. Petrochemical yield was found to be a function of the H/Ceff ratio, and so petrochemical yields increased as alcohols were added into the feedstock. In an isotopic study with co-feeds of 12C pine wood and 13C methanol, both 12C and 13C were found in all the hydrocarbon products indicating that methanol was involved in all the hydrocarbon formation reactions. However, the distribution of 12C and 13C across the products was not uniform. Higher levels of 13C were found in toluene, xylene, propylene, and butenes, while more 12C existed in naphthalene and ethylene.
Co-pyrolysis of olefins (ethylene and propylene) with a series of furanic compounds (furan, 2-methylfuran, furfural, and furfuryl alcohol) over ZSM-5 was carried out over the 450–600 °C temperature range [47]. While co-feeding of ethylene with furans did not change the product distribution significantly, the presence of propylene during CFP of furan and 2-methylfuran significantly increased the toluene aromatic selectivity and xylenes aromatic selectivity, respectively. Co-feeding of propylene with furfural and fufuryl alcohol also enhanced toluene selectivity. Diels–Alder cycloaddition reactions occurred when olefins were co-fed with furanics. Increasing temperature resulted in an increase in selectivity for benzene, toluene and CO, and decreasing temperature increased the coke yield and decreased the xylene yield.
Co-pyrolysis of corn stalk with food waste over HZSM-5 obviously changed the product distribution [49]. The synergistic effect between the two feedstocks during the co-CFP process significantly enhanced the production of aromatics.
Effect of Biomass Pretreatment
Drying: Simple drying in the relative absence of oxygen atmospheric conditions is the easiest pretreatment process to reduce the moisture of feedstock to below 10% [50]. The dry approach can be divided into nonreactive drying, reactive drying, and torrefaction [51]. Low temperature heat treatment leads to a shrinkage and reduced porosity of biomass, while severe heat treatment results in degradation and carbonization. The dry approach reduces biomass transportation costs and grinding energies, increases biomass stability, and improves conversion to liquid product [51].
Grinding: To increase the heat and mass transfer rates during CFP, biomass feedstocks need to be dried and ground into small pieces. Grinding specifications for biomass feeds in circulating beds, fluidized beds and rotating cone reactors are less than 6 mm, 2 mm, and 200 μm, respectively [52]. Minimizing the feedstock size increases contact between the feed material, catalyst, and reactor wall. However, cost of size reduction increases as biomass feed size decreases, and so some alternate strategies have also been developed. For example, Putun et al. [53] used a stainless steel basket to improve the heat and mass transfer to get higher volatile yields. A maximum liquid yield of 46.72% was achieved with particle sizes as large as 0.45–0.85 mm, heating rate of 500 °C/min, pyrolysis temperature of 500 °C, and sweeping gas flowrate of 400 cm3/min.
Torrefaction: Torrefaction pretreatment of pinewood has been shown to have a substantial effect on the bio-oil yield, properties, and distribution of pyrolysis products. Torrefaction of wood biomass can be performed in tube furnaces at relatively lower temperatures before catalytic pyrolysis reactions. The results showed the bio-oil yield decreased with increasing torrefaction temperature, while the biochar rapidly increased. Moreover, the aromatic hydrocarbon content increased while the content of acids and aldehydes sharply decreased [54].
The effect of torrefaction severity on CFP of corncobs was investigated over nanosized HZSM-5 in a semibatch pyroprobe reactor [55]. Torrefaction at 210–240 °C with a residence time of 40 min effectively improved the selectivity of BTX (benzene, toluene, and xylenes). While mild torrefactions at 210 and 240 °C had little impact on the aromatic yield, severe torrefactions at 270 and 300 °C led to a sharp decrease of aromatic yield and a significant increase of coke formation because of serious cross-linking and charring of corncob.
Torrefaction pretreatment of pinewood [56] significantly reduced the yields of acetic acid and guaiacol, while the aromatic yield was increased by 30% during catalytic pyrolysis. Compared with raw pinewood, the torrefied wood reduced coke deposition on ZSM-5 in the CFP process [57]. Torrefaction was also reported to increase BTX selectivity from CFP of hemicellulose and lignin; however, it had little impact on CFP of cellulose. The torrefaction on CFP of hemicellulose, cellulose, lignin over HZSM-5 in a tubular reactor resulted in the following rank order of structure change during torrefaction was hemicellulose > lignin > cellulose [57]. In addition, the aromatic yield of hemicellulose and lignin decreased after torrefaction that might because devolatilization and polycondensation of hemicellulose and lignin during torrefaction.
Effect of Indigenous Catalysts/Alkali and Alkaline Earth Metals
During biomass pyrolysis, inorganics, such as magnesium, potassium, and calcium, can catalyze biomass decomposition and promote char-forming reactions. Inorganic compounds are the main compounds of ash that constitutes as little as < 1 wt % in softwoods and up to 15 wt % in herbaceous biomass and agricultural residues [58]. To investigate the effect of potassium on CFP of biomass and properties of resulting bio-oil, numerous studies have compared low and high-metal contents of biomass feedstock. For example, the K content can be decreased by washing using acid or hot water. Higher K content feedstocks can be prepared by soaking biomass in potassium salt solutions, ion-exchange, or simply by mechanically mixing potassium salts with powdered biomass. The pyrolysis of lignocellulosic biomass is significantly altered by the potassium. Increased composition of potassium in the feedstock lowered the thermal decomposition temperature, promoted char formation, and led to agglomeration [59]. The presence of K during pyrolysis specifically facilitated certain chemical reactions, such as dehydration and demethoxylation, and suppressed the formation of levoglucosan. However, in another study, decomposition of levoglucosan was accelerated significantly by the alkali and alkaline earth metallic species (AAEM) inherent in the feed [60]. CFP of poplar wood with higher K contents—introduced by impregnation with K3PO4—selectively produced phenolic compounds [61,62]. Potassium inhibited the devolatilization of holocellulose to form organic volatile compounds. However, as K3PO4 contents were increased further, yields of the total phenolic compounds decreased.
Indigenous catalysts can enhance water–gas shift reaction, breakage, and restructuring of heteroatoms of tar. Regarding the bio-oil composition, indigenous catalysts promoted the conversion of sugars and the formation of carboxylic acids, aldehydes, and oxygenated aromatics [63]. In order to investigate the effect of AAEM species, Mallee wood was washed by water and a dilute acid solution [64]. While the bio-oil yield was not affected by the AAEM metals, the bio-oil composition was obviously affected. Inorganic salts and salt mixtures in switchgrass ash were investigated using a micropyrolyzer-GCMS, including their impact on the product distribution from cellulose pyrolysis [65]. Various concentrations of NaCl, KCl, MgCl2, CaCl2, Ca(OH)2, Ca(NO3)2, CaCO3, and CaHPO4 and switchgrass ash were added to pure cellulose prior to pyrolysis. The presence of mineral salts and higher pyrolysis temperatures enhanced the formation of low molecular weight species, e.g., formic acid, glycolaldehyde, and acetol, instead of leading to the formation of anhydro sugars, such as levoglucosan. Inorganic salts also affected the pyrolysis reaction kinetics [66]. Changes were observed in activation energies and pre-exponential factors for cellulose pyrolysis and were due to potassium content.
2.1.2. Waste Polymers
Regarding the subunits, Waste plastics can be divided to Polyethylene (PE), polypropylene (PP), polystyrene (PS), polyvinyl chloride (PVC), and polyethylene-terephthalate (PET). Not all kinds of waste plastics are suitable for CFP: PE, PP, and PS are good feedstock, while PVC and PET should be forbidden as hazardous gas or chemicals are generated during the for CFP process. Similar to Municipal biomass waste, the generation of waste plastics increases as the results of urbanization and is expect to be up to 0.25 billion tons by 2025 [67]. Waste tire is another important synthetic polymer source for CFP. Waste tire contains 60–65 wt % of rubber and 25–35 wt % of carbon black. Elevating H/Ceff ratio was proven to be an effective way to increase the carbon yield of aromatics and alleviating the formation of coke [47]. By approximation, about 1.79 million tons of waste tire were disposed in landfills without recycle in 2012. The development of pyrolysis technique has realized the conversion of waste tires to valuable products. Compared with biomass, waste polymers are more energy concentrated. For example, the energy contents of PET and tire crumb are 10,234 and 16,039 Btu/lb, respectively [68]. Regarding their structure, the oil yield was polystyrene and SBR (mainly part of tire) can be as high as 95 wt % [69] and 60 wt % [70], respectively. However, considering the material cost of waste polymer (~$150 per ton), the pyrolysis of pure waste polymer may not be economically favorable.
2.1.3. Polymers and Co-Pyrolysis of Polymers with Biomass
The large-scale, global production, usage and inappropriate management of plastics and tires has caused severe environmental problems. Chemical recycling of these materials, particularly pyrolysis conversion of polymers into useful chemicals, is an efficient and environmentally-friendly solution to address the accumulation of waste plastic and tires. During the pyrolysis process, polymers are degraded thermally in an inert atmosphere. However, direct thermal treatment of waste plastics and tires yields pyrolysis oil of inferior quality. Many catalysts used in the petrochemical industry, such as zeolites, have been directly borrowed for use in the CFP of waste synthetic polymers to improve the heating value and lower the carbon number of the products in the resulting pyrolysis oil.
Catalysts have been studied for the CFP of waste polymers in order to lower the reaction temperatures and optimize product distributions. HZSM-5 catalyzed pyrolysis of PP can reduce the BTX formation temperature from 700 °C for pure PP to 300 °C with three main compounds, i.e., alkenes, dienes, and aromatics, in the resulting oil [71]. These is a strong effect of both the polymer microstructure and feed mixture on the pyrolysis processes. For example, by mixing high density polyethylene (HDPE) with low density polyethylene (LDPE) and PP, the yield of condensable products was higher than for the thermal pyrolysis of pure HDPE. Conversely, inclusion of used tires in the feed decreased the yield of solid product by 33.5% (including unreacted polymer feedstock), as compared to a pure HDPE feed [72]. However, wide carbon number distributions were obtained in all cases, indicating no selectivity toward specific chemical products. In contrast, the pyrolysis of pure HDPE using HZSM-5 significantly increased yield of gaseous products, particularly C10–C16 hydrocarbons. It was observed that including PP in the feed mixture had no obvious effect on the product distributions in comparison to a pure HDPE and polyolefin mixture. The CFP of used tires significantly changed the carbon number distribution compared to that obtained from polyolefins.
Instead of performing pyrolysis of biomass or synthetic polymer independently, several co-pyrolysis studies have been reported in the literature [73,74]. As in standard pyrolysis reactions, co-pyrolysis processes are carried out in anaerobic conditions with temperatures around 400–600 °C [75]. Interactions during the pyrolysis process between the intermediates of lignocellulose and synthetic polymers can result in high carbon yields, aromatic hydrocarbons, aromatic selectivity, and heating/calorific value. At the same time, these reactions can decrease the amounts of solid residues such as the coke and bio-char produced. In practice, the H/Ceff ratio is a parameter that is strongly related to the carbon yield of aromatics and the formation of byproducts such as coke and bio-char [76,77]. The H/Ceff ratios of most biomass is less than 0.3, and these small values result in low yields of bio-oil liquid and severe coke formation [44]. Regarding the increase in the feedstock H/Ceff ratio, synthetic polymers can be mixed with biomass at various ratios and improvements in pyrolysis performance evaluated.
Any kind of virgin, residue, and/or waste lignocellulosic biomass can be applied in co-pyrolysis. For synthetic polymers feedstock, waste plastics and tires are the two main material categories most feasible due to cost and environmental considerations. Plastic waste is usually a mixture of materials composition including LDPE, HDPE, PET, PP, PS, and PVA, and waste tire materials generally contain 60–65 wt % rubber and 25–35 wt % carbon black [78]. In pyrolysis, co-feeding biomass with these synthetic polymer wastes can significantly reduce the amount of waste disposed in landfills, via incineration and marine environments [79], bringing benefits for the environment and making strides towards a low carbon society.
Mechanism of Catalytic Co-Pyrolysis
While the advantages of co-pyrolysis have been examined for both catalytic and non-catalytic pyrolysis, catalytic co-pyrolysis has gained more attention as it has demonstrated improvements in aromatic yield and selectivity. In general, advantages gained from co-pyrolysis come from both (i) the interaction of biomass and polymers during the thermal degradation and (ii) the interaction of pyrolytic volatiles at the catalytic sites [80]. The detailed mechanisms vary depending on the specific composition(s) of biomass and polymers used as well as the catalytic system.
A recent study showed that furan and furfural, which originate from the degradation of cellulose and hemicellulose in biomass, can react with olefins, such as PP and PE, through Diels-Alder reactions to form aromatics [49]. Subsequent dehydration of the aromatics can help drive the reaction and generate more aromatics [81]. At the same time, the olefins and alkanes originating from the polymer feedstock can also participate in the oxygenation reaction of cellulose and hemicellulose, preventing the formation of coke [82]. Instead of olefins present in PP and PE, styrene is the major active intermediate from the degradation of PS. The styrene intermediate can react with the allene derived from furans with successive alkylations to form indene. Subsequent reaction between allene and indene generates naphthalene and increases the aromatic yield [80]. By comparison of the experimental and theoretical carbon yield of products from CFP of biomass/polymer mixture with that of biomass or polymer alone, one can decide if there is chemical interaction between feedstocks. It was reported that synergetic effects between biomass and aliphatic polymer significantly enhanced selectivity to aromatics [83]. Polymer feedstock also aids the reduction of char formation during lignin degradation to phenolic compounds. Degradation of polymers though both random scission and chain-end scission reactions forms the radicals within the long carbon chains [84], and hydrogen transfer reactions may then convert these radical fragments into straight chain hydrocarbons. Hydrogen originating from the degraded polymers can react with biomass-derived oxygenates, as they are strong acceptors, and thus suppress char formation [85].
2.2. Reactors
In pyrolysis the thermochemical decomposition of feedstocks occurs under oxygen-constrained conditions, and this process can be used to produce pyrolysis oil (bio-oil), chemicals and (bio)char. While a mixture of products results from pyrolysis, the product yields and distributions are highly dependent on the equipment, process design, and operating parameters. Operating parameters include the type and the capacity of the reactor, pyrolysis temperature(s), solids residence time, carrier gas flowrate, vapor residence time, and biomass feedstock type and size [86], and these must be optimized for a given feedstock to control product yields and bio-oil properties.
Often the reactors used for pyrolysis at the lab and pilot-scales are different in design and operation than those used for commercial production. Among available pyrolysis units, fixed-bed reactors are very commonly used to investigate the effect of catalyst type(s) on CFP due to their low cost and easy operation. Recently, both lab-scale and pilot-scale pyrolysis oil production was examined using auger pyrolysis reactors as they are generally easy to operate and have low energy consumption and carrier gas requirements.
In designing a fast pyrolysis reactor, high heating rates, high heat transfer rates, and carefully controlled temperature are basic requirements. Moreover, the pyrolysis vapors need to be rapidly cooled or quenched to form the condensate oil product [86,87]. Four main reactor technologies are currently available for commercialization: Fluidized beds; circulating fluid beds; vacuum pyrolyzers; and ablative pyrolyzers (cyclonic and plate types).
While a wide variety of reactors have been tested, fluidized beds, and circulating fluidized beds are the most promising configurations for the ease of operation and higher quality products. Circulating fluid beds are energetically self-sustainable and can be used for very high throughputs due to high gas velocities. Heat is supplied by recirculation of heated sand or solid catalysts. To improve the conversion of biomass, solids recycling of partially reacted feed and fine sands or catalysts are necessary [88,89,90]. The char residence time is slightly higher than the gas residence time, and in-situ filtration of vapor [91] or post-treatment [92,93] of the pyrolysis oil is needed to reduce the char content caused by carryover/entrainment. The disadvantage for circulating fluid beds is the low thermal efficiency for heat transfer, as this is primarily dependent on gas-solid convective heat transfer.
Bubbling fluidized beds have great technical advantages in short residence times for vapors and larger gas-solid interface area. Scale-up of bubbling fluidized beds has been shown to be successful accomplished [94]. Bubbling fluidized beds generally have good temperature control. Shallow bed depths and high gas flow rates favor the production of higher quality pyrolysis oils. However, bubbling fluidized bed reactors suffer from several disadvantages [52]: (1) A low bed height-to-diameter ratio may cause transverse temperature and concentration gradients; (2) a high gas-to-biomass fed ratio can lead to low thermal efficiencies; and (3) a necessity to grind particles to less than 2–3 mm significantly increases the cost for feedstock processing.
As an alternative to fluidized beds, the conical spouted bed reactor (CSBR) has been developed and successfully used for pyrolysis of both biomass and waste plastics on both lab and pilot-scales [95,96,97,98]. The CSBR is superior to conventional spouted beds because the conical geometry can facilitate the use of high gas velocities, enhancing heat and mass transfer between phases and mitigating particle agglomeration. Moreover, CSBRs can be operated both in continuous and batch mode operation. Continuous mode offers good contact between phases, high heating rates, short gas residence times, and easy removal of carbon black with the cyclic solids movement. Compared to batch processing, continuous mode operation for the pyrolysis of scrap tires produced higher yields of light aromatics and lower yields of heavy liquids and tar [99].
In vacuum pyrolyzers, the lower pressures depress the boiling points that reduced the reactor temperature needed for conversion. Generally, side reactions can prevent these reactors while achieving carbon conversion at lower time and energy inputs. This is due to very short residence times that result from low operating pressures. Vacuum pyrolyzers also have an advantage because the biomass particle residence time is not coupled to that of the volatiles and so the residence time for the volatiles can be easily adjusted [100,101]. However, the major challenge in these systems is poor heat and mass transfer rates that makes equipment scale-up less technically feasible [52], and so they are most commonly used at lab and bench scales.
Ablative pyrolysis reactors have been investigated since the 1980s. In ablative reactors, reaction rates are not limited by heat transfer [102,103]. The ablative pyrolysis process is limited by the rate of heat supplied to the reactor instead of the rate of heat transfer into the biomass, as is the case with other reactors [102,103]. In reactors where the heat transfer rate into the biomass controls the reaction rates, there are subsequently requirements on the feedstock material size and shape requirements that necessitate without the need for energy and time-intensive feedstock preprocessing. An advantage of the ablative reactor is that it is large, whole tree wood chips—and even rods—that can be used as feedstock. In addition, inert gas is not required, so the processing equipment volume is smaller. Unfortunately, large amounts of tar are produced in ablative reactors due in part to the necessity to recycle the partially reacted solid particles in order to achieve sufficiently long residence times for the biomass. The moving parts in cone and plate type ablative pyrolysis reactors make them more mechanically complex, which can increase operating and maintenance costs. An additional drawback for ablative pyrolyzers is that they have a high heat loss, as the heated surface is at a temperature substantially above the required reaction temperature [104].
2.3. Vapor Cleaning and Quenching
Some char, which contains almost all of the ash from the biomass, is inevitably carried over into the vapor phase after the pyrolysis. The alkaline components of biomass ash are known to be cracking catalysts for the organic molecules in the vapor, with a consequent low yield of condensed liquid. Even in the cooled liquid products char still contributes to the instability problems by accelerating the “aging” reactions. Therefore, rapid and complete char separation is difficult, but is highly desirable. Char can be removed by hot vapor filtration or by filtration of the condensate using, for example, cartridge or rotary filters.
The collection of liquids has long been challenging due to the nature of the gas phase, which is mostly in the form of aerosols rather than a true vapor. Careful design and temperature control is necessary to avoid blockage from differential condensation of the heavy ends. The light ends collected can be used to reduce the viscosity of the condensate liquid. Some research groups have examined electrostatic precipitation to rapidly recover the aerosols [105]. In fluidized bed systems, the vapor/aerosol concentration can be very low, further increasing the difficulty of product separation due to the low vapor pressure.
2.4. Optimization of Bio-Oil Yield Using Statistical Method
For both the in-bed and in-situ catalytic vapor upgrading techniques, the feedstock is rapidly heated in a non-oxidizing atmosphere. Vapors are condensed in several steps resulting in dark brown pyrolysis liquid. More careful control of the design and operation of the pyrolysis reactor and condensers is needed in order to give high pyrolysis liquid yields. Key fast pyrolysis design parameters [52], including the feedstock type and treatment method, very high heating and heat transfer rates, carefully controlled temperatures around 450–550 °C, short vapor residence times obviously significantly affect the CFP behavior and product distribution. Moreover, these parameters are always interrelated, hampering the optimization of the CFP process. Many statistical methods have been developed to achieve the optimum condition.
Taguchi experimental design method can skip the correlation between the yield of bio-oil with the process parameters. It was employed to establish the optimal operation conditions to maximize the yield of the bio-oil produced from camphor tree wastes [106]. A 3.35 mm feedstock size, 40 rpm screw extruder rotational speed, and a 500 °C pyrolysis temperature can achieve the highest yield of bio-oil (60.2%).
Response surface methodology has been used to optimize the CFP processing parameters of red oak wood biomass on a lab scale auger pyrolyzer. Steel shot was used as the heat carrier and the heat carrier temperature, heat carrier mass flow rate, auger speed, and gas flow rate were included in the optimization. A circumscribed central composite design of experiments found maximum bio-oil yield (greater than 73 wt %) and minimum char yields could be obtained at high values of all of the tested parameters—high sweep gas flow rate (3.5 standard L/min), high heat carrier temperature (~600 °C), high auger speed (63 RPM), and high heat carrier mass flow rate (18 kg/h). In this study, a synergistic interaction effect was discovered for the heat carrier mass flow rate and auger speed [107].
2.5. Pyrolysis Oil Stability
2.5.1. Pyrolysis Oil Composition
The oil produced from pyrolysis is generally complex fluid, dark brown in color, and composed of a very large number of compounds. During the CFP pyrolysis process, a large number of reactions—including hydrolysis, dehydration, isomerization, dehydrogenation, aromatization, retro-condensation, and coking—occur due to the chemical complexity of the feedstock materials and high temperatures. The particular chemical composition and physical properties of any given pyrolysis oil is dependent upon the following [108]:
- (1)
- feedstock (including particle size, dirt, moisture, and protein content);
- (2)
- heat transfer rate;
- (3)
- extent of vapor dilution, residence time and temperature of vapors in the reactor;
- (4)
- efficiency of the condensation equipment;
- (5)
- char temperature during pyrolysis;
- (6)
- efficiency of the char removal system;
- (7)
- storage time and temperature; and
- (8)
- extent of contamination during pyrolysis and storage.
Organic Compounds
Crude pyrolysis oil is a complex mixture that contains more than 400 different compounds. These compounds can be broadly classified into volatile organic compounds (hydroxyacetaldehyde, formic acid, and acetic acid), furanic compounds, monophenols, sugars (fermentable and cross-linked), lignin oligomers, and water [109,110,111]. The organic composition of pyrolysis oil is correlated with the biomass feedstocks. Milne [112] and Diebold [113] made a comparison about the composition of fast pyrolysis oils derived from different feedstocks. The components were similar, while their contents varied significantly. The liquid mixtures are derived primarily from decomposition/fragmentation reactions of the three major components of lignocellulose [114]: cellulose, hemicellulose, and lignin. The guaiacyl derivatives, coniferaldehyde, and coniferyl alcohol are major products from softwood lignins, while guaiacyl and syringyl derivatives are derived from hardwood lignins. Grass lignins yield p-vinylphenol is a major compound [115]. Phenolic compounds in pyrolysis oils are mainly derived from pyrolysis of lignin. Bark tends to contain highly reactive tannins. The miscellaneous oxygenates, sugars, and furans are from the decomposition of cellulose and hemicellulose. Levoglucosan is mainly produced from cellulose [116,117]. The cellulose degree of polymerization and crystallinity are relevant to the pyrolysis oil composition. The biomass, having high protein contents, for example bark or grass, would be expected to produce pyrolysis oils with higher nitrogen contents.
Inorganic Compounds
Inorganic compounds, while their contents are very low, play a critical role in pyrolysis oil product selectivity. The inorganic or mineral content of biomass is not only found in the aqueous phase, but also in the organic phase and deposited solids. The alkali metal species and amounts present are primarily due to the feedstock composition, but the presence of alkali salts has a major impact on the reaction mechanisms. During pyrolysis, inorganic elements—especially K and Ca—catalyze biomass decomposition [58]. The addition of minor amounts of alkali materials to cellulose feedstock has been shown to shift the final product from levuglucosan to glycolaldehyde [118,119,120]. These materials with counter ions in solution including oxalates, silicates, carbonates, phosphates, chlorides, and sulfates are important considerations even after pyrolysis [121,122,123]. The char and inorganic compounds have been reported to catalyze polymerization reactions during bio-oil storage, resulting in increases in viscosity and average molecular weights [120].
2.5.2. Accelerated Aging
The storage properties of pyrolysis oil are critical to its usage as a transportation fuel. The raw pyrolysis oil is characterized by a number of deleterious properties such as high-water content, low heating value, high viscosity, immiscibility with hydrocarbon liquids, and high solids content. Since pyrolysis oil is not a product of chemical and thermodynamic equilibrium as a result of rapid quench, the active ingredients continue to react until equilibrium is reached [124]. All the reactions that occur during storage produce hydrates, hemi-acetals, water, resins, oligomers, and esters, etc. The pyrolysis oil will be oxidized if exposed to oxygen, forming more acids and peroxides that catalyze polymerization of unsaturated compounds. As all these processes progress with time, it is called “aging” [125]. These aging reactions lead to an increase in high molecular-mass lignin materials with a decrease in aldehydes, ketones, and lignin monomers. Etherification and esterification reactions occurring between hydroxyl (-OH), carbonyl (C=O), and carboxyl (C(=O)OH) groups are reported to be the primary chemical reactions taking place in pyrolytic oils [126]. Recently, aggregation of pyrolytic lignins was found to be the central process driving the aging processes [127]. Char particles can also accelerate the aging reactions. Polarity of the pyrolysis oil can even be changed by aging reactions.
It is necessary to evaluate property changes during storage and transportation to understand the chemical instability and the development of stabilization strategies. An “accelerated aging method” has been used to shorten the investigation time, as elevated temperatures have been reported to accelerate the aging reactions [17]. Czernik [126] investigated the effects of storage conditions on physical and chemical properties of fast pyrolysis oils from oak biomass. The oil samples were stored at temperatures between 37 and 90 °C in sealed glass vessels. Water content, viscosity and molecular weight of the oil increased with aging time and temperature. First-order reaction kinetics were used to predict changes in molecular weight and viscosity during storage. FTIR results indicate that etherification or esterification occurs during storage. Boucher et al. also investigated the stability of pyrolysis oil derived from softwood bark residue [128] at 40, 50, and 80 °C for different periods of time. The properties of the pyrolysis oil were dramatically altered when aged at 80 °C. For example, the molecular weight increase after aging for one week at 80 °C was equivalent to the effects seen after storage for one year at room temperature. The addition of aqueous phase to the pyrolysis oil should be less than 15%, otherwise the oil’s stability was lowered significantly. Oasmaa et al. [129] investigated the physicochemical property change for pyrolysis oil stored in darkness at 9 °C or at room temperature under light. The average molecular mass, viscosity, water content, density, and pour point of pyrolysis liquids increased with lengthened storage time. The average molecular mass was correlated with the viscosity, water-insolubles, and the high-molecular-mass (HMM) fraction of water-insolubles observed. The increase in lignin-derived HMM fraction of water-insoluble was caused by polymerization/condensation reactions of carbohydrate constituents, aldehydes, and ketones. Hilten and Das [130] utilized three stability ranking methods to compare oxidative and thermal stability of alcohol-stabilized and un-stabilized pyrolysis oil. Each method involved an accelerated aging procedure ranging from several minutes to 24 h. The C-O and C=O functional groups intensity increased after the aging process. The methanol stabilized oils showed better average stability. Ortega et al. [131] studied the aging of the oils made from hardwood and softwood feedstocks at room temperature. The viscosity increased significantly after five months. To explore aging characteristics in the absence of water loss, pyrolysis oils were aged in a water saturated environment at elevated temperatures (~ 65–85 °C). Oils aged at 65 °C after seven days showed increases in viscosity similar to the oils aged for five months at room temperature. The high water content and absence of char inhibit the occurrence of polymerizations.
To understand the chemistry of the aging process of pyrolysis oils, Ben and Ragauskas [132] investigated chemical structural changes during aging of loblolly pine residue pyrolysis oils at 80 °C for 60 h by in situ NMR method. The content of aliphatic C-O bonds and aromatic C-H bonds decreased during the aging process, while the contents of aliphatic C-C bonds, aromatic C-C, and C-O bonds increased. The HSQC NMR results indicated that the content of aromatic C-H bonds at ortho and para positions to hydroxyl groups decreased faster than that of aromatic C-H bonds at meta positions to hydroxyl groups. The content of levoglucosan decreased during aging. The condensation reaction to form aromatic C-O and C-C bonds could be initiated by the decomposition of the instable organic peroxides present in pyrolysis oils. The crosslinking reaction between formaldehyde and aromatic C-H bonds could also form new aromatic C-C bonds and consume aromatic C-H bond and carbonyl groups.
Hu et al. [133] investigated the roles of several typical compounds representing the sugars, sugar derivatives, and aromatics found in pyrolysis oil for their contribution to condensation reactions. Experiments were performed by increasing the temperature from 90 to 190 °C. The decomposition of glucose into volatile compounds with hydroxyl, carbonyl groups, or conjugated C=C bonds played a key role for the polymerization. Furfural, hydroxyl aldehyde and hydroxyl acetone can react to form polymers. The carboxylic acids played the role of catalyst toward polymerization. The phenolic compounds promoted the acid catalyzed reactions. The adding of methanol significantly suppressed the decomposition of glucose and the polymerization of other compounds.
Alsbou et al. [134] described the evaluation of an accelerated aging process on the physicochemical properties of pyrolysis oil from fast pyrolysis of ash and birch woods using pilot scale and lab scale reactors. The water content, viscosity, decomposition temperature, and ash content levels increased as the aging period lengthened. GC/MS analysis of the pyrolysis oil indicated the amount of olefinic containing compounds significantly reduced. FTIR showed a reduction in aldehyde and hydroxyl carbon signals indicating etherification and esterification occurred.
Aging in a broad temperature range (40–290 °C) was performed to determine stability and explore the aging mechanisms. The heat-treatment at temperatures above 60 °C significantly increased the content of HMM lignin with increasing aging time. FTIR results indicated aging reaction mechanisms for oils aged at temperatures between 40 and 80 °C and between 150 and 290 °C were different. Decomposition of sugar compounds above 200 °C was detected by differential scanning calorimetry (DSC), the reactions among furfural, 2-cyclopenten-1-one, 2-hydroxy-3-methyl-2-cyclopenten-1-one, hydroxybenzenes with available ortho/para sites, and the active sites on HMM lignin contributed to property changes as a result of heat treatment [135].
2.5.3. Additives to Stabilize Pyrolysis Oil
Various organics have been used to achieve stabilization of pyrolysis oil. Diebold et al. [125] added methanol, ethanol, acetone, ethyl acetate, and methyl isobutyl ketone to improve the stability of pyrolysis oil. The oils were aged in sealed glass vials for 10–150 h at 90 °C. The more severely aged samples have more built up gas pressure and some leakage was observed. These additives significantly decreased the rate of aging, as measured by the rate of increase of oil viscosity with time, by factors of seven to 18 times that observed with pyrolysis oil. The formation of hydrogen-bonding of pyrolysis oil with these additives and the chemical reactions between them caused the aging inhibition effect. Methanol was promising for its low cost and effectiveness to reduce aging rates.
Similar results were observed by other groups [128,136,137]. It is concluded that the addition of alcohols improved homogeneity, while significantly lowering the rate of increase in viscosity and molecular mass. The viscosity decrease was caused by a stabilizing effect of alcohols on the water-insoluble lignin-derived fraction. The formation of acetals due to reactions of alcohols with aldehydes, ketones, and anhydro sugars also slow down the aging reactions. A higher amount (> 10 wt %) of additive retarded the aging by almost a year.
Bakhshi et al. [138] found the adding of tetralin improved the stability of pyrolysis oil. The properties of the pyrolysis oil mixture kept unchanged with time. The free radicals existed in pyrolysis oil might be quenched by the donation of hydrogen from the tetralin. Meng et al. [139] observed that free radicals were preferentially located in the pyrolysis oil lignin fraction, especially in the higher molecular weight lignin. The pyrolysis oil radicals were present in a stable state for radical scavengers showed negligible effects on the condensation of the pyrolytic lignin.
Alcohols with different chain lengths, i.e., methanol, ethanol, 1-propanol, 2-propanol, and 1-octanol, were added to crude pyrolysis oil and their thermal stabilities at 200 °C were investigated [140]. Isothermal DSC results indicated that except 1-octanol, all the low molecular mass alcohols enhanced the stability of pyrolysis oil. However, 1-octanol was the most efficient in decreasing the viscosity and molecular weight of pyrolysis oil. FTIR spectra indicated reactive carbonyl and aldehyde groups were captured by the added monofunctional alcohols. Esterifications have been found to have contributed significantly to mitigate aging reactions.
2.6. Pyrolysis Oil Upgrading
The low heating value and chemical instability make the pyrolysis oil must be upgraded to be an efficient fuel source. Many physical and chemical pyrolysis oil upgrading technologies have been studied. These include filtration [58], catalytic cracking [141,142], hydrotreating [143,144], and steam reforming [145,146,147].
The presence of char particles in the pyrolysis oil accelerates the aging reactions in storage and end-use. Additionally, the presence of high concentrations of submicron char particles in pyrolysis oils will pontentially result in ash and alkali metal build-up and cloggin during injection and combustion when used as fuels for steam boilers, diesel engines, and turbine operations [120]. Char is entrained with organic vapors; however, particles larger than about 10 μm in diameter can be efficiently separated from the vapors by cyclonic separation. Hot-gas filtration can efficiently remove the smaller char particles, but fine char particulates pass through these filters as well. Agblevor et al. [58] reported hot gas filtration reduced the alkali metals content to less than 10 ppm, while cold filtration of the oils dissolved in acetone was ineffective in reducing the alkali metals content to acceptable levels. Likewise, leaching studies exhibited no leaching of alkali metals [58,120]. Microfiltration of the condensed pyrolysis oil can be used to further reduce the fine char content. Javaid et al. [148] used tubular ceramic membranes to remove char particles less than 10 μm from condensed pyrolysis oil. The process was conducted at temperatures ranging from 38 to 45 °C and at pressures ranging from one to three bars. A significant reduction in overall ash content of the pyrolysis oil was observed, while no significant changes in other propeties occured due to the microfiltration process. Removal of nano-sized char particles has been examined using nano-filtration methods. However, minerals solubilized by the acidic nature of pyrolysis oil cannot be removed by filtration and can contribute to aging related instabilities of the pyrolysis oil discussed previously [149].
Catalytic hydrotreatment is a promising approach to improve the properties of pyrolysis liquids. Elliott [144] presented a thorough review of developments in the field of pyrolysis oil catalytic hydroprocessing over the past few decades. Both precious metal catalysts and conventional catalysts developed for petroleum hydroprocessing have been tested. Conventional hydroprocessing techniques applied to petroleum feed stocks must be adjusted for pyrolysis oil hydroprocessing. Hydroprocessing is normally carried out at temperatures 250–450 °C. It is crucial that a catalyst be able to withstand water at the extreme conditions of high temperature and pressure. Deoxygenation increases with higher residence times; therefore, long liquid phase residence times are required. In general, liquid hourly space velocity (LHSV) should be in the order of 0.1–1.5 h−1 for flow reactors [150], and the favored residence time in batch reactors are 3–4 h [151,152].
The reactivity of oxygenated groups found in pyrolysis oil is dependent on hydrotreatment temperature. Olefins, aldehydes, and ketones can easily be reduced by hydrogen at low temperatures. These reactions improve the stability for pyrolysis oil by removing highly reactive groups. Alcohols are reacted at 250–300 °C by catalytic hydrogenation and carboxylic and phenolic ethers are also reduced to ~300 °C. However, thermal dehydration of alcohols forms olefins that may further polymerize under hydrotreatment conditions. Venderbosch et al. [143] recently developed an integral processing route for the improved hydrotreatment of pyrolysis oil. It includes a lower operating temperature step at 175 to 250 °C in which reactive components (olefins, aldehydes, ketones and alcohols) are reacted, and then the stabilized product fraction can be further processed by hydrotreatment at a high temperature.
Ketonization is the process of linking two carboxylic acids with eliminating carbon dioxide and water to form a ketone product. Ketonization of carboxylic acids in bio-oil can overcome its acidity and also improve the quality [153]. Lilga et al. [154] developed a novel process for a single step conversion of levulinic acid and ɣ-valerolactone to a biphasic liquid product and improve carbon yield in the fuel range. Metal oxides [155], such as CdO, CoO, Fe2O3, La2O3, TiO2, CeO2, ZrO2, V2O5, MoO3, and MnO2, supported on silicon oxides and pure silica have been investigated for the ketonization of acetic acid. While temperature significantly affected the conversion of acetic acid, there were differences in activities between these metal oxides with CdO, La2O3, CeO2, and MnO2 exhibiting higher activities. The iron redox state and mixed alkali doping were found to significantly impact the ketonization of key bio-oil oxygenates, e.g., acetic acid, acetol, formic acid, and levoglucosan [156]. A lower temperature (300 °C) reduction of iron oxide led to higher selectivity toward acetone and 2-butanone while coking was not severe. The presence of mixed alkali metals enhanced ketonization activity.
3. Catalyst Design and Product Distribution Optimization
Catalytic fast pyrolysis can be categorized into two main processes: In-bed and in-situ catalytic vapor upgrading. In-bed catalytic pyrolysis occurs in a reactor where biomass feedstock and catalysts are mixed together. Biomass is rapidly heated in the absence of air, vaporizes, and quickly condenses to liquid, together with by-products such as char and gas. With the in-situ vapor upgrading process, the feedstock is thermally cracked to produce pyrolytic vapors that pass through catalyst beds where they are upgraded into liquid, gaseous as well as solid byproducts through homogeneous gas phase reactions. The in-situ catalytic vapor upgrading technique offers greater flexibility in optimizing the production process because the catalytic temperature and pyrolysis temperature are independently controlled. In addition, for in-situ catalytic vapor upgrading process, higher temperatures favored formation of alkyl phenols and aromatics [157], and the biochar formed with in-situ vapor upgrading can be easily collected as a valuable byproduct. The disadvantage is the low yield of liquid oil product and higher amounts of water and gas than the in-bed technique [157]. It should be noted that the theoretical catalyst/feedstock ratio for in-situ vapor upgrading is much higher than the in-bed process because the feed consists of volatile bio-oil and non-condensable gas. Coke formation on the catalyst was significantly reduced with in-situ vapor upgrading, as compared to in-bed CFP.
A few comparative studies on lab-scale in-bed and in-situ catalytic vapor upgrading techniques have been reported [157,158,159,160]. A large catalyst/biomass ratio was usually used for these reactors to ensure a sufficient catalytic effect for both processes. The bench-scale CFP efforts using fixed bed reactors suffers from the drawbacks of much lower bio-oil yield and higher char yield due to slow heat transfer rates. A successful bench-scale comparative study carried out on a spouted-bed reactor gave similar results with a micro-scale experimental apparatus [158]. With limited prior efforts comparing vapor upgrading processes, it is difficult to develop general guidelines for the role of different mechanisms for contact between pyrolysis vapors and catalyst play on differences observed in product distributions. For example, in a comparison of in-situ CFP and in-situ vapor upgrading with HZSM-5 in a two-stage fluidized-bed/fixed-bed combination reactor, both technologies gave similar carbon yield of aromatics and olefins, while the in-situ vapor upgrading CFP produced nearly double the olefins [161]. However, in a different study comparing in-bed and in-situ catalytic vapor upgrading of lignin using HZSM-5, the in-situ catalytic vapor upgrading process exhibited higher selectivity to aromatics and produced more syngas and less CO2 than the in-bed process.
Catalysts play a pivotal role in maximizing bio-oil yield and optimizing product distribution. There are many excellent reviews [162,163,164,165,166] on catalysts used in the CFP of biomass and co-CFP of biomass with synthetic polymers. However, few reviews discerned the effect of different CFP process on the pyrolysis oil yield and product distributions while discussing the catalysts effect. The CFP process involves a complex reaction network sensitive to feedstock, pyrolysis technique, pyrolysis operational parameters, and catalyst(s) bulk properties. Thus, the catalysts used for in-bed and in-situ catalytic vapor upgrading will be discussed separately.
3.1. Catalysts for CFP
3.1.1. Zeolites
Zeolites, with crystalline aluminosilicate structures fabricated by silica tetrahedron and alumina tetrahedron through oxygen bridges, exhibit high surface areas, abundant acid sites, and high thermal and hydrothermal stability that facilitates cracking and dehydration through adsorption of the oxygen containing compound on acid sites, following by decomposition or bimolecular monomer dehydration, respectively [167]. The production of bio-oil from CFP of biomass and/or polymers involves a complex reaction network sensitive to the acidity, pore size, crystallinity, porosity, elemental composition, and crystal size, etc. The interplay of these parameters under the reaction conditions has been a challenge to improve catalyst design [89,168,169]. In comparison with non-catalytic fast pyrolysis, the use of zeolitic catalysts caused a decrease in the bio-oil yield due to enhanced formation of gases, as well as coke deposition on the catalyst. A borrowed theory of indirect hydrocarbon pools [170] in zeolites has been used to explain the complex reaction network inside the zeolite during catalytic pyrolysis.
Pore size of zeolites directly impacts liquid yield and production distribution. Table 1 lists typical zeolite catalysts using in the CFP process showing different structures and pore sizes. Generally, medium pore (5.2–5.9 Å) and large pore size zeolites produce more valuable compounds. Here, we will focus on the development of zeolite catalyst usage specifically in converting biomass and synthetic polymers to pyrolysis oil and fuel additives.
The effect of pore size and shape of zeolites on the CFP of glucose was investigated with a variety of zeolites (small pore ZK-5, SAPO-34, medium pore ferrierite, ZSM-23, MCM-22, SSZ-20, ZSM-11, ZSM-5, IM-5, TNU-9, and large pore SSZ-55, Beta zeolite, Y zeolite) [171]. The pore size directly affects the aromatic yield: Small pore zeolites did not produce any aromatics, but more CO, CO2, and coke; medium pore zeolites with pore sizes in the range of 5.2–5.9 Å produced the highest aromatic yields; and large pore zeolites formed low aromatic yields, high coke yield, and low oxygenate yields. Moreover, the internal pore space and steric hindrance may also contribute the aromatic production. Medium pore ZSM-5 and ZSM-11 with moderate internal pore space and steric hindrance have the highest aromatic yield and the least amount of coke. Based on the analysis of kinetic diameters of products and reactants, most of aromatic products and reactants can fit inside the pores of medium and large pore zeolites; polycyclic aromatics cannot enter smaller pores of zeolites, and therefore they may form by secondary reactions on crystal surface or either directly or via reaction of the smaller aromatics. In an in-situ catalytic upgrading of bio-oil vapor obtained from lignin, five types of high aluminum zeolites, i.e., H-Ferrierite, H-Mordenite, H-ZSM-5, H-Beta, and H-USY were tested [172]. H-ZSM-5 produced the highest yield of light oil while H-Beta exhibited the highest selectivity towards monoaromatic hydrocarbons.
Pore enlargement at high temperature was also found to affect the pyrolysis behavior of feedstock. The CFP of lignin over ZSM-5, mordenite, beta, and Y zeolite were investigated to determine the role of shape selectivity of zeolites [35]. Thermal distortion of zeolite pore structure at 650 °C effectively enlarge the pore sizes of the zeolites by 2.5–3.4 Å, as shown by crystallography. While monolignols could not be effectively converted by ZSM-5 and mordenite zeolites because of size exclusion or pore blockage, many less bulky lignin-derived oxygenates can diffuse into the pores of the zeolites and convert to other chemicals. The aromatic yield followed the order ZSM-5 > beta > mordenite > Y. Beta and Y zeolites were the most effective catalysts for deoxygenating lignin-derived oxygenates. ZSM-5 is the optimal catalyst for CFP of softwood because it can achieve satisfactory deoxygenation and aromatic production simultaneously. Beta zeolite was more promising for the CFP of hardwood feedstock to convert bulky oxygenates derived from syringyl lignin.
ZSM-5
ZSM-5 is used in the industrial transformation of methanol to olefins. Not only in fossil fuel processing industries, but also in CFP, ZSM-5 zeolite is the mostly investigated catalyst due to advantages of its acidity and pore structure. ZSM-5 zeolite has an MFI structure, a channel system composed by interconnected straight and sinusoidal 10-membered ring channels of ~5.0 Å diameter, which intersect to form larger void spaces of ~7.0 Å in diameter. The medium-pore structure of ZSM-5 blocks the access of big molecules. In most cases, the use of ZSM-5 leads to a decrease in both yield and oxygen content of the resulting oil and an increase in production of single-ring aromatics.
The acidity of the zeolite, expressed as silica-to-alumina ratio (SAR, or Si/Al ratio) affects the reactivity and yields. High acidities provide a higher affinity for carbon and water formation. Glucose, furan, and maple wood have been investigated over different types of ZSM-5 catalyst in semi-batch and fixed-bed reactors. A SAR of 30 was an optimum for the aromatic production from glucose conversion [173].
By comparing characterization and catalytic results of commercial and laboratory-synthesized ZSM-5 zeolites, crystallinity, and framework aluminum site accessibility were found to be critical to achieve high aromatic yields [174]. The acid sites of zeolites can be modified by chemical liquid deposition (CLD) and acid dealumination (AD). The optimized synthesized ZSM-5 catalyst exhibited a very high aromatic hydrocarbon yield. The CFP of pinewood over CLD and AD modified ZSM-5 led to increases of relative area ratio of benzene, toluene, and xylene (BTX) by 37.2% and 30.4%, respectively. The CLD by SiO2 preferentially covered strong acid sites, while AD removed part of superficial acid sites, and thus the yields of precursors of heavy coke, e.g., indenes and naphthalenes decreased significantly. The low cost and high catalytic performance made CLD modification of ZSM-5 promising in the CFP of biomass [175].
The effect of crystal sizes of ZSM-5 has been less investigated due to the complexity of zeolite crystals. Zheng et al. [35] hypothesized that the diffusion of reactants and products in the ZSM-5 pore channels may limit the catalytic performance. The CFP of cellulose, hemicellulose, lignin, pine, corncob, and straw over ZSM-5 with varying crystal size (2 μm, 200 nm and 50 nm) were conducted in a pyroprobe pyrolyzer. ZSM-5 with crystal size of 200 nm exhibited the maximum aromatic yield and minimum BTX selectivity, while the 50 nm ZSM-5 gave the highest yield of desired BTX products. They attributed the high performance of 200 nm crystal to the highest micropore surface area and maximum acid sites and Brønsted to Lewis acid sites ratio. The CFP of furan also indicated that decreasing crystallite size can increase furan conversion and improve selectivity to BTX and olefins [176].
Mesoporous ZSM-5 obtained by NaOH desilication resulted in enhanced activity, selectivity, and stability for conventional methanol to gasoline/olefin reactions. However, the effect of desilication on catalytic activity is more complex for biomass transformations. The desilicated ZSM-5 present higher selectivity to mono-aromatics and higher stability against coke deposition than the conventional ones. The conversion of biomass also increases because of enhanced mass transport and intracrystalline diffusion that favor aromatic production and lower coke formation [177]. Similar observations during the CFP of model compounds [176], e.g., furan was also reported: Increasing mesoporosity increased furan conversion and improved selectivity to BTX and olefins. As proven with cyclohexane as a probe molecule, coke was found inside the micropores of ZSM-5, and thus, the catalysts of coke formation can be largely reduced up to 65% by introducing mesoporosity. During in-situ red oak pyrolysis vapor upgrading, the usage of mesoporous ZSM-5 also enhanced formation of aromatics [177]. The mild desilication treatment increased the number of accessible Brønsted acid sites by ~50% by introducing mesoporosity while the elemental composition, element distribution, and crystallographic structure were not changed obviously. Desilication increased the aromatic yields from 23.9% of parent zeolite to 27.9% while keeping 67.4% selectivity to benzene, toluene, and xylene.
HZSM-5
HZSM-5, the proton form of ZSM-5, is the most widely investigated zeolite in CFP. HZSM-5 continues to gain much interest after a review [178] focused on the usage of HZSM-5 in CFP and pyrolysis oil upgrading was published in 2013. Jackson et al. [179] investigated HZSM-5, KZSM-5, AlMCM-41, solid phosphoric acid, and a commercial hydrodesulfurization catalyst Co/Mo/Al2O3 in the pyrolysis of Asian lignin. HZSM-5 zeolite performed best in producing deoxygenated organics because of its strong acidity: A deoxygenated liquid composed of simple aromatics (46.7%) and naphthalenic ring compounds (46.2%) was obtained. Mihalcik et al. [180] studied the CFP of lignocellulosic biomass samples comprising oak, corncob, corn stover, and switchgrass, as well as the fractional components of biomass, i.e., cellulose, hemicellulose, and lignin with H-mordenite, HZSM-5, HY, H-Beta, and H-Ferrierite, and also got the result that HZSM-5 was the most effective zeolite for producing aromatics. The pore size distribution of H-ZSM5 was proposed to favor the formation of aromatic hydrocarbons [181].
Fast pyrolysis of black-liquor lignin by HZSM-5 with Si/Al ratio of 25, 50, and 210 was investigated by Py–GC/MS (pyrolysis gas chromatography/mass spectrometry) from 550 to 900 °C [182]. The result indicated HZSM with SAR 25 was the most effective one for promoting the formation of aromatic monomers. A low number of acid sites (low Si/Al ratio) also enhances the production of phenol alkoxy species [181].
The usage of mesoporous HZSM-5 in CFP of oak in a microfluidized bed reactor at 500 °C led to doubled selectivity toward mono-aromatic compounds than conventional microporous HZSM-5 [183]. The mesopores played the role of “highways” where big molecules (e.g., levoglucosan) can diffuse to more accessible pore mouths. They also enhanced diffusion of and evacuation of catalytic products, which improved the selectivity of mono-aromatic hydrocarbons. It has been reported mesoporosity increased the aromatic yield, while it decreased the coke yield in CFP of beach wood and cellulose [184,185]. However, a maximum amount of mesopore volume exists for an optimal yield of bio-oil production. Too high amount of mesopore volume is deteriorate because the intermediate aromatic hydrocarbons continue to polymerize to form poly-cyclic aromatic hydrocarbons and coke [185].
MCM-22
MCM-22 is one kind of mesoporous zeolite containing two independent pore systems [186]. Both of the pore structures are accessible through rings composed of ten tetrahedral atoms. The pore size distribution plot obtained with argon shows two distinct peaks in the 6–7 Å region. Moreover, the unique three-step uptake profile of bulky 2,2-dimethylbutane observed, in the dynamic sorption experiment, also reflects the complex pore structure of MCM-22 [187]. These coexisting pore systems have facilitated the usage of MCM-22 zeolites to crack higher molecular-weight compounds to produce aromatic hydrocarbons in the fossil industry.
The effect of Si/Al ratio of MCM-22 has been investigated in the CFP process of acid-washed wheat [188]. MCM-22 with SAR 40 (lowest Si/Al ratio investigated in the work) showed the highest bio-oil deoxygenation activity and energy yield at 450 °C. The high acid site concentration of MCM-22 (SAR = 40) promoted side reactions, e.g., gaseous hydrocarbons formation by cracking and coke formation. Moreover, MCM-22 exhibited lower aromatization activity than ZSM-5, while it produced a high concentration of oxygenated aromatics.
MCM-36, a pillared zeolite obtained from MCM-22, exhibited a lower deactivation rate than its parent structure [189]. Catalytic transformations of levoglucosan with H-MCM-22 and pillared H-MCM-36 were carried out in a fixed bed reactor at 573 °K [190]. The acidity of the zeolite catalysts significantly affected the yield of the oil products, phases, and product distribution. Oxygenated species such as aldehydes and furfural were the main liquid products. MCM-36 showed higher activity in the formation of oxygenated species, except acetone, than over MCM-22.
ITQ-2, obtained by delamination of MCM-22, possesses higher external surface area and acidity than its parent MCM-22. The thinner structure facilitates the contact of reactants with catalytic sites and lets the products leave easily. Uemura et al. [191] compared the performance of both catalysts in the in-situ catalytic vapor upgrading of paddy husk. However, from the X-ray diffraction pattern of the synthesized zeolite catalysts, only a small amount of MCM-22 was exfoliated to form ITQ-2. Even in the presence of many impurities, ITQ-2 still exhibited a higher deoxygenation degree of oxygenated species and higher yields of aromatic hydrocarbons.
Zeolite Y
Zeolite Y is widely used in fluid catalytic cracking process, hydrocracking processes, and biomass catalytic conversion to fuel and chemicals. Ultra-stable Y zeolite (USY) is the most abundant zeolite catalyst used for the fluid catalytic cracking (FCC) process. The presence of extra-framework Al species in USY is an important source of Brønsted acidity and Lewis acidity. While the micropores of zeolite Y are relatively large, most reactions still occur on the zeolite surface.
Feedstock significantly affect the pyrolysis products of CFP over NaY zeolite [192]: Glucose is mainly converted into furans, aromatics, phenols, and light oxygenates; cellulose and cellobiose are mainly converted to benzene, toluene, and xylene. Most furan compounds form on the surface of NaY by dehydration, fragmentation/retro-aldol condensation, decarbonylation, or decarboxylation. The pyrolysis intermediates of anhydro-oligosaccharides dominate the formation of aromatics and phenols in the zeolite pores. The pyrolysis of lignin rich biomass, e.g., corn straw fermentation residue with NaY zeolite, promoted the formation of phenols [193].
Comparing HZSM-5, Na-ZSM5, H-USY, and H-Beta, H-USY has the largest pore volume and performed best in CFP processing of alkaline lignin [181]. A high yield of liquid (75 wt %) was obtained over H-USY, which had a low SAR of 7. The carbon yield of aromatic hydrocarbons was as high as around 40 wt % at 650 °C.
The effect of introduction of mesopores on catalytic co-pyrolysis of torrefied cork oak and high density polyethylene was investigated over mesoporous and microporous HY catalysts [194]. The catalytic co-pyrolysis over micropore HY and Meso-HY with higher acidities produced a larger amount of mono aromatic hydrocarbons. In addition, Meso-HY showed stronger synergy for the formation of MAHs than micropore HY owing to its larger pore allowing the HDPE reaction intermediates diffuse into the pore of the catalysts.
The quick deactivation of zeolite Y was reported in CFP process of hydrogen chloride pretreated lignin [195]. SEM pictures showed heavy coke formation that blocked pore structures and poisoned the active sites of catalysts. A calcination in air at high temperatures above 550 °C can recover part of the catalytic activity. Catalyst regeneration after a few catalytic cycles and mild oxidation conditions are beneficial to reuse the spent catalysts.
Metal Doping of Zeolites for In-Bed CFP Processes
Zeolite texture, acidity, and porosity are critical parameters for the distribution of products in the resulting pyrolysis oil. Additionally, the doping of zeolites with metals is an efficient method for tuning the activity/selectivity of catalyst active sites. A significant amount of work has been done examining CFP using meta-modified zeolite catalysts.
Fe/ZSM-5 was effective in increasing the production of aromatic hydrocarbons, partially monocyclic aromatic hydrocarbons [196]. XRD results indicated that the structure of ZSM-5 was maintained after the incorporation of Fe. Temperature has a significant effect on CFP of wood sawdust over Fe/ZSM-5: Aromatic hydrocarbons yields increased with an increase of temperature from 500 to 600 °C, and then decreased with further temperature increase. Fe/ZSM-5 catalyst demonstrated better activity in the conversion of oxygenates and formation of (MAHs) than the ZSM-5 catalyst. Fe may contribute to the formation of MAHs and simultaneously hindered the further polymerization reaction of MAHs and other oxygenates. Fe/ZSM-5 was used to catalytically crack beech sawdust vapors in a fixed bed tubular reactor at 500 °C [197]. Compared to non-catalytic pyrolysis, Fe/ZSM-5 significantly reduced oxygen content from the organic phase and increased the yield of desirable products such as phenolics and aromatic compounds.
Lamellar and pillared ZSM-5 materials, modified with Mg and Zn oxides, were investigated on a lab-scale downdraft fixed-bed reactor for in-situ catalytic upgrading of eucalyptus woodchips fast-pyrolysis vapors at atmospheric pressure and a temperature of 500 °C [198]. MgO and ZnO highly dispersed over ZSM-5 supports, thus significantly decreased their textural properties and the concentration of the Brønsted and Lewis acid sites present in the parent zeolite, as detected by FTIR using pyridine. This tailoring of ZSM-5 activity avoided an excessive cracking of the bio-oil, leading to a higher yield of the organic compounds and a lower content of undesired polyaromatic hydrocarbons and coke. The obtained liquid exhibited higher quality in terms of H/C and O/C ratios, and larger heating values, though the liquid yield decreased.
Fe/HZSM-5 with a low Fe loading (1.4 wt %) was effective in producing aromatic hydrocarbons from cellulose, cellobiose, lignin, and switchgrass by CFP using a microscale pyrolysis reactor [199]. The carbon yield of selected aromatics (benzene, toluene, o,p-xylenes, ethylbenzene, 1,2,4-trimethylbenzene, naphthalene, and 2-methylnapthalene) from cellulose, cellobiose and switchgrass were ~18%, 25% and ~17%, respectively. The introduction of Fe favored the production of benzene and naphthalenes and hampered the yield of p-xylene, ethylbenzene, and trimethylbenzene.
Zn, Ga, Ni, Co, Mg, and Cu modified HZSM-5 have been investigated for the production of aromatics [200]. Results showed that these metal-modified catalysts yielded higher amounts of non-condensable gas at the expense of the liquid yields compared to the parent HZSM-5. Ga/HZSM-5 catalysts produced the highest yields of bio-oil and the lowest amount of coke. Zn/HZSM-5 exhibited the highest selectivity toward single-ring aromatics, whereas Ni/HZSM-5 was most selective to benzene production and C10+ polycyclic aromatic hydrocarbons. Zn/HZSM-5 was the most selective for the production of aromatic hydrocarbons, particularly toluene and xylenes, while Co/HZSM-5 highly favored formation of indene.
Co/HZSM-5, Mo/HZSM-5, and Ni/HZSM-5 (5.0 wt %) prepared by wet impregnation can be used as catalysts for in-situ upgrading of Shengli lignite pyrolysis vapors in a drop tube reactor at 600 °C [201]. The bi-functional metal-loaded HZSM-5 showed comparable catalytic activity and the oxygen contents were reduced to below 7.1%. Ni/HZSM-5 produced high-quality tars with the highest aromatics content at 94.2% (based on area%). Compared with non-modified HZSM-5, the introduction of metal causes the increase of aromatics and the decrease of organic oxygen species. More information on metal doped zeolites and their usage in the in-bed and in-situ catalytic vapor upgrading processes can be found in Table 2 and Table 3, respectively.
3.1.2. SAPO
SAPO-34 has a structure similar to chabazite, comprised of 3-dimensional interconnected channels with circular 8-ring apertures. SAPO-34 has been industrialized for its high selectivity for ethylene and propene in the methanol-to-olefin process. While SAPO-type materials have expanded the usage of zeolite catalysis into synthetic fuels, synthesis of fine chemicals and pharmaceuticals, etc., its successful usage in fast pyrolysis is rare.
Furan compounds are widely used as organic solvents or reagents for the production of medicines, resins, fuel additives, etc. Lange et al. [222] have recently reviewed the promising usage of furfural offers as a rich platform for lignocellulosic biofuels, including methylfuran and methyltetrahydrofuran, valerate esters, ethylfurfuryl, and ethyltetrahydrofurfuryl ethers, as well as various C10–C15 coupling products. Furfural, 5-hydroxymethyl-furfural and furandicarboxylic acid were in the list of the top 15 target chemicals that can be generated from carbohydrates [223]. Furan compounds are mainly produced from biomass hydrolysis-dehydration technology with acidic catalysts [224,225,226,227] and biomass pyrolysis with organic/inorganic acids and acidic salts. However, these acid catalysts are difficult to be recycled and the volatile and acidic property also causes environmental problems. The CFP of cellulose over SAPO type catalysts is effective and more environmentally friendly. The mild acidity of SAPO caused by the framework composition may facilitate the formation of furans [170]. Pore diameters of SAPO-34 and SAPO-18 are 0.38 nm and 0.37 nm, respectively, far smaller than the kinetic diameter of oligosaccharide and levoglucosan (0.67 nm), making the dehydration of sugar compounds only capable of occurring on the surface of SAPO-34 and SAPO-18. Furthermore, those formed furans, e.g., 5-methylfurfural (0.59 nm), also cannot enter pores of SAPO-34/18 and undergo further conversion to form aromatics and thus exhibited a higher selectivity of furans than ZSM-5. The yield of sugar compound decreased significantly in the CFP process of cellulose over metal/SAPO catalysts because of the direct conversion of oligosaccharides to furans [228]. The mild acidity of AlCu-SAPO-34 and ZrCu-SAPO-18 promoted furan formation at 500–600 °C: Their furans selectivity can be as high as 56.94% and 63.86%, respectively.
3.1.3. Alkali and Alkaline Earth Metallic (AAEM) Species
As have been discussed in feedstock pretreatment section, alkali and alkaline earth metallic species can also be used as catalysts for the CFP of biomass. KCl and CaCl2 were found to be capable of increasing the total content of phenols from pyrolysis of alkali lignin [229]. CaCl2 promoted the convention rate of alkali lignin by 3.5%. However, KCl increased the yield of residual carbon by 6.2% compared to the non-catalytic pyrolysis. Similar observations were also found by other groups. FeCl3 was also reported as capable of facilitating the formation of bio-char [230]. MgCl2 can significantly increase viscosity and average molecular weight of resulting oil obtained from yellow pine as the magnesium content increases [231]. The water content also increased due to a dehydration reaction induced by magnesium.
Magnesium oxide (MgO) was tested in a circulating fluidized bed pilot scale unit and was compared to a commercially available ZSM-5 catalyst [232]. The 2DGC-TOFMS analyses of the produced bio-oils indicated MgO enhanced ketonization and aldol condensation reactions. Compared with ZSM-5, MgO prompted deoxygenation mainly via the formation of CO2, accompanied with a significant increase of coke. No alkali metals were found to deposit on MgO, while alkali metals gradually substituted the proton functionalities of ZSM-5, indicating different deactivation mechanisms between acidic and basic catalysts. CaO can efficiently remove chlorine during CFP of sewage sludge [233].
Metal oxides have also been used for vapor upgrading of biomass and polymers. ZnO, CaO, Fe2O3, and MgO were used for the CFP of a poplar wood-polypropylene composite (WPP) [234]. CaO has strong basicity that facilitates the removal of oxygen by eliminating carboxylic acids and phenols from the products. The less basic MgO showed weaker deoxygenation ability, but exhibited high chain scission activities that significantly enhanced alkene yields. ZnO produced the highest alkene yields, increased ketone and phenol yields while reducing carboxylic acids. Fe2O3 was found to be an enhanced formation of aromatics. Furthermore, ZnO enhanced the synergistic effect between poplar wood and PP pyrolysis of WPP: Yields of carboxylic acids, phenols, and light alkenes were increased, while carbonyl-containing oxygenates such as aldehydes, ketones, and furans were reduced.
The Mg-Al mixed oxides derived from hydrotalcite precursors are active materials for the CFP of biomass. A series of Mg-Al mixed oxides, synthesized by simultaneous co-precipitation with different Mg/Al ratios, act as catalysts for the in-situ catalytic vapor upgrading of wheat straw [235]. The reactor pyrolysis zone and catalytic section were maintained at 550 and 450 °C, respectively. The biomass to catalyst ratio was fixed at five. The removal of oxygen and gas production depends on their Mg/Al ratio. The Al-Mg mixed oxide (Mg/Al =4.0) catalyst showed similar deoxygenation activity and bio-oil energy yield with the ZSM-5 zeolite. The catalysts promoted the aldol condensation and ketonization, leading to the removal of oxygen in the form of H2O and CO2.
3.1.4. Transition Metal and Other Metal Based Catalysts
Metal oxides, particularly transition metal oxides, are widely investigated in CFP considering their redox properties, low cost, and high efficiency. The redox properties could potentially catalyze the thermal decomposition of biomass and pyrolysis intermediates to form more stable products. Selective production of valuable chemical intermediates, such as ketones can be achieved from CFP of microalgae over ZrO2 and TiO2 [236]. In the absence of catalysts, the products were mainly long chain carboxylic acids, e.g., tetradecanoic and hexadecanoic acids. ZrO2 enhanced the formation of ketones, especially 16-hentriacontanone and 14-heptacosanone, which were formed via ketonization reactions of palmitic and myristic acids present in the algae.
Sulfated TiO2, ZrO2, and SnO2 were used for the CFP process of cellulose [237]. The content of levoglucosan and hydroxyacetaldehyde significantly decreased or were even completely eliminated. The selectivity toward furan compounds was significantly affected by metal oxides: SnO2 showed the highest selectivity toward formation of 5-methyl furfural; TiO2 and ZrO2 favored the formation of furfural and furan, respectively.
Metal oxide were effective for deoxygenation of the pyrolysis vapors to form hydrocarbons. Metal oxide/activated carbon (5 wt %) catalysts prepared by impregnation have been used to upgrade fast pyrolysis vapors obtained from CFP of Jatropha curcas waste residue at 873 K [238]. Ce/AC promoted aromatics formation and produced the highest hydrocarbon yield (86.56%), significantly higher than 11.32% yield without catalysts. The aliphatic hydrocarbon yield was in the order of Ce/AC > AC > Pd/AC > Ni/AC. The metal oxide/activated carbon catalysts obviously reduced the oxygen-containing compounds content. One advantage of these catalysts was the low yield (0.62–7.80%) of toxic polycyclic aromatic hydrocarbons.
Jackson et al. [179] used Co/Mo/Al2O3 in the pyrolysis of Asian lignin at 600 °C in flowing helium. The sand bed produced a liquid phase with 97% oxygenated aromatics and a gas phase with 18 vol% CO, 16 vol% CO2, and 12 vol% CH4. In contrast, the Co/Mo/Al2O3 hydrotreating catalyst produced a liquid consisting of 21% aromatics, 75% oxygenated aromatics, and a hydrogen rich gas phase. Mullen and Boateng [239] investigated the CFP of lignin and found that CoO/MoO3 enhanced the production of aromatic hydrocarbons through a direct deoxygenation of methoxyphenol units.
La2O3 contributed less in improving oil yield and reducing water content, however, it was significantly effective to remove chlorine from the bio-oil during CFP of sewage sludge at 450 °C in a fixed-bed reactor [233].
Metal carbides and metal nitrides are composed of carbon/nitrogen and is a less electronegative element. Four kinds of noble metal-like catalysts W2C/AC, W2N/AC, Mo2C/AC, and Mo2N/AC were used to upgrade the fuel properties of biomass (pine wood, microcrystalline cellulose, xylan, and milled wood lignin) pyrolysis vapors with catalyst-to-biomass ratios of 0.5–2.0 [240]. Generally, W-based catalysts outperformed the Mo-based catalysts, and the W2C/AC performance was comparable Pd/AC. W2C/AC and W2N/AC promoted the decomposition of lignin to generate monomeric phenolic compounds. During the CFP of holocellulose, anhydro sugars, and linear aldehydes contents decreased, while furans, cyclopentanones, and linear ketones increased. W2C/AC and W2N/AC catalysts can convert aldehyde, C=C bond, and carboxyl group through decarbonylation, hydrogenation, decarboxylation, and other reactions, and thus increased the stability of bio-oil.
A sequentially connected two-stage system consisting of a fluidized bed pyrolyzer and a fluidized bed catalytic reactor was used for the CFP of cedar chips over Ni2P catalysts [241]. The resulting pyrolysis oil mainly consisted of phenolic compounds. Ni2P supported on SiO2 reduced the oxygen content of bio-oil to about half the original levels. The deoxygenation activity of Ni2P was found to be higher than that of Ni/SiO2, Pd/C, SiO2–Al2O3, and an FCC-catalyst. Ni2P/ZSM-5 eliminated oxygen in the bio-oil with smaller reduction in the oil yield than Ni2P/SiO2.
Red mud (RM), a side product generated in the industrial production of alumina, has gained focus for its catalytic effects in CFP recently. RM is highly basic and mainly composed of iron oxide, unleached residual alumina, titanium oxide, and silica [242]. Red mud supported nickel catalysts (Ni/RM) were successfully used to hydrodeoxygenate pinyon-juniper catalytic pyrolysis oil. 40%Ni/RM was optimal to achieve the maximum organic liquid yield (68.6%). Compared to the commercial Ni/SiO2-Al2O3, 40%Ni/RM produced significantly higher organic liquid yield, while gas and coke yields were lower [243]. Ni/RM was also used to catalytically convert water-soluble low-molecular-weight oxygenates obtained from pyrolysis of lignocellulosic biomass to produce C6 to C14 hydrocarbons. Several model compounds, such as acetic acid, propionic acid, furfural, butanone, pentanone, heptanone, and 2,6-dimethyl-4-heptanone have been found to be involved in the formation of alkanes [244]. Aldehydes and ketones can react with furans to yield straight or branched-chain hydrocarbons. Even without the introduction of other metals, RM has showed similar catalytic properties in a fractional catalytic pyrolysis of Pinyon and juniper wood [245]. Bromine from the non-metallic fractions of waste printed circuit boards was reported to be capable of being fixated by co-pyrolysis with RM [246]. Compared to the single usage of Fe2O3, Na2O, Al2O3, and TiO2 for the bromine fixation, synergistic effects of metal oxides in RM were found in fixing bromine.
3.1.5. Dual Catalyst Systems
Due to the quite different properties and pyrolysis behaviors of biomass and waste synthetic polymers, dual-catalyst beds are often used for catalytic fast co-pyrolysis. As has been discussed in the CFP of biomass, zeolite, especially HZSM-5, is one of the most efficient catalysts. The medium pores of HZSM-5 facilitate the rearrangements of small pyrolytic molecules; however, the reaction of bulky pyrolysis intermediates/reactants are hampered because of diffusional limitations. Metal oxides are active and efficient in deoxygenations and in tar cracking reactions during gasification of biomass [247]. By the combination usage of metal oxide and zeolite, a dual catalytic stage system formed by two catalytic beds, might be optimal for the catalytic fast co-pyrolysis of biomass and waste synthetic polymers.
LLDPE was found can enhance the production of aromatic and aliphatic hydrocarbons via catalytic fast co-pyrolysis with xylan over dual beds of CaO and HZSM-5 [248]. Acids formation was inhibited by converting into ketones by CaO. The pyrolysis intermediates of LLDPE promoted the Diels-Alder reactions of furans and the non-furanic compounds may participate in the hydrocarbon pool reactions, resulting in the formation of aromatic and aliphatic hydrocarbons. An increased production of aromatics and olefins was also reported from the co-pyrolysis of bamboo residual and waste tire over dual beds of CaO and HZSM-5 [249]. A maximum yield of hydrocarbons could be obtained at HZSM-5 to CaO mass ratio of 3:2. Interestingly, HZSM-5/MgO with the same mass ratio of 3:2 was optimal to significantly increase the yield of aromatics for the co-CFP of bamboo residual and waste lubricating oil [250].
Dual catalyst beds of two zeolites, though rarely used, have been reported to successfully enhance the production of aromatic hydrocarbons. In the catalytic fast co-pyrolysis of bagasse and bio-plastic dual beds of USY and HZSM-5 produced a much higher aromatic yield than either of the single catalyst beds [251].
The layout schemes of feedstock and catalysts affects bio-oil yield and aromatics selectivity. The layout has been investigated for the catalytic co-pyrolysis of glucose and HDPE over MgCl2 and HZSM-5 [252]. Experimental results indicated the HDPE/HZSM-5/glucose/HZSM-5 arrangement, as shown in Figure 3, which performed best in increasing bio-oil yield and aromatics selectivity. Furan compounds produced from pyrolysis of pine sawdust over MgCl2 transfer a short distance to react with short-chain olefins and form aromatics. Therefore, an appropriate ratio between the amounts of furans and short-chain olefins existed for increasing bio-oil yield and aromatics selectivity. An optimum oil yield of 20.6% and aromatics selectivity of 95.9% (based on peak area %) were obtained at 600 °C with a biomass/HDPE ratio of 1:2, and a feedstock to catalyst ratio of 1:1.
The catalytic fast co-pyrolysis of bamboo residual (BR) and waste lubricating oil (WLO) was performed over dual catalytic beds of MgO and HZSM-5 [250]. Figure 4 presented the feedstocks placement for CFP of BR and WLO over dual catalytic beds, and the samples were placed in the quartz tube to form a dual catalytic bed system. The feedstocks to overall catalysts mass ratio is 1:2, and the pyrolyzer was heated at 2000 °C/s to an optimal temperature of 600 °C, at which yields of furans and phenols were high. MgO promoted deacidification via ketonization and aldol condensation reactions. MgO exhibited high selectivity toward light phenols, e.g., phenol and 4-methyl-phenol. A HZSM-5/MgO mass ratio of 3:2 largely accelerated the formation of aromatics during catalytic co-CFP of BR and WLO. Moreover, the WLO percentage played a vital role in the yield of hydrocarbons, and the maximum yield of 70.30% could be attained at the percentage of 60%.
3.2. Catalyst Deactivation and Regeneration
3.2.1. Catalyst Deactivation in Biomass Pyrolysis
One of most challenging issues in biomass pyrolysis, catalyzed by zeolite, is the catalyst deactivation, which is affected by various of factors, such as the properties of synthesized catalysts, reaction conditions, and the feedstock composition [253]. Coking, poisoning, and sintering are the major three reasons for the catalyst deactivation. Coke deposition caused pore blockage, pore volume decrease, active site poisoning, and therefore catalyst deactivation was observed in the CFP process of lignin using HUSY zeolite [254]. Even with high heating rates and catalyst-to-feed ratios to avoid thermal decomposition biomass compounds before they enter the pores of zeolite catalyst, the yield of coke can be as high as 30 wt % over ZSM-5 [255].
Ma et al. [195] reported a FAU (SAR = 15) zeolite underwent a rapid deactivation in the pretreated lignin pyrolysis. During catalyst deactivation, there was excessive coke deposition in the zeolite pore structure resulted in both pore blockage and active site poisoning. Coke deposition is related to the decomposition of the oxygenates derived from biomass pyrolysis and the re-polymerization of phenolic oxygenates [256]. Coke on deactivated zeolites has been found on different sites: Coke trapped inside micropores, coke precursors in the mesopore structures, and external coke formed on zeolite crystal surface. Coke mainly form through two pathways [257]. The first is that the thermal coke forms from the polymerization of reactants and subsequent deposition on the zeolite outer surface; and the second is catalytic coke that forms in zeolite’s pores due to interactions of reactants or products with acid sites. The microporous cokes are much more toxic than the other two. Both kinds of coke lead to deactivation by covering acid sites and blocking pores. Similar deactivation routes were revealed in a hydrodeoxygenation of raw bio-oil reaction: Thermal lignin from alkylmethoxy phenols deposited on the catalyst mesopores at low temperature, and aromatic coke accumulated in the catalyst micropores at high temperature via condensation reactions of polycyclic aromatic hydrocarbons [258]. It is worthy to note that in HDO reactions, the oxidation of surface-active metal also serves as a major controlling factor for catalyst deactivation besides the coke formation [259].
Temperature was found to significantly affect the coke formation. Furan, an intermediate compound of CFP of biomass, has been used as a model compound to investigate coke deposition, which leads to ZSM-5 deactivation [260]. The compositions of coke obtained at different pyrolysis temperatures and reaction times were analyzed, as well as the organics of hydrofluoric acid washed catalysts. FTIR and TGA results indicated high pyrolysis temperatures that lead to the formation of coke species with high aromaticity. Coke was mainly formed via condensation and rearrangement procedures below 200 °C; while above 200 °C, polyaromatic components of coke were mainly formed by hydrogen transfer, besides the condensation and rearrangement.
Alkali metals of biomass have been found substituted by the proton functionalities of zeolites acid sites, which contributes to catalyst deactivation. Paasikallio et al. [261] carried out a four-day pilot-scale CFP of pine sawdust using HZSM-5 catalyst a spray drying process. The micropore area and volume continuously decreased and a linear correlation of coke formation with reaction was found. The catalyst particles were physically damaged, as well as the chemical composition changed: Contents of Ca, K, Mg, and P linearly increased with time-on stream and reached 1.1 wt % after the reaction, indicating that biomass alkalis overtook the proton functionalities of acid sites on the HZSM-5 catalyst. In a comparison of natural MgO catalysts with ZSM-5 in a circulating fluidized-bed pilot scale unit, no alkali metals were found to deposit on MgO, indicating different deactivation mechanisms between acidic and basic catalysts in biomass fast pyrolysis [232].
To decrease coke formation, several strategies have been used, including introducing meso-porous structure or decrease zeolite crystal size to enhance diffusion. Desilicated zeolites with mesoporous structures have shown higher selectivity toward monoaromatics and longer stability based on coke deposition than microporous zeolites [183]. The open mesopores produced after desilication can facilitate big molecules, e.g., levoglucosan to diffuse to pore mouths, where Brønsted acid sites catalyze the conversion of larger molecules to small fractions. These fractions diffused and formed aromatics in the micropores. An alkali treatment made for enlarged pores and hindered catalyst deactivation. However, there is some disagreement on the effect of mesoporous structure. The mass transportation properties of mesoporous ZSM-5 exhibited no apparent conventional ZSM-5. This led to the assumption that ZSM-5 deactivation was mainly caused by the coverage of active sites due to coke formation, but not the blockage of the transportation pathways, e.g., mesopore structures [176].
Surface modification technique has been used on zeolite to mitigate the catalyst deactivation and increase the pyrolysis oil yield during CFP. Chemical liquid deposition (CLD) with SiO2 over ZSM-5 catalysts can cover strong acid sites; acid dealumination (AD) favors the removal of surface acid sites. The yield of indenes and naphthalenes, which are important precursors of coke, decreases significantly during CFP of pinewood after CLD or AD treatment. Moreover, CLD and AD modified ZSM-5 catalysts boosted the production of BTX by 37.2% and 30.4%, respectively. CLD modification method over ZSM-5 seems to be more preferable in the catalytic pyrolysis of biomass because of low environmental cost and superior catalytic performance [175]. Zhang et al. [262] applied the CLD method to modify ZMS-5 using 3-triethoxysilylpropylamine for the catalytic conversion of furan. The coke yield decreased from 44.1% to 26.7%. In contrast to the original one, the modified ZSM-5 catalyst also produced higher yields of aromatics and olefins.
Pre-coking zeolites was also an effective way to inhibit the coke formation. Wang et al. [263] pre-coked (0–5.4%) HZSM-5 zeolite and applied it to conduct the CFP of mushroom waste. The yields of hydrocarbons increased at first and then decreased with the increasing of pre-coked percentage with a maximum value of 53.5%. Zhang et al. [264] reported that spent HZSM-5 zeolites could reduce the coke yield, but lower the total yield of products compared to the fresh zeolite and regenerated sample. In addition, the spent zeolite favors the formation of products with higher hydrocarbon, but lower oxygen contents.
3.2.2. Catalyst Deactivation in Polymer Pyrolysis
The study of catalyst deactivation and regeneration is especially important in the catalytic pyrolysis of plastic due to the consideration of catalyst cost on the industrial application of polymer (co)pyrolysis. The Zn promoted zeolites shown a good stability during the catalytic cracking of low-density polyethylene. The Zn-ZSM-11 sample maintained a stable performance during the 11 reaction cycles, with average yields of 67 wt % gaseous hydrocarbons and 28 wt % of liquid hydrocarbons; while the activity of Zn-BETA shown a different trend: The yield of gaseous hydrocarbons decreased from 64 wt % to 41 wt % in the eighth cycle. Both Brønsted and Lewis acid sites on Zn-BETA zeolite are completely lost due to the coke deposition. Zn-ZSM-11 shows a lower coke yield because of the shape selectivity effect [265].
The deactivation behavior of the HZSM5 and HUSY catalysts was studied during the catalytic pyrolysis of polyethylene [266]. HUSY exhibits a fast activity deactivation, while the small pore size in HZSM5 inhibits coke deposition. HZSM is recommended in plastic pyrolysis since the deactivation rate was extremely low and thus, more efficient for regeneration. However, the usage of zeolite catalyst in plastic pyrolysis only maximized the production of volatile hydrocarbon [267]. A study was conducted to assess the hydrogen production from different plastics (polypropylene, polyethylene, polyethylene terephthalate and polystyrene) in a two-step reaction system (pyrolysis and in-line reforming). The deactivation rate was dependent on feed stock, and it was faster in the case of polystyrene and polyethylene terephthalate than those observed for the polyolefins and the plastic mixture [268].
Zeaiter [269] thoroughly examined the catalytic decomposition of waste polyethylene bottles to determine the possibility of continuous operation based on two zeolite catalysts: HUSY and HBeta. It was found that the thermo-catalytic pyrolysis of HDPE over HUSY and HBeta zeolites is a promising waste conversion method that would generate valuable amounts of chemicals and fuel-like products. However, both HUSY and HBeta showed deactivation and their activities started to decrease with the continuous feed of waste HDPE to the reactor.
Like the biomass pyrolysis, catalytic polyethylene pyrolysis also has two different reaction mechanisms: A reversible deactivation by coke deposition and an irreversible deactivation. The coke deposited on the external surface of catalysts can be easily removed by thermal treatment at 550 °C in oxygen-containing atmosphere. Permanent deactivation is caused by changes to the zeolites structure at high temperatures, formation of stable coke species and/or poisoning of the acid sites by metals originating from the polymer itself [270]. Mullen et al. [271] reported the co-pyrolysis of HDPE and switchgrass could mitigate the coke formation over HZSM-5 catalysts. Olefins generated from the catalytic pyrolysis of HDPE react with furanic compounds that originated from switchgrass to produce aromatic hydrocarbons and therefore reduce coke deposition. However, the blending effect on catalyst deactivation was vanished with the increasing exposure of pyrolysis vapors to the catalysts; although the yield of aromatic hydrocarbons kept higher for the blend than that for the HDPE or switchgrass alone.
3.2.3. Catalyst Regeneration
Catalyst deactivation by coke deposition is the major barrier in the catalytic pyrolysis of biomass and polymers. The coke can be removed by calcination of the spent catalyst in an oxygen-containing gas or water stream environment at high temperatures [195,254,272], which restores the porosity and activity to a large extent. However, recovery of catalytic activity may be incomplete and attributed to irreversible changes in catalyst structure that occur during the deactivation and/or regeneration processes [195,273]. Vitolo et al. [272] investigated the stability of HZSM-5 catalyst for the upgrading of wood pyrolysis oils in repeated reacting-regenerating cycles, and they found an irreversible loss of catalytic activity after five cycles. The Brønsted acid sites were gradually destroyed by the repeated regeneration treatments. With the exception of simple calcination, catalyst regeneration process may also contain washing and reducing steps. For example, used catalysts with adsorbed organic species have been washed by organic solvents, dried in an oven, and then the calcine samples are reduced in a H2-containing atmosphere [274]. Spent Pt/H-MFI-90 catalysts with adhered reaction impurities were each washed with ethanol and methanol three times, calcined in air at 400 °C for 10 h and reduced in a continuous hydrogen flow [275]. The catalytic conversion of guaiacol could be partially restored at a conversion rate of 35%.
The oxygen concentration significantly affects the regeneration of spent catalysts. For the regeneration of deactivated ZSM-5 after CFP, the best oxygen concentration was 15%, with the catalysts steadily producing aromatics and olefins after 30 reaction-regeneration cycles [276]. Steam can be introduced to decrease the internal temperature of catalyst particle. When 15% oxygen and 5% steam were used as the regeneration agents, the reaction temperature was reduced efficiently, and the hydrocarbons yield was increased by 31.3% in comparison to 15% oxygen only. Interestingly, at 2.2%, the retained coke hydrocarbons yield was 27.4% higher than for completely regenerated catalyst. This result might be due to a partial modification of ZSM-5 pore structures.
Temperature is another factor influencing the regeneration efficiency. Spent ZSM-5 (SAR = 30) used for biomass CFP could be fully regenerated at 650 °C and 700 °C within 20 min in a 4% O2 atmosphere, whereas regeneration at 550 °C and 600 °C required longer regeneration times. In the case of regeneration at 500 °C, there may be coke species that are not removed and the catalyst activity never be fully restored [277]. Higher regeneration temperatures are more effective for restoring Brønsted acid sites and catalyst mesoporosity by rapidly removing aromatic coke deposits. Additionally, regeneration at 650 °C and 700 °C led to a slightly higher total porosity, as compared to the pristine catalyst. This effect was attributed to the formation of additional mesoporosity from catalyst steaming.
4. Summary and Outlook
This review focuses on using catalytic fast pyrolysis to produce high grade pyrolysis oil from biomass and co-pyrolysis of biomass with plastic waste. The use of specific kinds of catalysts, microporous and mesoporous materials, transition metal oxides, and AAEM, are shown to allow for substantial optimization of the product distribution. Co-pyrolysis of biomass with waste synthetic polymers over single catalyst or dual catalysts beds can significantly improve both the quantity and quality of pyrolysis oil, with a significant impact on improved heating values. The low cost and easy availability of biomass and waste polymers makes this simple and effective technique very promising as a route to obtain high-grade pyrolysis oil. Furthermore, catalytic fast pyrolysis and co-pyrolysis reduces the volume of both biomass and synthetic polymer wastes. Pyrolysis of biomass and co-pyrolysis of biomass with polymer wastes has significant potential to simultaneously provide a renewable fuel oil and chemical source, reduce the cost and environmental impact of biomass and polymer waste disposal, and increase energy source diversity and security.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- EIA. International Energy Annual 2001 Edition; U.S. Department of Energy: Washington, DC, USA, 2003.
- Wessner, C. What is the best guess as to when the world’s oil reserves will run out? Popul. Sci. 1999. [Google Scholar]
- Appenzeller, T. The End of Cheap Oil, National Geographic. Available online: https://www.nationalgeographic.com/environment/global-warming/end-cheap-oil/ (accessed on 15 July 2018).
- EIA. Annual Energy Review 2010; Energy Information Administration: Washington, DC, USA, 2011.
- Chow, J.; Kopp, R.J.; Portney, P.R. Energy resources and global development. Science 2003, 302, 1528–1531. [Google Scholar] [CrossRef] [PubMed]
- EIA. Annual Energy Outlook 2014 with Projections to 2040; Enery Information Adminstration: Washington, DC, USA, 2014.
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, C.; Reyes-Sosa, F.M.; Díez, B. Enzymatic hydrolysis of biomass from wood. Microb. Biotechnol. 2016, 9, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Bulushev, D.A.; Ross, J.R.H. Catalysis for conversion of biomass to fuels via pyrolysis and gasification: A review. Catal. Today 2011, 171, 1–13. [Google Scholar] [CrossRef]
- Li, X.; Chen, G.; Liu, C.; Ma, W.; Yan, B.; Zhang, J. Hydrodeoxygenation of lignin-derived bio-oil using molecular sieves supported metal catalysts: A critical review. Renew. Sustain. Energy Rev. 2017, 71, 296–308. [Google Scholar] [CrossRef]
- Li, X.; Luo, X.; Jin, Y.; Li, J.; Zhang, H.; Zhang, A.; Xie, J. Heterogeneous sulfur-free hydrodeoxygenation catalysts for selectively upgrading the renewable bio-oils to second generation biofuels. Renew. Sustain. Energy Rev. 2018, 82, 3762–3797. [Google Scholar] [CrossRef]
- Filiciotto, L.; Balu, A.M.; Van der Waal, J.C.; Luque, R. Catalytic insights into the production of biomass-derived side products methyl levulinate, furfural and humins. Catal. Today 2018, 302, 2–15. [Google Scholar] [CrossRef]
- Morgan, H.M.; Bu, Q.; Liang, J.; Liu, Y.; Mao, H.; Shi, A.; Lei, H.; Ruan, R. A review of catalytic microwave pyrolysis of lignocellulosic biomass for value-added fuel and chemicals. Bioresour. Technol. 2017, 230, 112–121. [Google Scholar] [CrossRef]
- Fan, L.; Zhang, Y.; Liu, S.; Zhou, N.; Chen, P.; Cheng, Y.; Addy, M.; Lu, Q.; Omar, M.M.; Liu, Y.; et al. Bio-oil from fast pyrolysis of lignin: Effects of process and upgrading parameters. Bioresour. Technol. 2017, 241, 1118–1126. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, H.; Chen, S.; Wu, J. Catalytic co-pyrolysis of lignocellulosic biomass with polymers: A critical review. Green Chem. 2016, 18, 4145–4169. [Google Scholar] [CrossRef]
- Lin, F.; Waters, C.L.; Mallinson, R.G.; Lobban, L.L.; Bartley, L.E. Relationships between biomass composition and liquid products formed via pyrolysis. Front. Energy Res. 2015, 3, 45. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U.; Steele, P.H. Pyrolysis of wood/biomass for bio-oil: A critical review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Garrett, D.E.; Mallan, G.M. Pyrolysis Process for Solid Wastes. U.S. Patent 4,153,514, 8 May 1979. [Google Scholar]
- Mao, J.-D.; Johnson, R.; Lehmann, J.; Olk, D.; Neves, E.G.; Thompson, M.; Schmidt-Rohr, K. Abundant and stable char residues in soils: Implications for soil fertility and carbon sequestration. Environ. Sci. Technol. 2012, 46, 9571–9576. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Cao, X.; Zhao, L.; Wang, H.; Yu, H.; Gao, B. Removal of Cu, Zn, and Cd from aqueous solutions by the dairy manure-derived biochar. Environ. Sci. Pollut. Res. 2013, 20, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Wawra, A.; Friesl-Hanl, W.; Jäger, A.; Puschenreiter, M.; Soja, G.; Reichenauer, T.; Watzinger, A. Investigations of microbial degradation of polycyclic aromatic hydrocarbons based on 13 c-labeled phenanthrene in a soil co-contaminated with trace elements using a plant assisted approach. Environ. Sci. Pollut. Res. 2018, 25, 6364–6377. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yi, Y.; Fang, Z.; Tsang, E.P. Effects of biochar on phytotoxicity and translocation of polybrominated diphenyl ethers in ni/fe bimetallic nanoparticle-treated soil. Environ. Sci. Pollut. Res. 2018, 25, 2570–2579. [Google Scholar] [CrossRef]
- Fellmann, T.; Witzke, P.; Weiss, F.; Van Doorslaer, B.; Drabik, D.; Huck, I.; Salputra, G.; Jansson, T.; Leip, A. Major challenges of integrating agriculture into climate change mitigation policy frameworks. Mitig. Adapt. Strategy Glob. Chang. 2018, 23, 451–468. [Google Scholar] [CrossRef]
- Tan, X.; Liu, Y.; Zeng, G.; Wang, X.; Hu, X.; Gu, Y.; Yang, Z. Application of biochar for the removal of pollutants from aqueous solutions. Chemosphere 2015, 125, 70–85. [Google Scholar] [CrossRef]
- Jaiswal, A.K.; Elad, Y.; Paudel, I.; Graber, E.R.; Cytryn, E.; Frenkel, O. Linking the belowground microbial composition, diversity and activity to soilborne disease suppression and growth promotion of tomato amended with biochar. Sci. Rep. 2017, 7, 44382. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Reinoso, F. The role of carbon materials in heterogeneous catalysis. Carbon 1998, 36, 159–175. [Google Scholar] [CrossRef]
- Dickerson, T.; Soria, J. Catalytic fast pyrolysis: A review. Energies 2013, 6, 514–538. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.D.; Jensen, P.A.; Knudsen, K.G.; Jensen, A.D. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Kaiqi, S.; Shuangxi, S.; Qiang, H.; Xuwen, L.; Lan, J.; Ya, L. Review of catalytic pyrolysis of biomass for bio-oil. In Proceedings of the 2011 International Conference on Materials for Renewable Energy. Environment, Shanghai, China, 20–22 May 2011; pp. 317–321. [Google Scholar] [CrossRef]
- Rosendahl, L. Biomass Combustion Science, Technology and Engineering; Elsevier Science: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Carpenter, D.; Westover, T.L.; Czernik, S.; Jablonski, W. Biomass feedstocks for renewable fuel production: A review of the impacts of feedstock and pretreatment on the yield and product distribution of fast pyrolysis bio-oils and vapors. Green Chem. 2014, 16, 384–406. [Google Scholar] [CrossRef]
- Kan, T.; Strezov, V.; Evans, T.J. Lignocellulosic biomass pyrolysis: A review of product properties and effects of pyrolysis parameters. Renew. Sustain. Energy Rev. 2016, 57, 1126–1140. [Google Scholar] [CrossRef]
- Wang, K.; Mante, O.D.; Peters, J.E.; Dayton, D.C. Influence of the feedstock on catalytic fast pyrolysis with a solid acid catalyst. Energy Technol. 2017, 5, 183–188. [Google Scholar] [CrossRef]
- Yu, Y.; Li, X.; Su, L.; Zhang, Y.; Wang, Y.; Zhang, H. The role of shape selectivity in catalytic fast pyrolysis of lignin with zeolite catalysts. Appl. Catal. A 2012, 447–448, 115–123. [Google Scholar] [CrossRef]
- French, R.; Czernik, S. Catalytic pyrolysis of biomass for biofuels production. Fuel Process. Technol. 2010, 91, 25–32. [Google Scholar] [CrossRef]
- Zhang, J.; Choi, Y.S.; Yoo, C.G.; Kim, T.H.; Brown, R.C.; Shanks, B.H. Cellulose-hemicellulose and cellulose-lignin interactions during fast pyrolysis. ACS Sustain. Chem. Eng. 2015, 3, 293–301. [Google Scholar] [CrossRef]
- Wu, S.; Shen, D.; Hu, J.; Zhang, H.; Xiao, R. Cellulose-lignin interactions during fast pyrolysis with different temperatures and mixing methods. Biomass Bioenergy 2016, 90, 209–217. [Google Scholar] [CrossRef]
- Mukkamala, S.; Wheeler, M.C.; van Heiningen, A.R.P.; DeSisto, W.J. Formate-assisted fast pyrolysis of lignin. Energy Fuels 2012, 26, 1380–1384. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as renewable raw material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Xue, Y.; Sharma, A.; Bai, X. Lignin valorization through thermochemical conversion: Comparison of hardwood, softwood and herbaceous lignin. ACS Sustain. Chem. Eng. 2016, 4, 6608–6617. [Google Scholar] [CrossRef]
- Jiang, W.; Wu, S.; Lucia, L.A.; Chu, J. A comparison of the pyrolysis behavior of selected β-o-4 type lignin model compounds. J. Anal. Appl. Pyrolysis 2017, 125, 185–192. [Google Scholar] [CrossRef]
- Guo, D.; Wu, S.; Lyu, G.; Guo, H. Effect of molecular weight on the pyrolysis characteristics of alkali lignin. Fuel 2017, 193, 45–53. [Google Scholar] [CrossRef]
- Zhang, H.; Cheng, Y.-T.; Vispute, T.P.; Xiao, R.; Huber, G.W. Catalytic conversion of biomass-derived feedstocks into olefins and aromatics with zsm-5: The hydrogen to carbon effective ratio. Energy Environ. Sci. 2011, 4, 2297–2307. [Google Scholar] [CrossRef]
- Horne, P.A.; Nugranad, N.; Williams, P.T. Catalytic coprocessing of biomass-derived pyrolysis vapours and methanol. J. Anal. Appl. Pyrolysis 1995, 34, 87–108. [Google Scholar] [CrossRef]
- Xue, Y.; Zhou, S.; Bai, X. Role of hydrogen transfer during catalytic copyrolysis of lignin and tetralin over hzsm-5 and hy zeolite catalysts. ACS Sustain. Chem. Eng. 2016, 4, 4237–4250. [Google Scholar] [CrossRef]
- Cheng, Y.-T.; Huber, G.W. Production of targeted aromatics by using diels-alder classes of reactions with furans and olefins over zsm-5. Green Chem. 2012, 14, 3114–3125. [Google Scholar] [CrossRef]
- Zhang, H.; Carlson, T.R.; Xiao, R.; Huber, G.W. Catalytic fast pyrolysis of wood and alcohol mixtures in a fluidized bed reactor. Green Chem. 2012, 14, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhong, Z.; Min, M.; Ding, K.; Xie, Q.; Ruan, R. Catalytic fast co-pyrolysis of biomass and food waste to produce aromatics: Analytical Py-GC/MS study. Bioresour. Technol. 2015, 189, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Badger, P.C. Processing cost analysis for biomass feedstocks, ORNL/TM-2002/199; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1996.
- Westover, T.L.; Phanphanich, M.; Clark, M.L.; Rowe, S.R.; Egan, S.E.; Zacher, A.H.; Santosa, D. Impact of thermal pretreatment on the fast pyrolysis conversion of southern pine. Biofuels 2013, 4, 45–61. [Google Scholar] [CrossRef]
- Bridgwater, A.V.; Peacocke, G.V.C. Fast pyrolysis processes for biomass. Renew. Sustain. Energy Rev. 2000, 4, 1–73. [Google Scholar] [CrossRef]
- Uzun, B.B.; Pütün, A.E.; Pütün, E. Rapid pyrolysis of olive residue. 1. Effect of heat and mass transfer limitations on product yields and bio-oil compositions. Energy Fuels 2007, 21, 1768–1776. [Google Scholar] [CrossRef]
- Chen, D.; Li, Y.; Deng, M.; Wang, J.; Chen, M.; Yan, B.; Yuan, Q. Effect of torrefaction pretreatment and catalytic pyrolysis on the pyrolysis poly-generation of pine wood. Bioresour. Technol. 2016, 214, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.; Zhao, Z.; Huang, Z.; Zhao, K.; Wei, G.; Wang, X.; He, F.; Li, H. Catalytic fast pyrolysis of biomass pretreated by torrefaction with varying severity. Energy Fuels 2014, 28, 5804–5811. [Google Scholar] [CrossRef]
- Zhang, H.; Shao, S.; Jiang, Y.; Vitidsant, T.; Reubroycharoen, P.; Xiao, R. Improving hydrocarbon yield by two-step pyrolysis of pinewood in a fluidized-bed reactor. Fuel Process. Technol. 2017, 159, 19–26. [Google Scholar] [CrossRef]
- Zheng, A.; Jiang, L.; Zhao, Z.; Huang, Z.; Zhao, K.; Wei, G.; Wang, X.; He, F.; Li, H. Impact of torrefaction on the chemical structure and catalytic fast pyrolysis behavior of hemicellulose, lignin, and cellulose. Energy Fuels 2015, 29, 8027–8034. [Google Scholar] [CrossRef]
- Agblevor, F.A.; Besler, S. Inorganic compounds in biomass feedstocks. 1. Effect on the quality of fast pyrolysis oils. Energy Fuels 1996, 10, 293–298. [Google Scholar] [CrossRef]
- Zhou, L.; Jia, Y.; Nguyen, T.-H.; Adesina, A.A.; Liu, Z. Hydropyrolysis characteristics and kinetics of potassium-impregnated pine wood. Fuel Process. Technol. 2013, 116, 149–157. [Google Scholar] [CrossRef]
- Hu, S.; Jiang, L.; Wang, Y.; Su, S.; Sun, L.; Xu, B.; He, L.; Xiang, J. Effects of inherent alkali and alkaline earth metallic species on biomass pyrolysis at different temperatures. Bioresour. Technol. 2015, 192, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Zhang, Z.B.; Yang, X.C.; Dong, C.Q.; Zhu, X.F. Catalytic fast pyrolysis of biomass impregnated with k3po4 to produce phenolic compounds: Analytical py-gc/ms study. J. Anal. Appl. Pyrolysis 2013, 104, 139–145. [Google Scholar] [CrossRef]
- Zhang, Z.B.; Lu, Q.; Ye, X.N.; Li, W.T.; Hu, B.; Dong, C.Q. Production of phenolic-rich bio-oil from catalytic fast pyrolysis of biomass using magnetic solid base catalyst. Energy Convers. Manag. 2015, 106, 1309–1317. [Google Scholar] [CrossRef]
- Fermoso, J.; Hernando, H.; Jiménez-Sánchez, S.; Lappas, A.A.; Heracleous, E.; Pizarro, P.; Coronado, J.M.; Serrano, D.P. Bio-oil production by lignocellulose fast-pyrolysis: Isolating and comparing the effects of indigenous versus external catalysts. Fuel Process. Technol. 2017, 167, 563–574. [Google Scholar] [CrossRef]
- Mourant, D.; Wang, Z.; He, M.; Wang, X.S.; Garcia-Perez, M.; Ling, K.; Li, C.-Z. Mallee wood fast pyrolysis: Effects of alkali and alkaline earth metallic species on the yield and composition of bio-oil. Fuel 2011, 90, 2915–2922. [Google Scholar] [CrossRef]
- Patwardhan, P.R.; Satrio, J.A.; Brown, R.C.; Shanks, B.H. Influence of inorganic salts on the primary pyrolysis products of cellulose. Bioresour. Technol. 2010, 101, 4646–4655. [Google Scholar] [CrossRef] [PubMed]
- Trendewicz, A.; Evans, R.; Dutta, A.; Sykes, R.; Carpenter, D.; Braun, R. Evaluating the effect of potassium on cellulose pyrolysis reaction kinetics. Biomass Bioenergy 2015, 74, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.B.; Schultz, L.; Glarborg, P.; Skaarup-Jensen, L.; Dam-Johansen, K.; Frandsen, F.; Henriksen, U. Devolatilization characteristics of large particles of tyre rubber under combustion conditions. Fuel 2006, 85, 1335–1345. [Google Scholar] [CrossRef]
- Leung, D.; Yin, X.; Zhao, Z.; Xu, B.; Chen, Y. Pyrolysis of tire powder: Influence of operation variables on the composition and yields of gaseous product. Fuel Process. Technol. 2002, 79, 141–155. [Google Scholar] [CrossRef]
- Isoda, T.; Nakahara, T.; Kusakabe, K.; Morooka, S. Catalytic cracking of polyethylene-liquefied oil over amorphous aluminosilicate catalysts. Energy. Fuels 1998, 12, 1161–1167. [Google Scholar] [CrossRef]
- Mastral, A.; Murillo, R.; Garcıa, T.; Navarro, M.; Callen, M.; López, J. Study of the viability of the process for hydrogen recovery from old tyre oils. Fuel Process. Technol. 2002, 75, 185–199. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Q.; Zhou, Z.; Yang, J.; Qi, F.; Pan, Y. Online study on the pyrolysis of polypropylene over the hzsm-5 zeolite with photoionization time-of-flight mass spectrometry. Energy Fuels 2015, 29, 1090–1098. [Google Scholar] [CrossRef]
- Palza, H.; Aravena, C.; Colet, M. Role of the catalyst in the pyrolysis of polyolefin mixtures and used tires. Energy Fuels 2017, 31, 3111–3120. [Google Scholar] [CrossRef]
- Chattopadhyay, J.; Kim, C.; Kim, R.; Pak, D. Thermogravimetric characteristics and kinetic study of biomass co-pyrolysis with plastics. Korean J. Chem. Eng. 2009, 25, 1047. [Google Scholar] [CrossRef]
- Paradela, F.; Pinto, F.; Gulyurtlu, I.; Cabrita, I.; Lapa, N. Study of the co-pyrolysis of biomass and plastic wastes. Clean Technol. Environ. Policy 2009, 11, 115–122. [Google Scholar] [CrossRef]
- Mushtaq, F.; Mat, R.; Ani, F.N. A review on microwave assisted pyrolysis of coal and biomass for fuel production. Renew. Sustain. Energy Rev. 2014, 39, 555–574. [Google Scholar] [CrossRef]
- Zhou, C.-H.; Xia, X.; Lin, C.-X.; Tong, D.-S.; Beltramini, J. Catalytic conversion of lignocellulosic biomass to fine chemicals and fuels. Chem. Soc. Rev. 2011, 40, 5588–5617. [Google Scholar] [CrossRef]
- Zhang, Y.; Bi, P.; Wang, J.; Jiang, P.; Wu, X.; Xue, H.; Liu, J.; Zhou, X.; Li, Q. Production of jet and diesel biofuels from renewable lignocellulosic biomass. Appl. Energy 2015, 150, 128–137. [Google Scholar] [CrossRef]
- Martínez, J.D.; Puy, N.; Murillo, R.; García, T.; Navarro, M.V.; Mastral, A.M. Waste tyre pyrolysis—A review. Renew. Sustain. Energy Rev. 2013, 23, 179–213. [Google Scholar] [CrossRef]
- Cho, M.-H.; Jung, S.-H.; Kim, J.-S. Pyrolysis of mixed plastic wastes for the recovery of benzene, toluene, and xylene (btx) aromatics in a fluidized bed and chlorine removal by applying various additives. Energy Fuels 2010, 24, 1389–1395. [Google Scholar] [CrossRef]
- Xue, Y.; Kelkar, A.; Bai, X. Catalytic co-pyrolysis of biomass and polyethylene in a tandem micropyrolyzer. Fuel 2016, 166, 227–236. [Google Scholar] [CrossRef]
- Li, X.; Zhang, H.; Li, J.; Su, L.; Zuo, J.; Komarneni, S.; Wang, Y. Improving the aromatic production in catalytic fast pyrolysis of cellulose by co-feeding low-density polyethylene. Appl. Catal. A 2013, 455, 114–121. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, J.; Xiao, R.; Shen, D.; Jin, B.; Xiao, G.; Chen, R. Co-catalytic pyrolysis of biomass and waste triglyceride seed oil in a novel fluidized bed reactor to produce olefins and aromatics integrated with self-heating and catalyst regeneration processes. RSC Adv. 2013, 3, 5769–5774. [Google Scholar] [CrossRef]
- Dorado, C.; Mullen, C.A.; Boateng, A.A. H-zsm5 catalyzed co-pyrolysis of biomass and plastics. ACS Sustain. Chem. Eng. 2014, 2, 301–311. [Google Scholar] [CrossRef]
- Artetxe, M.; Lopez, G.; Amutio, M.; Elordi, G.; Bilbao, J.; Olazar, M. Light olefins from hdpe cracking in a two-step thermal and catalytic process. Chem. Eng. J. 2012, 207–208, 27–34. [Google Scholar] [CrossRef]
- Serrano, D.P.; Aguado, J.; Escola, J.M.; Rodríguez, J.M.; San Miguel, G. An investigation into the catalytic cracking of ldpe using py-gc/ms. J. Anal. Appl. Pyrolysis 2005, 74, 370–378. [Google Scholar] [CrossRef]
- Brassard, P.; Godbout, S.; Raghavan, V. Pyrolysis in auger reactors for biochar and bio-oil production: A review. Biosyst. Eng. 2017, 161, 80–92. [Google Scholar] [CrossRef]
- Diebold, J.P.; Bridgwater, A.V. Overview of fast pyrolysis of biomass for the production of liquid fuels. In Developments in Thermochemical Biomass Conversion; Bridgwater, A.V., Boocock, D.G.B., Eds.; Springer: The Netherlands, 1997; pp. 5–23. [Google Scholar]
- Boateng, A.A.; Daugaard, D.E.; Goldberg, N.M.; Hicks, K.B. Bench-scale fluidized-bed pyrolysis of switchgrass for bio-oil production. Ind. Eng. Chem. Res. 2007, 46, 1891–1897. [Google Scholar] [CrossRef]
- Aho, A.; Kumar, N.; Eränen, K.; Salmi, T.; Hupa, M.; Murzin, D.Y. Catalytic pyrolysis of biomass in a fluidized bed reactor: Influence of the acidity of h-beta zeolite. Process. Saf. Environ. Prot. 2007, 85, 473–480. [Google Scholar] [CrossRef]
- Zhang, H.; Xiao, R.; Wang, D.; He, G.; Shao, S.; Zhang, J.; Zhong, Z. Biomass fast pyrolysis in a fluidized bed reactor under n2, co2, co, ch4 and h2 atmospheres. Bioresour. Technol. 2011, 102, 4258–4264. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, E.; Hogendoorn, K.J.A.; Wang, X.; Westerhof, R.J.M.; Kersten, S.R.A.; van Swaaij, W.P.M.; Groeneveld, M.J. Fast pyrolysis of biomass in a fluidized bed reactor: In situ filtering of the vapors. Ind. Eng. Chem. Res. 2009, 48, 4744–4756. [Google Scholar] [CrossRef]
- Lee, K.-H.; Kang, B.-S.; Park, Y.-K.; Kim, J.-S. Influence of reaction temperature, pretreatment, and a char removal system on the production of bio-oil from rice straw by fast pyrolysis, using a fluidized bed. Energy Fuels 2005, 19, 2179–2184. [Google Scholar] [CrossRef]
- Kang, B.-S.; Lee, K.H.; Park, H.J.; Park, Y.-K.; Kim, J.-S. Fast pyrolysis of radiata pine in a bench scale plant with a fluidized bed: Influence of a char separation system and reaction conditions on the production of bio-oil. J. Anal. Appl. Pyrolysis 2006, 76, 32–37. [Google Scholar] [CrossRef]
- Rüdisüli, M.; Schildhauer, T.J.; Biollaz, S.M.A.; van Ommen, J.R. Scale-up of bubbling fluidized bed reactors—A review. Powder Technol. 2012, 217, 21–38. [Google Scholar] [CrossRef]
- Fernandez-Akarregi, A.R.; Makibar, J.; Lopez, G.; Amutio, M.; Olazar, M. Design and operation of a conical spouted bed reactor pilot plant (25kg/h) for biomass fast pyrolysis. Fuel Process. Technol. 2013, 112, 48–56. [Google Scholar] [CrossRef]
- Aguado, R.; Olazar, M.; San José, M.J.; Aguirre, G.; Bilbao, J. Pyrolysis of sawdust in a conical spouted bed reactor. Yields and product composition. Ind. Eng. Chem. Res. 2000, 39, 1925–1933. [Google Scholar] [CrossRef]
- Amutio, M.; Lopez, G.; Alvarez, J.; Olazar, M.; Bilbao, J. Fast pyrolysis of eucalyptus waste in a conical spouted bed reactor. Bioresour. Technol. 2015, 194, 225–232. [Google Scholar] [CrossRef]
- Makibar, J.; Fernandez-Akarregi, A.R.; Díaz, L.; Lopez, G.; Olazar, M. Pilot scale conical spouted bed pyrolysis reactor: Draft tube selection and hydrodynamic performance. Powder Technol. 2012, 219, 49–58. [Google Scholar] [CrossRef]
- López, G.; Olazar, M.; Aguado, R.; Bilbao, J. Continuous pyrolysis of waste tyres in a conical spouted bed reactor. Fuel 2010, 89, 1946–1952. [Google Scholar] [CrossRef]
- Garcìa-Pérez, M.; Chaala, A.; Pakdel, H.; Kretschmer, D.; Roy, C. Vacuum pyrolysis of softwood and hardwood biomass: Comparison between product yields and bio-oil properties. J. Anal. Appl. Pyrolysis 2007, 78, 104–116. [Google Scholar] [CrossRef]
- Roy, C.; Chaala, A.; Darmstadt, H. The vacuum pyrolysis of used tires: End-uses for oil and carbon black products. J. Anal. Appl. Pyrolysis 1999, 51, 201–221. [Google Scholar] [CrossRef]
- Di Blasi, C. Heat transfer mechanisms and multi-step kinetics in the ablative pyrolysis of cellulose. Chem. Eng. Sci. 1996, 51, 2211–2220. [Google Scholar] [CrossRef]
- Helleur, R.; Popovic, N.; Ikura, M.; Stanciulescu, M.; Liu, D. Characterization and potential applications of pyrolytic char from ablative pyrolysis of used tires. J. Anal. Appl. Pyrolysis 2001, 58–59, 813–824. [Google Scholar] [CrossRef]
- Peacocke, G.V.C.; Bridgwater, A.V. Ablative plate pyrolysis of biomass for liquids. Biomass Bioenergy 1994, 7, 147–154. [Google Scholar] [CrossRef]
- Daugaard, D.; Jones, S.T.; Dalluge, D.L.; Brown, R.C. Selective Temperature Quench and Electrostatic Recovery of Bio-Oil Fractions. U.S. Patent 8,992,736, 31 March 2015. [Google Scholar]
- Chen, Y.-C.; Pan, Y.-N.; Hsieh, K.-H. Process optimization of fast pyrolysis reactor for converting forestry wastes into bio-oil with the taguchi method. Procedia Environ. Sci. 2011, 10, 1719–1725. [Google Scholar] [CrossRef]
- Brown, J.N.; Brown, R.C. Process optimization of an auger pyrolyzer with heat carrier using response surface methodology. Bioresour. Technol. 2012, 103, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Diebold, J.P. A Review of the Chemical and Physical Mechanisms of the Storage Stability of Fast Pyrolysis Bio-Oils; National Renewable Energy Laboratory: Golden, CO, USA, 2000.
- Piskorz, J.; Scott, D.S.; Radlein, D. Composition of oils obtained by fast pyrolysis of different woods. In Pyrolysis Oils from Biomass; American Chemical Society: Washington, DC, USA, 1988; Volume 376, pp. 167–178. [Google Scholar]
- Ed, J.S.; Thomas, A.M. Pyrolysis Oils from Biomass; American Chemical Society: Washington, DC, USA, 1988; Volume 376, p. 372. [Google Scholar]
- Garcìa-Pérez, M.; Chaala, A.; Pakdel, H.; Kretschmer, D.; Rodrigue, D.; Roy, C. Multiphase structure of bio-oils. Energy Fuels 2005, 20, 364–375. [Google Scholar] [CrossRef]
- Milne, T.A.A.; Davis, M.; Deutch, S.; Johnson, D. A Review of the Chemical Composition of Fast Pyrolysis Oils, in Developments in Thermal Biomass Conversion; Blackie Academic and Professional: London, UK, 1997. [Google Scholar]
- Diebold, J.P. A Review of the Toxicity of Biomass Pyrolysis Liquids Formed at Low Temperatures; National Renewable Energy Laboratory: Golden, CO, USA, 1997; p. 35. [Google Scholar]
- Branca, C.; Giudicianni, P.; Di Blasi, C. Gc/ms characterization of liquids generated from low-temperature pyrolysis of wood. Ind. Eng. Chem. Res. 2003, 42, 3190–3202. [Google Scholar] [CrossRef]
- Saiz-Jimenez, C.; De Leeuw, J.W. Lignin pyrolysis products: Their structures and their significance as biomarkers. Org. Geochem. 1986, 10, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Kawamoto, H.; Murayama, M.; Saka, S. Pyrolysis behavior of levoglucosan as an intermediate in cellulose pyrolysis: Polymerization into polysaccharide as a key reaction to carbonized product formation. J. Wood Sci. 2003, 49, 469–473. [Google Scholar] [CrossRef]
- Shafizadeh, F.; Furneaux, R.H.; Cochran, T.G.; Scholl, J.P.; Sakai, Y. Production of levoglucosan and glucose from pyrolysis of cellulosic materials. J. Appl. Polym. Sci. 1979, 23, 3525–3539. [Google Scholar] [CrossRef]
- Piskorz, J.; Radlein, D.; Scott, D.S. On the mechanism of the rapid pyrolysis of cellulose. J. Anal. Appl. Pyrolysis 1986, 9, 121–137. [Google Scholar] [CrossRef]
- Evans, R.J.; Milne, T.A. Molecular characterization of the pyrolysis of biomass. Energy Fuels 1987, 1, 123–137. [Google Scholar] [CrossRef]
- Agblevor, F.A.; Besler, S.; Evans, R.J. Inorganic Compounds in Biomass Feedstocks: Their Role in Char Formation and Effect on the Quality of Fast Pyrolysis Oils; National Renewable Energy Laboratory: Golden, CO, USA, 1994. [Google Scholar]
- French, R.J.; Milne, T.A. Vapor phase release of alkali species in the combustion of biomass pyrolysis oils. Biomass Bioenergy 1994, 7, 315–325. [Google Scholar] [CrossRef]
- Dayton, D. The fate of alkali metal during biomass thermochemical conversion. In Developments in Thermochemical Biomass Conversion; Bridgwater, A.V., Boocock, D.G.B., Eds.; Springer: The Netherlands, 1997; pp. 1263–1277. [Google Scholar]
- French, R.J.; Dayton, D.C.; Milne, T.A. The Direct Observation of Alkali Vapor Species in Biomass Combustion and Gasification; National Renewable Energy Laboratory: Golden, CO, USA, 1994. [Google Scholar]
- Lu, Q.; Yang, X.-L.; Zhu, X.-F. Analysis on chemical and physical properties of bio-oil pyrolyzed from rice husk. J. Anal. Appl. Pyrolysis 2008, 82, 191–198. [Google Scholar] [CrossRef]
- Diebold, J.P.; Czernik, S. Additives to lower and stabilize the viscosity of pyrolysis oils during storage. Energy Fuels 1997, 11, 1081–1091. [Google Scholar] [CrossRef]
- Czernik, S.; Johnson, D.K.; Black, S. Stability of wood fast pyrolysis oil. Biomass Bioenergy 1994, 7, 187–192. [Google Scholar] [CrossRef]
- Fratini, E.; Bonini, M.; Oasmaa, A.; Solantausta, Y.; Teixeira, J.; Baglioni, P. Sans analysis of the microstructural evolution during the aging of pyrolysis oils from biomass. Langmuir 2005, 22, 306–312. [Google Scholar] [CrossRef]
- Boucher, M.E.; Chaala, A.; Pakdel, H.; Roy, C. Bio-oils obtained by vacuum pyrolysis of softwood bark as a liquid fuel for gas turbines. Part ii: Stability and ageing of bio-oil and its blends with methanol and a pyrolytic aqueous phase. Biomass Bioenergy 2000, 19, 351–361. [Google Scholar] [CrossRef]
- Oasmaa, A.; Kuoppala, E. Fast pyrolysis of forestry residue. 3. Storage stability of liquid fuel. Energy Fuels 2003, 17, 1075–1084. [Google Scholar] [CrossRef]
- Hilten, R.N.; Das, K.C. Comparison of three accelerated aging procedures to assess bio-oil stability. Fuel 2010, 89, 2741–2749. [Google Scholar] [CrossRef]
- Ortega, J.V.; Renehan, A.M.; Liberatore, M.W.; Herring, A.M. Physical and chemical characteristics of aging pyrolysis oils produced from hardwood and softwood feedstocks. J. Anal. Appl. Pyrolysis 2011, 91, 190–198. [Google Scholar] [CrossRef]
- Ben, H.; Ragauskas, A.J. In situ nmr characterization of pyrolysis oil during accelerated aging. ChemSusChem 2012, 5, 1687–1693. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, Y.; Mourant, D.; Gunawan, R.; Lievens, C.; Chaiwat, W.; Gholizadeh, M.; Wu, L.; Li, X.; Li, C.-Z. Polymerization on heating up of bio-oil: A model compound study. AlChE J. 2013, 59, 888–900. [Google Scholar] [CrossRef]
- Alsbou, E.; Helleur, B. Accelerated aging of bio-oil from fast pyrolysis of hardwood. Energy Fuels 2014, 28, 3224–3235. [Google Scholar] [CrossRef]
- Zhang, L.; Chaparro Sosa, A.; Walters, K.B. Impacts of thermal processing on the physical and chemical properties of pyrolysis oil produced by a modified fluid catalytic cracking pyrolysis process. Energy Fuels 2016, 30, 7367–7378. [Google Scholar] [CrossRef]
- Mante, O.D.; Agblevor, F.A. Storage stability of biocrude oils from fast pyrolysis of poultry litter. Waste Manag. 2012, 32, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Oasmaa, A.; Kuoppala, E.; Selin, J.-F.; Gust, S.; Solantausta, Y. Fast pyrolysis of forestry residue and pine. 4. Improvement of the product quality by solvent addition. Energy Fuels 2004, 18, 1578–1583. [Google Scholar] [CrossRef]
- Adjaye, J.D.; Sharma, R.K.; Bakhshi, N.N. Characterization and stability analysis of wood-derived bio-oil. Fuel Process. Technol. 1992, 31, 241–256. [Google Scholar] [CrossRef]
- Meng, J.; Smirnova, T.I.; Song, X.; Moore, A.; Ren, X.; Kelley, S.; Park, S.; Tilotta, D. Identification of free radicals in pyrolysis oil and their impact on bio-oil stability. RSC Adv. 2014, 4, 29840–29846. [Google Scholar] [CrossRef]
- Zhang, L.; Luo, Y.; Wijayapala, R.; Walters, K.B. Alcohol stabilization of low water content pyrolysis oil during high temperature treatment. Energy Fuels 2017, 31, 13666–13674. [Google Scholar] [CrossRef]
- Vitolo, S.; Seggiani, M.; Frediani, P.; Ambrosini, G.; Politi, L. Catalytic upgrading of pyrolytic oils to fuel over different zeolites. Fuel 1999, 78, 1147–1159. [Google Scholar] [CrossRef]
- Zhang, Q.; Chang, J.; Wang, T.; Xu, Y. Upgrading bio-oil over different solid catalysts. Energy Fuels 2006, 20, 2717–2720. [Google Scholar] [CrossRef]
- Venderbosch, R.H.; Ardiyanti, A.R.; Wildschut, J.; Oasmaa, A.; Heeres, H.J. Stabilization of biomass-derived pyrolysis oils. J. Chem. Technol. Biotechnol. 2010, 85, 674–686. [Google Scholar] [CrossRef]
- Elliott, D.C. Historical developments in hydroprocessing bio-oils. Energy Fuels 2007, 21, 1792–1815. [Google Scholar] [CrossRef]
- Rioche, C.; Kulkarni, S.; Meunier, F.C.; Breen, J.P.; Burch, R. Steam reforming of model compounds and fast pyrolysis bio-oil on supported noble metal catalysts. Appl. Catal. B Environ. 2005, 61, 130–139. [Google Scholar] [CrossRef]
- Takanabe, K.; Aika, K.-I.; Seshan, K.; Lefferts, L. Sustainable hydrogen from bio-oil—Steam reforming of acetic acid as a model oxygenate. J. Catal. 2004, 227, 101–108. [Google Scholar] [CrossRef]
- Chiaramonti, D.; Bonini, M.; Fratini, E.; Tondi, G.; Gartner, K.; Bridgwater, A.V.; Grimm, H.P.; Soldaini, I.; Webster, A.; Baglioni, P. Development of emulsions from biomass pyrolysis liquid and diesel and their use in engines—Part 1: Emulsion production. Biomass Bioenergy 2003, 25, 85–99. [Google Scholar] [CrossRef]
- Javaid, A.; Ryan, T.; Berg, G.; Pan, X.; Vispute, T.; Bhatia, S.R.; Huber, G.W.; Ford, D.M. Removal of char particles from fast pyrolysis bio-oil by microfiltration. J. Membr. Sci. 2010, 363, 120–127. [Google Scholar] [CrossRef]
- Naske, C.D.; Polk, P.; Wynne, P.Z.; Speed, J.; Holmes, W.E.; Walters, K.B. Postcondensation filtration of pine and cottonwood pyrolysis oil and impacts on accelerated aging reactions. Energy Fuels 2011, 26, 1284–1297. [Google Scholar] [CrossRef]
- McCall, M.J.; Brandvold, T.A. Fuel and Fuel Blending Components from Biomass Derived Pyrolysis Oil. U.S. Patent 8,329,969, 11 December 2012. [Google Scholar]
- Gutierrez, A.; Kaila, R.K.; Honkela, M.L.; Slioor, R.; Krause, A.O.I. Hydrodeoxygenation of guaiacol on noble metal catalysts. Catal. Today 2009, 147, 239–246. [Google Scholar] [CrossRef]
- Gagnon, J.; Kaliaguine, S. Catalytic hydrotreatment of vacuum pyrolysis oils from wood. Ind. Eng. Chem. Res. 1988, 27, 1783–1788. [Google Scholar] [CrossRef]
- Pham, T.N.; Sooknoi, T.; Crossley, S.P.; Resasco, D.E. Ketonization of carboxylic acids: Mechanisms, catalysts, and implications for biomass conversion. Acs Catal. 2013, 3, 2456–2473. [Google Scholar] [CrossRef]
- Lilga, M.A.; Padmaperuma, A.B.; Auberry, D.L.; Job, H.M.; Swita, M.S. Ketonization of levulinic acid and γ-valerolactone to hydrocarbon fuel precursors. Catal. Today 2018, 302, 80–86. [Google Scholar] [CrossRef]
- Gliński, M.; Kijeński, J.; Jakubowski, A. Ketones from monocarboxylic acids: Catalytic ketonization over oxide systems. Appl. Catal. A Gen. 1995, 128, 209–217. [Google Scholar] [CrossRef]
- Weber, J.; Thompson, A.; Wilmoth, J.; Batra, V.S.; Janulaitis, N.; Kastner, J.R. Effect of metal oxide redox state in red mud catalysts on ketonization of fast pyrolysis oil derived oxygenates. Appl. Catal. B Environ. 2019, 241, 430–441. [Google Scholar] [CrossRef]
- Fan, L.; Chen, P.; Zhou, N.; Liu, S.; Zhang, Y.; Liu, Y.; Wang, Y.; Omar, M.M.; Peng, P.; Addy, M.; et al. In-situ and ex-situ catalytic upgrading of vapors from microwave-assisted pyrolysis of lignin. Bioresour. Technol. 2018, 247, 851–858. [Google Scholar] [CrossRef]
- Gamliel, D.P.; Du, S.; Bollas, G.M.; Valla, J.A. Investigation of in situ and ex situ catalytic pyrolysis of miscanthus × giganteus using a pygc-ms microsystem and comparison with a bench-scale spouted-bed reactor. Bioresour. Technol. 2015, 191, 187–196. [Google Scholar] [CrossRef]
- Luo, G.; Resende, F.L.P. In-situ and ex-situ upgrading of pyrolysis vapors from beetle-killed trees. Fuel 2016, 166, 367–375. [Google Scholar] [CrossRef]
- Wang, K.; Johnston, P.A.; Brown, R.C. Comparison of in-situ and ex-situ catalytic pyrolysis in a micro-reactor system. Bioresour. Technol. 2014, 173, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Xiao, R.; Zhang, H. Ex-situ catalytic fast pyrolysis of biomass over hzsm-5 in a two-stage fluidized-bed/fixed-bed combination reactor. Bioresour. Technol. 2017, 243, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, H.; Karim, A.M.; Sun, J.; Wang, Y. Catalytic fast pyrolysis of lignocellulosic biomass. Chem. Soc. Rev. 2014, 43, 7594–7623. [Google Scholar] [CrossRef] [PubMed]
- Ennaert, T.; Van Aelst, J.; Dijkmans, J.; De Clercq, R.; Schutyser, W.; Dusselier, M.; Verboekend, D.; Sels, B.F. Potential and challenges of zeolite chemistry in the catalytic conversion of biomass. Chem. Soc. Rev. 2016, 45, 584–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, H.; Lim, J.K.; Hameed, B.H. Recent progress on biomass co-pyrolysis conversion into high-quality bio-oil. Bioresour. Technol. 2016, 221, 645–655. [Google Scholar] [CrossRef]
- Venderbosch, R.H. A critical view on catalytic pyrolysis of biomass. ChemSusChem 2015, 8, 1306–1316. [Google Scholar] [CrossRef]
- Galadima, A.; Muraza, O. In situ fast pyrolysis of biomass with zeolite catalysts for bioaromatics/gasoline production: A review. Energy Convers. Manag. 2015, 105, 338–354. [Google Scholar] [CrossRef]
- Corma, A. From microporous to mesoporous molecular sieve materials and their use in catalysis. Chem. Rev. 1997, 97, 2373–2420. [Google Scholar] [CrossRef]
- Imran, A.; Bramer, E.; Seshan, K.; Brem, G. Catalytic flash pyrolysis of biomass using different types of zeolite and online vapor fractionation. Energies 2016, 9, 187. [Google Scholar] [CrossRef]
- Aho, A.; Kumar, N.; Eränen, K.; Salmi, T.; Hupa, M.; Murzin, D.Y. Catalytic pyrolysis of woody biomass in a fluidized bed reactor: Influence of the zeolite structure. Fuel 2008, 87, 2493–2501. [Google Scholar] [CrossRef]
- Carlson, T.R.; Jae, J.; Lin, Y.-C.; Tompsett, G.A.; Huber, G.W. Catalytic fast pyrolysis of glucose with hzsm-5: The combined homogeneous and heterogeneous reactions. J. Catal. 2010, 270, 110–124. [Google Scholar] [CrossRef]
- Jae, J.; Tompsett, G.A.; Foster, A.J.; Hammond, K.D.; Auerbach, S.M.; Lobo, R.F.; Huber, G.W. Investigation into the shape selectivity of zeolite catalysts for biomass conversion. J. Catal. 2011, 279, 257–268. [Google Scholar] [CrossRef]
- Kurnia, I.; Karnjanakom, S.; Bayu, A.; Yoshida, A.; Rizkiana, J.; Prakoso, T.; Abudula, A.; Guan, G. In-situ catalytic upgrading of bio-oil derived from fast pyrolysis of lignin over high aluminum zeolites. Fuel Process. Technol. 2017, 167, 730–737. [Google Scholar] [CrossRef]
- Foster, A.J.; Jae, J.; Cheng, Y.-T.; Huber, G.W.; Lobo, R.F. Optimizing the aromatic yield and distribution from catalytic fast pyrolysis of biomass over zsm-5. Appl. Catal. A 2012, 423–424, 154–161. [Google Scholar] [CrossRef]
- Hoff, T.C.; Gardner, D.W.; Thilakaratne, R.; Wang, K.; Hansen, T.W.; Brown, R.C.; Tessonnier, J.P. Tailoring zsm-5 zeolites for the fast pyrolysis of biomass to aromatic hydrocarbons. ChemSusChem 2016, 9, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shao, S.; Luo, M.; Xiao, R. The comparison of chemical liquid deposition and acid dealumination modified zsm-5 for catalytic pyrolysis of pinewood using pyrolysis-gas chromatography/mass spectrometry. Bioresour. Technol. 2017, 244, 726–732. [Google Scholar] [CrossRef]
- Gou, J.; Wang, Z.; Li, C.; Qi, X.; Vattipalli, V.; Cheng, Y.-T.; Huber, G.; Conner, W.C.; Dauenhauer, P.J.; Mountziaris, T.J.; et al. The effects of zsm-5 mesoporosity and morphology on the catalytic fast pyrolysis of furan. Green Chem. 2017, 19, 3549–3557. [Google Scholar] [CrossRef]
- Hoff, T.C.; Gardner, D.W.; Thilakaratne, R.; Proano-Aviles, J.; Brown, R.C.; Tessonnier, J.-P. Elucidating the effect of desilication on aluminum-rich zsm-5 zeolite and its consequences on biomass catalytic fast pyrolysis. Appl. Catal. A 2017, 529, 68–78. [Google Scholar] [CrossRef]
- Tan, S.; Zhang, Z.; Sun, J.; Wang, Q. Recent progress of catalytic pyrolysis of biomass by hzsm-5. Chin. J. Catal. 2013, 34, 641–650. [Google Scholar] [CrossRef]
- Jackson, M.A.; Compton, D.L.; Boateng, A.A. Screening heterogeneous catalysts for the pyrolysis of lignin. J. Anal. Appl. Pyrolysis 2009, 85, 226–230. [Google Scholar] [CrossRef]
- Mihalcik, D.J.; Mullen, C.A.; Boateng, A.A. Screening acidic zeolites for catalytic fast pyrolysis of biomass and its components. J. Anal. Appl. Pyrolysis 2011, 92, 224–232. [Google Scholar] [CrossRef]
- Ma, Z.; Troussard, E.; van Bokhoven, J.A. Controlling the selectivity to chemicals from lignin via catalytic fast pyrolysis. Appl. Catal. A 2012, 423–424, 130–136. [Google Scholar] [CrossRef]
- Shen, D.; Zhao, J.; Xiao, R.; Gu, S. Production of aromatic monomers from catalytic pyrolysis of black-liquor lignin. J. Anal. Appl. Pyrolysis 2015, 111, 47–54. [Google Scholar] [CrossRef]
- Jia, L.Y.; Raad, M.; Hamieh, S.; Toufaily, J.; Hamieh, T.; Bettahar, M.M.; Mauviel, G.; Tarrighi, M.; Pinard, L.; Dufour, A. Catalytic fast pyrolysis of biomass: Superior selectivity of hierarchical zeolites to aromatics. Green Chem. 2017, 19, 5442–5459. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Zhou, G.; Wang, W.; Wang, C.; Komarneni, S.; Wang, Y. Catalytic fast pyrolysis of biomass with mesoporous zsm-5 zeolites prepared by desilication with naoh solutions. Appl. Catal. A 2014, 470, 115–122. [Google Scholar] [CrossRef]
- Gamliel, D.P.; Cho, H.J.; Fan, W.; Valla, J.A. On the effectiveness of tailored mesoporous mfi zeolites for biomass catalytic fast pyrolysis. Appl. Catal. A 2016, 522, 109–119. [Google Scholar] [CrossRef]
- Leonowicz, M.E.; Lawton, J.A.; Lawton, S.L.; Rubin, M.K. Mcm-22: A molecular sieve with two independent multidimensional channel systems. Science 1994, 264, 1910–1913. [Google Scholar] [CrossRef] [PubMed]
- Roth, W.J.; Kresge, C.T.; Vartuli, J.C.; Leonowicz, M.E.; Fung, A.S.; McCullen, S.B. Mcm-36: The first pillared molecular sieve with zeoliteproperties. Stud. Surf. Sci. Catal. 1995, 94, 301–308. [Google Scholar] [CrossRef]
- Hernando, H.; Fermoso, J.; Ochoa-Hernández, C.; Opanasenko, M.; Pizarro, P.; Coronado, J.M.; Čejka, J.; Serrano, D.P. Performance of mcm-22 zeolite for the catalytic fast-pyrolysis of acid-washed wheat straw. Catal. Today 2018, 304, 30–38. [Google Scholar] [CrossRef]
- Yang, S.-T.; Kim, J.-Y.; Kim, J.; Ahn, W.-S. CO2 capture over amine-functionalized mcm-22, mcm-36 and itq-2. Fuel 2012, 97, 435–442. [Google Scholar] [CrossRef]
- Käldström, M.; Kumar, N.; Heikkilä, T.; Murzin, D.Y. Pillared h-mcm-36 mesoporous and h-mcm-22 microporous materials for conversion of levoglucosan: Influence of varying acidity. Appl. Catal. A 2011, 397, 13–21. [Google Scholar] [CrossRef]
- Naqvi, S.R.; Uemura, Y.; Yusup, S.; Sugiura, Y.; Nishiyama, N. In situ catalytic fast pyrolysis of paddy husk pyrolysis vapors over mcm-22 and itq-2 zeolites. J. Anal. Appl. Pyrolysis 2015, 114, 32–39. [Google Scholar] [CrossRef]
- Wang, W.; Shi, Y.; Cui, Y.; Li, X. Catalytic fast pyrolysis of cellulose for increasing contents of furans and aromatics in biofuel production. J. Anal. Appl. Pyrolysis 2018, 131, 93–100. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, R.; Song, W. Influence of hsapo-34, hzsm-5, and nay on pyrolysis of corn straw fermentation residue via py-gc/ms. J. Anal. Appl. Pyrolysis 2016, 122, 183–190. [Google Scholar] [CrossRef]
- Lee, H.W.; Kim, Y.-M.; Lee, B.; Kim, S.; Jae, J.; Jung, S.-C.; Kim, T.-W.; Park, Y.-K. Catalytic copyrolysis of torrefied cork oak and high density polyethylene over a mesoporous hy catalyst. Catal. Today 2018, 307, 301–307. [Google Scholar] [CrossRef]
- Ma, Z.; Ghosh, A.; Asthana, N.; van Bokhoven, J. Visualization of structural changes during deactivation and regeneration of fau zeolite for catalytic fast pyrolysis of lignin using nmr and electron microscopy techniques. ChemCatChem 2018, 10, 4431–4437. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, X.; Chen, L.; Zhao, B.; Yang, S.; Xie, X. Comparision of catalytic fast pyrolysis of biomass to aromatic hydrocarbons over zsm-5 and fe/zsm-5 catalysts. J. Anal. Appl. Pyrolysis 2016, 121, 342–346. [Google Scholar] [CrossRef]
- Saraçoğlu, E.; Uzun, B.B.; Apaydın-Varol, E. Upgrading of fast pyrolysis bio-oil over fe modified zsm-5 catalyst to enhance the formation of phenolic compounds. Int. J. Hydrogen Energy 2017, 42, 21476–21486. [Google Scholar] [CrossRef]
- Fermoso, J.; Hernando, H.; Jana, P.; Moreno, I.; Přech, J.; Ochoa-Hernández, C.; Pizarro, P.; Coronado, J.M.; Čejka, J.; Serrano, D.P. Lamellar and pillared zsm-5 zeolites modified with mgo and zno for catalytic fast-pyrolysis of eucalyptus woodchips. Catal. Today 2016, 277, 171–181. [Google Scholar] [CrossRef]
- Schultz, E.L.; Mullen, C.A.; Boateng, A.A. Aromatic hydrocarbon production from eucalyptus urophylla pyrolysis over several metal-modified zsm-5 catalysts. Energy Technol. 2017, 5, 196–204. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, F.; Yang, X.; Huang, Y.; Liu, C.; Zheng, Z.; Gu, J. Study on aromatics production via the catalytic pyrolysis vapor upgrading of biomass using metal-loaded modified h-zsm-5. J. Anal. Appl. Pyrolysis 2017, 126, 169–179. [Google Scholar] [CrossRef]
- Liu, T.-L.; Cao, J.-P.; Zhao, X.-Y.; Wang, J.-X.; Ren, X.-Y.; Fan, X.; Zhao, Y.-P.; Wei, X.-Y. In situ upgrading of shengli lignite pyrolysis vapors over metal-loaded hzsm-5 catalyst. Fuel Process. Technol. 2017, 160, 19–26. [Google Scholar] [CrossRef]
- Mullen, C.A.; Tarves, P.C.; Boateng, A.A. Role of potassium exchange in catalytic pyrolysis of biomass over zsm-5: Formation of alkyl phenols and furans. ACS Sustain. Chem. Eng. 2017, 5, 2154–2162. [Google Scholar] [CrossRef]
- Campanella, A.; Harold, M.P. Fast pyrolysis of microalgae in a falling solids reactor: Effects of process variables and zeolite catalysts. Biomass Bioenergy 2012, 46, 218–232. [Google Scholar] [CrossRef]
- Till, Z.; Varga, T.; Sója, J.; Miskolczi, N.; Chován, T. Kinetic identification of plastic waste pyrolysis on zeolite-based catalysts. Energy Convers. Manag. 2018, 173, 320–330. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, M.; Shao, J.; Yang, H.; Zeng, K.; Chen, Y.; Luo, J.; Agblevor, F.A.; Chen, H. The conversion of biomass to light olefins on fe-modified zsm-5 catalyst: Effect of pyrolysis parameters. Sci. Total Environ. 2018, 628–629, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Li, J.; Feng, Y.; Wang, W.; Zhang, X.; Chen, Q.; Komarneni, S.; Wang, Y. Thermally stable phosphorus and nickel modified zsm-5 zeolites for catalytic co-pyrolysis of biomass and plastics. RSC Adv. 2015, 5, 30485–30494. [Google Scholar] [CrossRef]
- Vichaphund, S.; Aht-ong, D.; Sricharoenchaikul, V.; Atong, D. Production of aromatic compounds from catalytic fast pyrolysis of jatropha residues using metal/hzsm-5 prepared by ion-exchange and impregnation methods. Renew. Energy 2015, 79, 28–37. [Google Scholar] [CrossRef]
- Thangalazhy-Gopakumar, S.; Adhikari, S.; Gupta, R.B. Catalytic pyrolysis of biomass over h+zsm-5 under hydrogen pressure. Energy Fuels 2012, 26, 5300–5306. [Google Scholar] [CrossRef]
- Mullen, C.A.; Boateng, A.A. Production of aromatic hydrocarbons via catalytic pyrolysis of biomass over fe-modified hzsm-5 zeolites. ACS Sustain. Chem. Eng. 2015, 3, 1623–1631. [Google Scholar] [CrossRef]
- Karnjanakom, S.; Suriya-umporn, T.; Bayu, A.; Kongparakul, S.; Samart, C.; Fushimi, C.; Abudula, A.; Guan, G. High selectivity and stability of mg-doped al-mcm-41 for in-situ catalytic upgrading fast pyrolysis bio-oil. Energy Convers. Manag. 2017, 142, 272–285. [Google Scholar] [CrossRef]
- Rizkiana, J.; Guan, G.; Widayatno, W.; Yang, J.; Hao, X.; Matsuoka, K.; Abudula, A.J.R.A. Mg-modified ultra-stable y type zeolite for the rapid catalytic co-pyrolysis of low-rank coal and biomass. RSC Adv. 2016, 6, 2096–2105. [Google Scholar] [CrossRef]
- Iliopoulou, E.F.; Stefanidis, S.D.; Kalogiannis, K.G.; Delimitis, A.; Lappas, A.A.; Triantafyllidis, K.S. Catalytic upgrading of biomass pyrolysis vapors using transition metal-modified zsm-5 zeolite. Appl. Catal. B Environ. 2012, 127, 281–290. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, H.; Liu, X.; Zhu, S.; Hu, L.; Zhang, Q. Upgrading of bio-oil from catalytic pyrolysis of pretreated rice husk over fe-modified zsm-5 zeolite catalyst. Fuel Process. Technol. 2018, 175, 17–25. [Google Scholar] [CrossRef]
- Balasundram, V.; Ibrahim, N.; Kasmani, R.; Isha, R.; Hamid, M.K.A.; Hasbullah, H.; Ali, R.R. Catalytic upgrading of sugarcane bagasse pyrolysis vapours over rare earth metal (ce) loaded hzsm-5: Effect of catalyst to biomass ratio on the organic compounds in pyrolysis oil. Appl. Energy 2018, 220, 787–799. [Google Scholar] [CrossRef]
- Vichaphund, S.; Aht-ong, D.; Sricharoenchaikul, V.; Atong, D. Effect of cv-zsm-5, ni-zsm-5 and fa-zsm-5 catalysts for selective aromatic formation from pyrolytic vapors of rubber wastes. J. Anal. Appl. Pyrolysis 2017, 124, 733–741. [Google Scholar] [CrossRef]
- Ren, X.-Y.; Cao, J.-P.; Zhao, X.-Y.; Yang, Z.; Liu, T.-L.; Fan, X.; Zhao, Y.-P.; Wei, X.-Y. Catalytic upgrading of pyrolysis vapors from lignite over mono/bimetal-loaded mesoporous hzsm-5. Fuel 2018, 218, 33–40. [Google Scholar] [CrossRef]
- Li, P.; Li, D.; Yang, H.; Wang, X.; Chen, H. Effects of fe-, zr-, and co-modified zeolites and pretreatments on catalytic upgrading of biomass fast pyrolysis vapors. Energy Fuels 2016, 30, 3004–3013. [Google Scholar] [CrossRef]
- Ren, X.-Y.; Cao, J.-P.; Zhao, X.-Y.; Shen, W.-Z.; Wei, X.-Y. Increasing light aromatic products during upgrading of lignite pyrolysis vapor over co-modified hzsm-5. J. Anal. Appl. Pyrolysis 2018, 130, 190–197. [Google Scholar] [CrossRef]
- Lu, Q.; Guo, H.-Q.; Zhou, M.-X.; Zhang, Z.-X.; Cui, M.-S.; Zhang, Y.-Y.; Yang, Y.-P.; Zhang, L.-B. Monocyclic aromatic hydrocarbons production from catalytic cracking of pine wood-derived pyrolytic vapors over ce-mo2n/hzsm-5 catalyst. Sci. Total Environ. 2018, 634, 141–149. [Google Scholar] [CrossRef]
- Adam, J.; Antonakou, E.; Lappas, A.; Stöcker, M.; Nilsen, M.H.; Bouzga, A.; Hustad, J.E.; Øye, G. In situ catalytic upgrading of biomass derived fast pyrolysis vapours in a fixed bed reactor using mesoporous materials. Microporous Mesoporous Mater. 2006, 96, 93–101. [Google Scholar] [CrossRef]
- Antonakou, E.; Lappas, A.; Nilsen, M.H.; Bouzga, A.; Stöcker, M. Evaluation of various types of al-mcm-41 materials as catalysts in biomass pyrolysis for the production of bio-fuels and chemicals. Fuel 2006, 85, 2202–2212. [Google Scholar] [CrossRef]
- Lange, J.-P.; van der Heide, E.; van Buijtenen, J.; Price, R. Furfural—A promising platform for lignocellulosic biofuels. ChemSusChem 2012, 5, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US department of energy’s “top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Yemiş, O.; Mazza, G. Acid-catalyzed conversion of xylose, xylan and straw into furfural by microwave-assisted reaction. Bioresour. Technol. 2011, 102, 7371–7378. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Liu, S.; Abu-Omar, M.M.; Mosier, N.S. Selective conversion of biomass hemicellulose to furfural using maleic acid with microwave heating. Energy Fuels 2012, 26, 1298–1304. [Google Scholar] [CrossRef]
- Li, Y.; Lu, X.; Yuan, L.; Liu, X. Fructose decomposition kinetics in organic acids-enriched high temperature liquid water. Biomass Bioenergy 2009, 33, 1182–1187. [Google Scholar] [CrossRef]
- Chheda, J.N.; Román-Leshkov, Y.; Dumesic, J.A. Production of 5-hydroxymethylfurfural and furfural by dehydration of biomass-derived mono- and poly-saccharides. Green Chem. 2007, 9, 342–350. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Y.; Chen, Z.; Zhu, D.; Yang, H.; Liu, P.; Li, T.; Chen, H. Catalytic fast pyrolysis of cellulose to produce furan compounds with sapo type catalysts. J. Anal. Appl. Pyrolysis 2018, 129, 53–60. [Google Scholar] [CrossRef]
- Wang, W.-L.; Ren, X.-Y.; Li, L.-F.; Chang, J.-M.; Cai, L.-P.; Geng, J. Catalytic effect of metal chlorides on analytical pyrolysis of alkali lignin. Fuel Process. Technol. 2015, 134, 345–351. [Google Scholar] [CrossRef]
- Wang, W.-L.; Ren, X.-Y.; Chang, J.-M.; Cai, L.-P.; Shi, S.Q. Characterization of bio-oils and bio-chars obtained from the catalytic pyrolysis of alkali lignin with metal chlorides. Fuel Process. Technol. 2015, 138, 605–611. [Google Scholar] [CrossRef]
- Hwang, H.; Oh, S.; Choi, I.-G.; Choi, J.W. Catalytic effects of magnesium on the characteristics of fast pyrolysis products—Bio-oil, bio-char, and non-condensed pyrolytic gas fractions. J. Anal. Appl. Pyrolysis 2015, 113, 27–34. [Google Scholar] [CrossRef]
- Kalogiannis, K.G.; Stefanidis, S.D.; Karakoulia, S.A.; Triantafyllidis, K.S.; Yiannoulakis, H.; Michailof, C.; Lappas, A.A. First pilot scale study of basic vs acidic catalysts in biomass pyrolysis: Deoxygenation mechanisms and catalyst deactivation. Appl. Catal. B Environ. 2018, 238, 346–357. [Google Scholar] [CrossRef]
- Park, H.J.; Heo, H.S.; Park, Y.-K.; Yim, J.-H.; Jeon, J.-K.; Park, J.; Ryu, C.; Kim, S.-S. Clean bio-oil production from fast pyrolysis of sewage sludge: Effects of reaction conditions and metal oxide catalysts. Bioresour. Technol. 2010, 101, S83–S85. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhang, Z.; Zhang, Z.; Sun, J.; Wang, Q.; Pittman, C.U. Catalytic fast pyrolysis of a wood-plastic composite with metal oxides as catalysts. Waste Manag. 2018, 79, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Navarro, R.M.; Guil-Lopez, R.; Fierro, J.L.G.; Mota, N.; Jiménez, S.; Pizarro, P.; Coronado, J.M.; Serrano, D.P. Catalytic fast pyrolysis of biomass over mg-al mixed oxides derived from hydrotalcite-like precursors: Influence of mg/al ratio. J. Anal. Appl. Pyrolysis 2018, 134, 362–370. [Google Scholar] [CrossRef]
- Anand, V.; Gautam, R.; Vinu, R. Non-catalytic and catalytic fast pyrolysis of schizochytrium limacinum microalga. Fuel 2017, 205, 1–10. [Google Scholar] [CrossRef]
- Lu, Q.; Xiong, W.-M.; Li, W.-Z.; Guo, Q.-X.; Zhu, X.-F. Catalytic pyrolysis of cellulose with sulfated metal oxides: A promising method for obtaining high yield of light furan compounds. Bioresour. Technol. 2009, 100, 4871–4876. [Google Scholar] [CrossRef]
- Kaewpengkrow, P.; Atong, D.; Sricharoenchaikul, V. Selective catalytic fast pyrolysis of jatropha curcas residue with metal oxide impregnated activated carbon for upgrading bio-oil. Int. J. Hydrogen Energy 2017, 42, 18397–18409. [Google Scholar] [CrossRef]
- Mullen, C.A.; Boateng, A.A. Catalytic pyrolysis-gc/ms of lignin from several sources. Fuel Process. Technol. 2010, 91, 1446–1458. [Google Scholar] [CrossRef]
- Lu, Q.; Zhou, M.-X.; Li, W.-T.; Wang, X.; Cui, M.-S.; Yang, Y.-P. Catalytic fast pyrolysis of biomass with noble metal-like catalysts to produce high-grade bio-oil: Analytical py-gc/ms study. Catal. Today 2018, 302, 169–179. [Google Scholar] [CrossRef]
- Koike, N.; Hosokai, S.; Takagaki, A.; Nishimura, S.; Kikuchi, R.; Ebitani, K.; Suzuki, Y.; Oyama, S.T. Upgrading of pyrolysis bio-oil using nickel phosphide catalysts. J. Catal. 2016, 333, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Sushil, S.; Batra, V.S. Catalytic applications of red mud, an aluminium industry waste: A review. Appl. Catal. B Environ. 2008, 81, 64–77. [Google Scholar] [CrossRef]
- Jahromi, H.; Agblevor, F.A. Hydrodeoxygenation of pinyon-juniper catalytic pyrolysis oil using red mud-supported nickel catalysts. Appl. Catal. B Environ. 2018, 236, 1–12. [Google Scholar] [CrossRef]
- Agblevor, F.A.; Jahromi, H. Aqueous-phase synthesis of hydrocarbons from furfural reactions with low-molecular-weight biomass oxygenates. Energy Fuels 2018, 32, 8552–8562. [Google Scholar] [CrossRef]
- Yathavan, B.K.; Agblevor, F.A. Catalytic pyrolysis of pinyon–juniper using red mud and hzsm-5. Energy Fuels 2013, 27, 6858–6865. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.; Yang, J.; Liang, S.; Liu, K.; Xiao, K.; Deng, H.; Hu, J.; Xiao, B. Improving bromine fixation in co-pyrolysis of non-metallic fractions of waste printed circuit boards with bayer red mud. Sci. Total Environ. 2018, 639, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.A.; Akay, G. Effect of cao on tar production and dew point depression during gasification of fuel cane bagasse in a novel downdraft gasifier. Fuel Process. Technol. 2013, 106, 654–660. [Google Scholar] [CrossRef]
- Ding, K.; Zhong, Z.; Wang, J.; Zhang, B.; Fan, L.; Liu, S.; Wang, Y.; Liu, Y.; Zhong, D.; Chen, P.; et al. Improving hydrocarbon yield from catalytic fast co-pyrolysis of hemicellulose and plastic in the dual-catalyst bed of cao and hzsm-5. Bioresour. Technol. 2018, 261, 86–92. [Google Scholar] [CrossRef]
- Wang, J.; Zhong, Z.; Ding, K.; Zhang, B.; Deng, A.; Min, M.; Chen, P.; Ruan, R. Co-pyrolysis of bamboo residual with waste tire over dual catalytic stage of cao and co-modified hzsm-5. Energy 2017, 133, 90–98. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, B.; Zhong, Z.; Ding, K.; Deng, A.; Min, M.; Chen, P.; Ruan, R. Catalytic fast co-pyrolysis of bamboo residual and waste lubricating oil over an ex-situ dual catalytic beds of mgo and hzsm-5: Analytical py-gc/ms study. Energy Convers. Manag. 2017, 139, 222–231. [Google Scholar] [CrossRef]
- Zhang, H.; Likun, P.K.W.; Xiao, R. Improving the hydrocarbon production via co-pyrolysis of bagasse with bio-plastic and dual-catalysts layout. Sci. Total Environ. 2018, 618, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Pan, Z.; Zhang, C.; Wang, D.; Xie, Y.; Zhang, R. Segmented catalytic co-pyrolysis of biomass and high-density polyethylene for aromatics production with mgcl2 and hzsm-5. J. Anal. Appl. Pyrolysis 2018, 134, 209–217. [Google Scholar] [CrossRef]
- Wang, S.; Dai, G.; Yang, H.; Luo, Z. Lignocellulosic biomass pyrolysis mechanism: A state-of-the-art review. Prog. Energy Combust. Sci. 2017, 62, 33–86. [Google Scholar] [CrossRef]
- Ma, Z.; van Bokhoven, J.A. Deactivation and regeneration of h-usy zeolite during lignin catalytic fast pyrolysis. ChemCatChem 2012, 4, 2036–2044. [Google Scholar] [CrossRef]
- Carlson, T.R.; Vispute, T.P.; Huber, G.W. Green gasoline by catalytic fast pyrolysis of solid biomass derived compounds. ChemSusChem 2008, 1, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Arregi, A.; Lopez, G.; Amutio, M.; Artetxe, M.; Barbarias, I.; Bilbao, J.; Olazar, M. Role of operating conditions in the catalyst deactivation in the in-line steam reforming of volatiles from biomass fast pyrolysis. Fuel 2018, 216, 233–244. [Google Scholar] [CrossRef]
- Rezaei, P.S.; Shafaghat, H.; Daud, W.M.A.W. Production of green aromatics and olefins by catalytic cracking of oxygenate compounds derived from biomass pyrolysis: A review. Appl. Catal. A 2014, 469, 490–511. [Google Scholar] [CrossRef]
- Jahromi, H.; Agblevor, F.A. Hydrodeoxygenation of aqueous-phase catalytic pyrolysis oil to liquid hydrocarbons using multifunctional nickel catalyst. Ind. Eng. Chem. Res. 2018, 57, 13257–13268. [Google Scholar] [CrossRef]
- Cordero-Lanzac, T.; Palos, R.; Hita, I.; Arandes, J.M.; Rodríguez-Mirasol, J.; Cordero, T.; Bilbao, J.; Castaño, P. Revealing the pathways of catalyst deactivation by coke during the hydrodeoxygenation of raw bio-oil. Appl. Catal. B Environ. 2018, 239, 513–524. [Google Scholar] [CrossRef]
- Zhang, H.; Shao, S.; Xiao, R.; Shen, D.; Zeng, J. Characterization of coke deposition in the catalytic fast pyrolysis of biomass derivates. Energy Fuels 2014, 28, 52–57. [Google Scholar] [CrossRef]
- Paasikallio, V.; Lindfors, C.; Kuoppala, E.; Solantausta, Y.; Oasmaa, A.; Lehto, J.; Lehtonen, J. Product quality and catalyst deactivation in a four day catalytic fast pyrolysis production run. Green Chem. 2014, 16, 3549–3559. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, M.; Xiao, R.; Shao, S.; Jin, B.; Xiao, G.; Zhao, M.; Liang, J. Catalytic conversion of biomass pyrolysis-derived compounds with chemical liquid deposition (cld) modified zsm-5. Bioresour. Technol. 2014, 155, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhong, Z.; Ding, K.; Xue, Z. Catalytic fast pyrolysis of mushroom waste to upgraded bio-oil products via pre-coked modified hzsm-5 catalyst. Bioresour. Technol. 2016, 212, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhong, Z.-P.; Wang, X.-B.; Ding, K.; Song, Z.-W. Catalytic upgrading of fast pyrolysis biomass vapors over fresh, spent and regenerated zsm-5 zeolites. Fuel Process. Technol. 2015, 138, 430–434. [Google Scholar] [CrossRef]
- Lerici, L.C.; Renzini, M.S.; Sedran, U.; Pierella, L.B. Tertiary recycling of low-density polyethylene by catalytic cracking over zsm-11 and beta zeolites modified with Zn2+: Stability study. Energy Fuels 2013, 27, 2202–2208. [Google Scholar] [CrossRef]
- Marcilla, A.; Beltrán, M.I.; Hernández, F.; Navarro, R. Hzsm5 and husy deactivation during the catalytic pyrolysis of polyethylene. Appl. Catal. A 2004, 278, 37–43. [Google Scholar] [CrossRef]
- Anuar Sharuddin, S.D.; Abnisa, F.; Wan Daud, W.M.A.; Aroua, M.K. A review on pyrolysis of plastic wastes. Energy Convers. Manag. 2016, 115, 308–326. [Google Scholar] [CrossRef]
- Barbarias, I.; Lopez, G.; Artetxe, M.; Arregi, A.; Bilbao, J.; Olazar, M. Valorisation of different waste plastics by pyrolysis and in-line catalytic steam reforming for hydrogen production. Energy Convers. Manag. 2018, 156, 575–584. [Google Scholar] [CrossRef]
- Zeaiter, J. A process study on the pyrolysis of waste polyethylene. Fuel 2014, 133, 276–282. [Google Scholar] [CrossRef]
- Serrano, D.P.; Aguado, J.; Rodríguez, J.M.; Peral, A. Catalytic cracking of polyethylene over nanocrystalline hzsm-5: Catalyst deactivation and regeneration study. J. Anal. Appl. Pyrolysis 2007, 79, 456–464. [Google Scholar] [CrossRef]
- Mullen, C.A.; Dorado, C.; Boateng, A.A. Catalytic co-pyrolysis of switchgrass and polyethylene over hzsm-5: Catalyst deactivation and coke formation. J. Anal. Appl. Pyrolysis 2018, 129, 195–203. [Google Scholar] [CrossRef]
- Vitolo, S.; Bresci, B.; Seggiani, M.; Gallo, M.G. Catalytic upgrading of pyrolytic oils over hzsm-5 zeolite: Behaviour of the catalyst when used in repeated upgrading-regenerating cycles. Fuel 2001, 80, 17–26. [Google Scholar] [CrossRef]
- Yildiz, G.; Lathouwers, T.; Toraman, H.E.; van Geem, K.M.; Marin, G.B.; Ronsse, F.; van Duren, R.; Kersten, S.R.A.; Prins, W. Catalytic fast pyrolysis of pine wood: Effect of successive catalyst regeneration. Energy Fuels 2014, 28, 4560–4572. [Google Scholar] [CrossRef]
- Cheng, S.; Wei, L.; Zhao, X.; Julson, J. Application, deactivation, and regeneration of heterogeneous catalysts in bio-oil upgrading. Catalysts 2016, 6, 195. [Google Scholar] [CrossRef]
- Hellinger, M.; Baier, S.; Mortensen, P.M.; Kleist, W.; Jensen, A.D.; Grunwaldt, J.-D. Continuous catalytic hydrodeoxygenation of guaiacol over pt/SiO2 and pt/h-mfi-90. Catalysts 2015, 5, 1152–1166. [Google Scholar] [CrossRef]
- Shao, S.; Zhang, H.; Xiao, R.; Li, X.; Cai, Y. Controlled regeneration of zsm-5 catalysts in the combined oxygen and steam atmosphere used for catalytic pyrolysis of biomass-derivates. Energy Convers. Manag. 2018, 155, 175–181. [Google Scholar] [CrossRef]
- Yung, M.M.; Starace, A.K.; Griffin, M.B.; Wells, J.D.; Patalano, R.E.; Smith, K.R.; Schaidle, J.A. Restoring zsm-5 performance for catalytic fast pyrolysis of biomass: Effect of regeneration temperature. Catal. Today 2018. [Google Scholar] [CrossRef]
Figure 1.
Strategies for the production of fuels from biomass, reproduced with permission from [7]. Copyright American Chemical Society, 2006.
Figure 1.
Strategies for the production of fuels from biomass, reproduced with permission from [7]. Copyright American Chemical Society, 2006.

Figure 2.
Molecular structures of four β-O-4 lignin dimer structures with different substituents at the Cα and Cβ positions, reproduced with permission from [42], Copyright Elsevier, 2018.
Figure 2.
Molecular structures of four β-O-4 lignin dimer structures with different substituents at the Cα and Cβ positions, reproduced with permission from [42], Copyright Elsevier, 2018.

Figure 3.
Schematic of the pyrolysis system for the catalytic co-pyrolysis of glucose and high density polyethylene (HDPE), reproduced with permission from [252]. Copyright Elsevier, 2018.
Figure 3.
Schematic of the pyrolysis system for the catalytic co-pyrolysis of glucose and high density polyethylene (HDPE), reproduced with permission from [252]. Copyright Elsevier, 2018.

Figure 4.
Schematic layout of feedstock and catalysts for the catalytic co-pyrolysis of bamboo residual and waste lubricating oil, reproduced with permission from [250]. Copyright Elsevier, 2018.
Figure 4.
Schematic layout of feedstock and catalysts for the catalytic co-pyrolysis of bamboo residual and waste lubricating oil, reproduced with permission from [250]. Copyright Elsevier, 2018.


{kind=link}
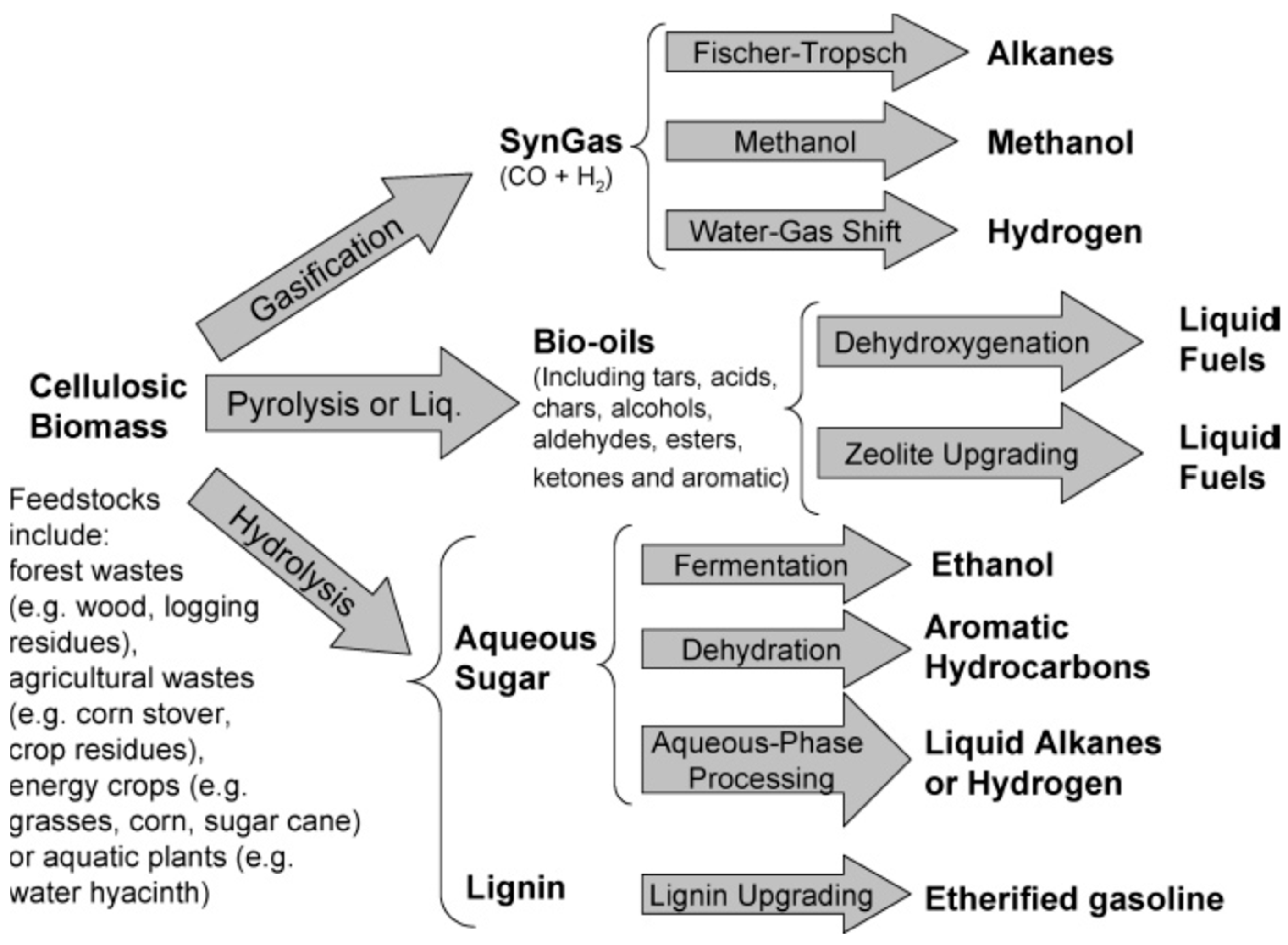{kind=link}
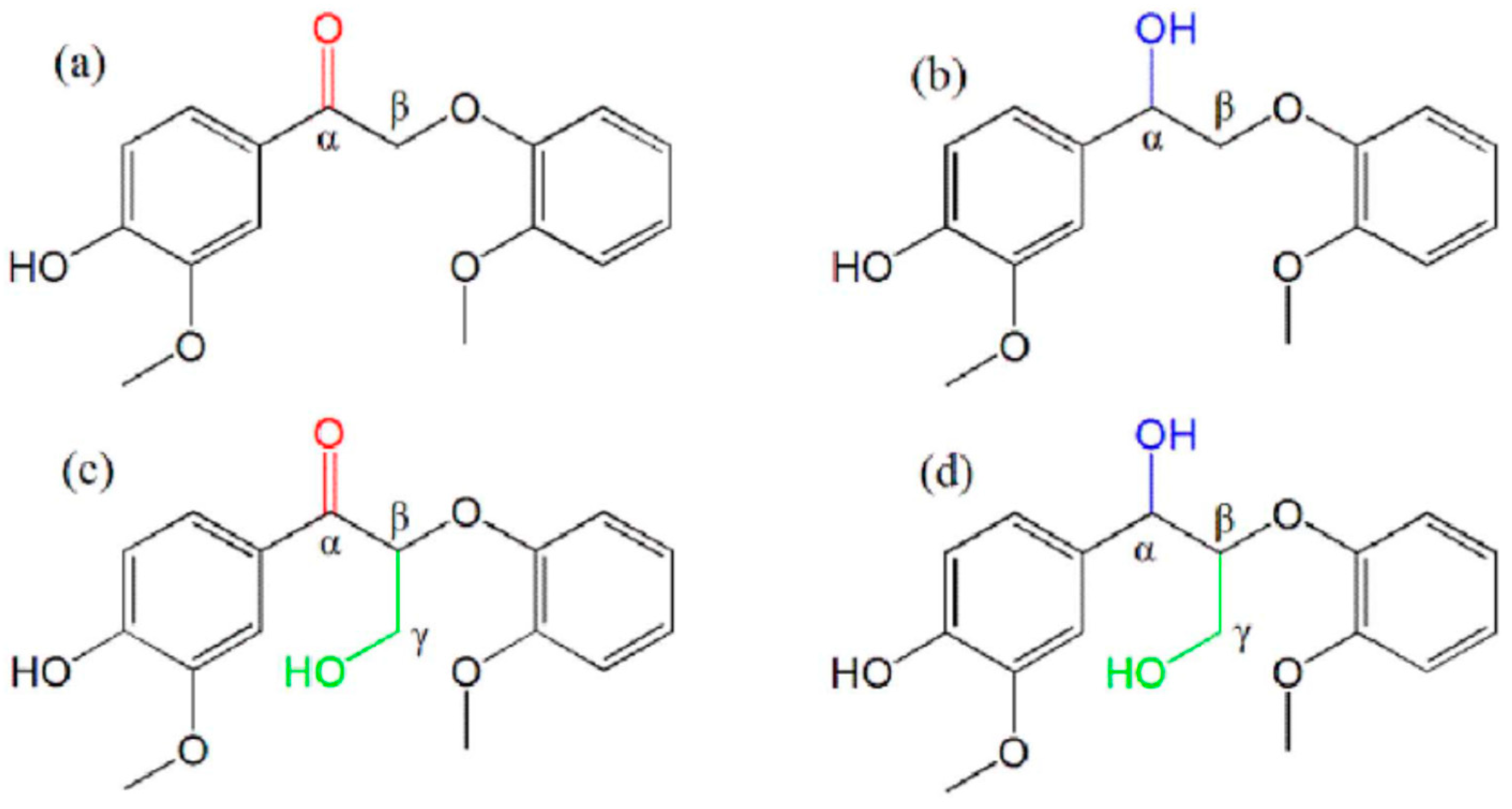{kind=link}
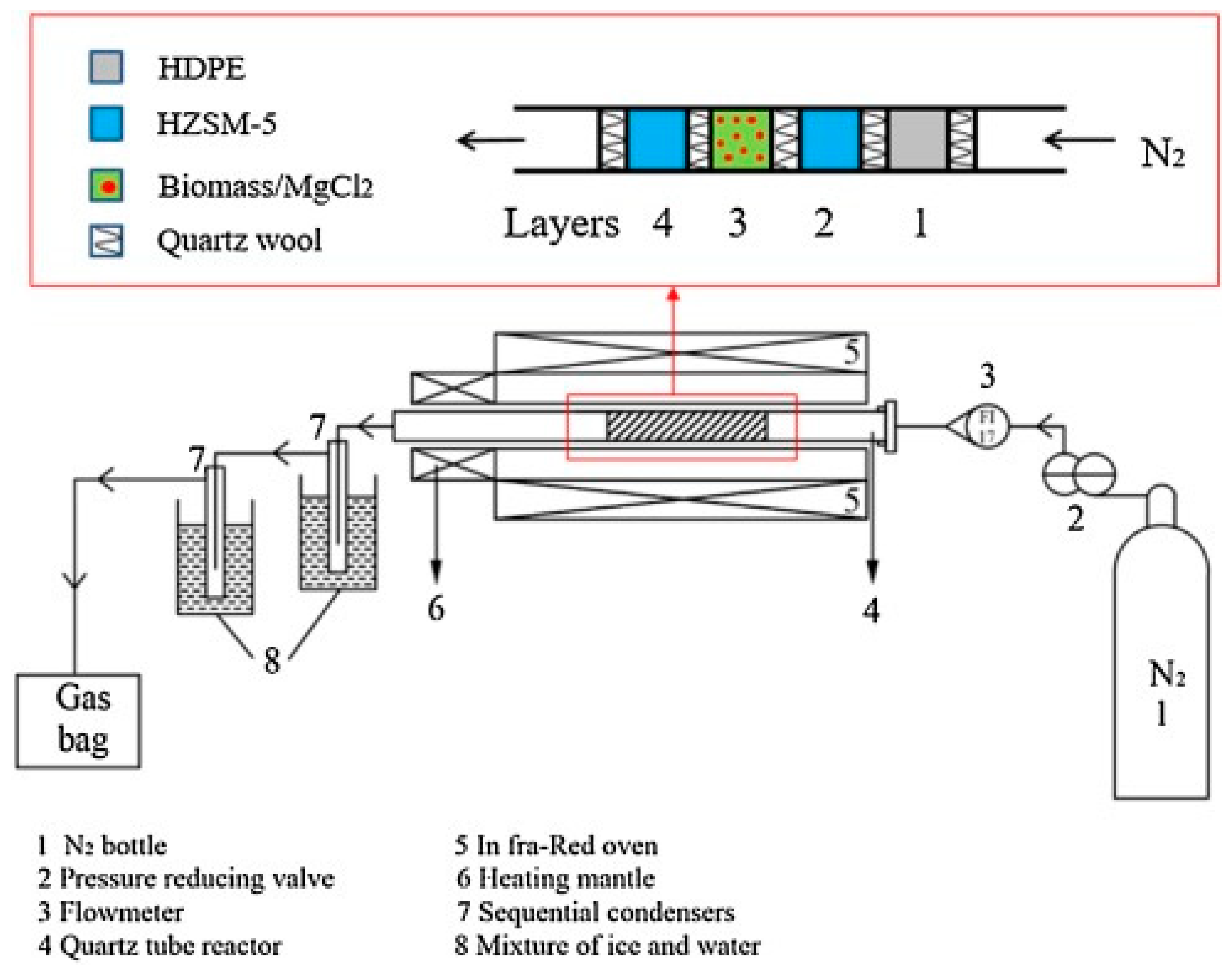{kind=link}
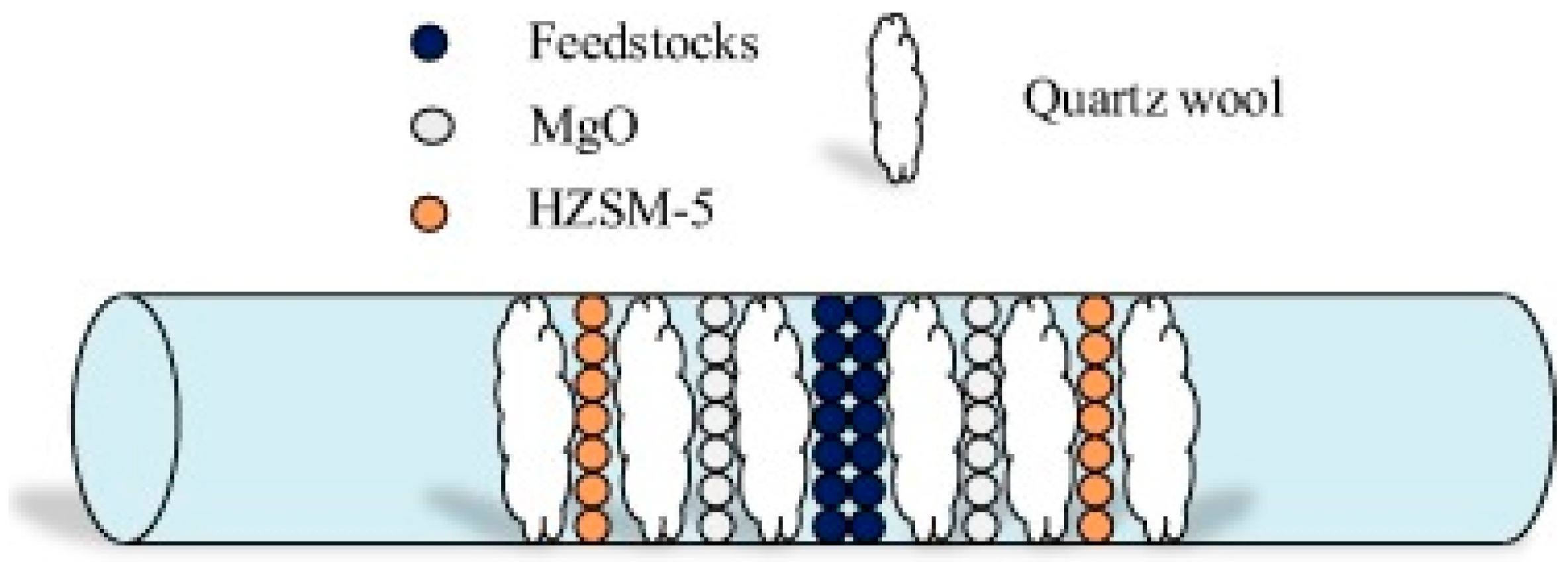{kind=link}
Table 1.
Pore structures of different zeolites used in catalytic fast pyrolysis process.
| Zeolite | Framework Type | Pore Dimensions, Å | Ring Sizes |
|---|---|---|---|
| ZK-5 | KFI | 8-ring 3.9 × 3.9; 8-ring 3.9 × 3.9 | 8, 6, 4 |
| ZSM-23 | MTT | 10-ring 4.5 × 5.2 | 10, 6, 5 |
| Ferrierite | FER | 10-ring 4.2 × 5.4; 8-ring 3.5 × 4.8 | 10, 8, 6, 5 |
| MCM-22 | MWW | 10-ring 4.0 × 5.5; 10-ring 4.1 × 5.1 | 10, 6, 5, 4 |
| ZSM-5 | MFI | 10-ring 5.1 × 5.1; 10-ring 5.3 × 5.6 | 10, 6, 5, 4 |
| ZSM-11 | MEL | 10-ring 5.3 × 5.4 | 10, 8, 6, 5, 4 |
| Y | FAU | 12-ring 7.4 × 7.4 | 12, 6, 4 |
| Beta | BEA | 12-ring 6.6 × 6.7; 12-ring 5.6 × 5.6 | 12, 6, 5, 4 |
| Mordenite | MOR | 12-ring 6.5 × 7.0; 8-ring 3.4 × 4.8; 8-ring 2.6 × >5.7 | 12, 8, 5, 4 |
Data from Chemistry of Zeolites and Related Porous Materials: Synthesis and Structure, Ruren Xu, Wenqin Pang, Jihong Yu, Qisheng Huo, Jiesheng Chen John, Wiley and Sons, 29 May 2009 and http://www.iza-structure.org/databases/.
Table 2.
Metal doped zeolite used for in-bed CFP.
| Catalysts | Pyrolysis Conditions | Key Findings | Ref. |
|---|---|---|---|
| K/ZSM-5 | Catalysts prepared by ion-exchange method. Feedstock: lignin and switch grass. Optimum catalyst/feedstock ratio = 2.5. Pyrolysis temperature = 500 °C. | K/ZSM-5 produced more valuable oxygenated compounds, particularly alkyl phenols and 2-methylfuran than noncatalytic pyrolysis or catalytic pyrolysis with HZSM-5. Low K/ZSM-5 loading favored the formation of 2-methyl furan. | [202] |
| Fe-, Cu-, and Ni/ZSM-5 | Catalysts prepared by ion-exchange method. Feedstock: duckweed and microalgae. Weight hourly space velocity (g feed gas/g cat h) = 13 and 56 h−1. Pyrolysis temperature = 500 °C. | Pyrolytic liquid yields and qualities obtained by metal-modified ZSM-5 were lower than HZSM-5. | [203] |
| Fe/ZSM-5 | Catalysts prepared by impregnation method. Feedstock: wood sawdust. Catalyst/feedstock ratio = 10. Pyrolysis temperature = 500–800 °C. | Fe/ZSM-5 maintained the structure of ZSM-5 and exhibited better activity in the conversion of oxygenates and formation of monocyclic aromatic hydrocarbons (MAHs) than the parent zeolite. | [196] |
| Ce-, Cu-, Fe2+-, Fe3+-Mg, Ni-, Sn- and Zn loaded on ZSM-5 and zeolite Y | Catalysts prepared by impregnation method. Feedstock: waste plastics. Catalyst/feedstock ratio = 1:9. Pyrolysis temperature = 430–500 °C. | Surface areas of zeolites decreased with introduction of metal. The catalyst efficiency increased in the order of Cu < Ce < Mg < Ni < Fe(III) < Fe(II) < Zn < Sn. | [204] |
| Fe/ZSM-5 | Catalyst prepared by impregnation method. Feedstock: hemicelluloses, cellulose, lignin and corn stalk. Catalyst/biomass = 0.5, 1, 2 and 4. Pyrolysis temperature = 600 °C. | Fe/ZSM-5 increased the production of light olefins and carbon yield of light olefins. For CFP of cellulose, 3% Fe/ZSM-5 was optimum. Total light olefins decreased in order of cellulose > corn stalk > hemicelluloses > lignin. The selectivity of C2H4 was more than 60% regardless of feedstock. | [205] |
| P/Ni/ZSM-5 | Catalysts were prepared by sequential impregnation method. Feedstock: pine wood and low-density polyethylene. Catalyst/feedstock ratio = 15. Pyrolysis temperature = 550 °C. | P/Ni-modified ZSM-5 significantly increased the yield of olefins and aromatic hydrocarbons and decreased the yields of low-value alkanes and undesired char/coke, as compared to ZSM-5. P/Ni/ZSM-5 exhibited improved hydrothermal stability and maintained comparable aromatic yields in co-feed CFP for up to 9 h. | [206] |
| Ni-, Co-, Mo-, Ga-, and Pd/HZSM-5 | Catalysts prepared by ion-exchange and impregnation methods. Feedstock: Jatropha residues. Biomass/catalyst ratios = 1:1, 1:5, and 1:10. Pyrolysis temperature = 500 °C. | Metal/HZSM-5 reduced contents of oxygenates (0.6–4.0%) and nitrogenates (1.8–4.6%). Aromatic selectivity was significantly increased up to 91–97%. The catalysts produced by metal-ion exchange showed slightly higher aromatic selectivity than the impregnated ones. | [207] |
| Ni-, Co-, Mo-, and Pt/HZSM-5 | Catalysts prepared by impregnation method. Pyrolysis hydrogen pressure ranged from 100 to 400 psi. Feedstock: pine wood. Biomass/catalyst ratios = 1:9. Pyrolysis temperature = 650 °C. | Except for Mo/HZSM-5, the aromatic hydrocarbons yield was not affected by hydrogen pressure. The presence of metal increased aromatic yield. Metal has no obvious effect on the aromatic selectivity. | [208] |
| Ni-, Zn-, and Ga/HZSM-5 | Catalyst prepared with Ga by total ion exchange. Catalysts prepared by ion exchange method. Feed stock: Eucalyptus urophylla. Biomass/catalyst ratios = 1:5 and 1:10. Pyrolysis temperature = 600 °C. | Ga-HZSM-5 increased the yield of aromatic hydrocarbons while Ni- and Zn- modified catalysts decreased the yield. Ga-HZSM-5 exhibited high selectivity toward xylenes. Ni/HZSM-5 produced more methane. Zn/HZSM-5 was the most selective for toluene production. | [199] |
| Fe/HZSM-5 | Catalysts prepared by ion exchange method. Feedstock: cellulose; cellobiose and lignin. Biomass/catalyst ratios = 1:5 and 1:10. Pyrolysis temperature = 500 °C. | Fe/HZSM-5 increased the yield of aromatic hydrocarbons for all three feedstocks examined. Fe-HZSM-5 favored the formation of naphthalenes while the selectivities toward p-xylene, ethylbenzene and trimethylbenzene decreased. | [209] |
| Mg-Al/MCM-41 | Catalysts prepared by impregnation method. Feedstock: cellulose and lignin. Biomass/catalyst ratios = 0.4:1. Pyrolysis temperature = 600 °C. | Mg/Al/MCM-41 presented high selectivity to aromatic hydrocarbons. Highest catalytic activity and stable reusability exhibited by 1 wt % Mg/Al-MCM-41. | [210] |
| K-, Na-, Mg-, Ce-, Cu-, and Fe/ultra-stable zeolite Y | Catalysts prepared by impregnation method. Feedstock: coal and cedar wood. Biomass/catalyst ratios = 1:1. Pyrolysis temperature = 600 °C. | Introduction of metal mitigated the deposition of coke on the zeolite surface. Mg/USY promoted hydrocarbon formation and showed good stability. | [211] |
Table 3.
Metal doped zeolites used for in-situ catalytic vapor upgrading.
| Catalysts | Pyrolysis Conditions | Key Findings | Ref. |
|---|---|---|---|
| Zn-, Ga-, Ni-, Co-, Mg- and Cu/ZSM-5 | Catalysts prepared by impregnation method. Feedstock: Yunnan pine. Biomass/catalyst ratio = 1:2. Pyrolysis temperature = 450 °C. Catalytic upgrading temperature = 500 °C. | Presence of M-ZSM-5 decreased oil yields and increased non-condensable gas amount. Ga/ZSM-5 produced the highest oil yields and the lowest amount of coke. Zn/ZSM-5 favored the formation of single-ring aromatics especially toluene and xylenes. Ni/ZSM-5 yielded more polycyclic aromatic hydrocarbons, e.g., C10+ polycyclic aromatic hydrocarbons. Co/ ZSM-5 was the most selective for indene production. | [200] |
| Ni- and Co/ZSM-5 | Catalysts prepared by wet impregnation method. Feedstock: Lignocel HBS 150–500. Catalyst/feedstock ratio = 1.5/0.7. Pyrolysis temperature = 500 °C. | Reduced metallic Ni and Co formed during pyrolysis which favor hydrogen transfer reactions. Produced oil was enriched in aromatics and phenols. NiO/ZSM-5 was more reactive than Co3O4/ZSM-5 in decreasing the organic phase and increasing the gaseous products. | [212] |
| Fe/ZSM-5 | Catalysts prepared by impregnation method. Feedstock: beech sawdust. Catalyst/biomass ratio = 0.5/5. Pyrolysis and catalytic upgrading temperature = 500 °C. | Bio-oil yields decreased in the presence of catalysts. Fe/ZSM-5 catalyst reveal a significant enhancement quality of the pyrolysis products in comparison with non-catalytic experiment. Catalyst increased oxygen removal from the organic phase of bio-oil and further developed the production of desirable products such as phenolics and aromatic compounds. | [197] |
| Fe/ZSM-5 | Catalysts prepared by wet impregnation method. Feedstock: rice husk. Catalyst/biomass ratio = 5:1. Pyrolysis temperature = 600 °C. Pyrolysis and catalytic upgrading temperature = 550 °C. | Increased Fe loading increased hydrocarbons content at the expense of decreasing yield of bio-oil. Optimum was 4% Fe/ZSM-5 to obtain high yield of upgraded bio-oil. | [213] |
| Ce/HZSM-5 | Catalysts prepared by impregnation method. Feedstock: sugarcane bagasse. Catalyst/biomass ratio = 1.5, 1, 1.5 and 2. Pyrolysis and catalytic upgrading temperature = 500 °C. | Introduction of Ce increased pyrolysis oil yield and decreased coke yield, in comparison to HZSM-5. The optimal ratio of 1.5/1 catalyst/biomass produced an 2.45% content of C6–C8 hydrocarbons. | [214] |
| Co, Ni, Mo, Ga, and Pd-modified HZSM-5 | Catalysts prepared by both ion-exchange and wet impregnation methods. Feedstock: Jatropha residues. Biomass/catalyst ratio = 1:1, 1:5, and 1:10. Pyrolysis and catalytic upgrading temperature = 500 °C. | Catalysts enhanced hydrocarbon production, particularly aromatics, and reduced oxygen and nitrogen containing compound contents. Mo/HZSM-5 showed the highest aromatics (97%) with low PAHs selectivity. | [207] |
| Ni-ZSM-5 | Catalysts was prepared by ion-exchange method. Feedstock: rubber waste. Rubber/catalyst ratio = 1:1, 1:5 and 1:10. Pyrolysis and catalytic upgrading temperature = 500 °C. | Catalyst enhanced the aromatic production, especially benzene, toluene and xylene compounds. | [215] |
| Co, Mo–Co and Ni–Co modified HZSM-5 | Catalysts prepared by impregnation method. Feedstock: lignite. Pyrolysis and catalytic upgrading temperature = 600 °C. | Light aromatics (e.g., benzene, toluene, ethylbenzene, xylene and naphthalene) production was significantly increased by Mo–Co or Ni–Co/HZSM-5. Yields of benzene, ethylbenzene and m, p-xylene followed the order Ni/Co-HZSM-5 >Mo/Co-HZSM-5 > Co-HZSM-5 > HZSM-5. Further NaOH treatment of bimetallic catalysts inhibited the coke formation. | [216] |
| Fe- Zr-, and Co- HZSM-5 | Catalysts were prepared by impregnation method. Feedstock: Pine sawdust. Catalyst/biomass ratio = 0.5. Pyrolysis temperature 550 °C. Catalytic upgrading temperature = 450–650 °C. | Fe- and Zr/HZSM-5 produced over 45% aromatic hydrocarbons in the resulting bio-oil. However, Co/HZSM-5 performed worse than HZSM-5 by producing the highest gas and coke yields. Zr/HZSM-5 enhanced the formation of benzene and its derivatives. Fe/HZSM-5 produced more naphthalene and its derivatives, and increased Fe loading facilitated deoxygenation and hydrocarbon formation. | [217] |
| Co/HZSM-5 | Catalysts prepared by impregnation method. Feedstock: Baiyinhua lignite. Catalyst/ lignite ratio = 1. Pyrolysis temperature = 400 to 700 °C. Catalytic upgrading temperature = 600 °C. | A significant reduction of the oxygen content and increase in total content of light aromatics was observed for 3% Co-HZSM5 at 600°C. | [218] |
| Fe-, Ce-, La-, Cu-, and Cr- Mo2N/HZSM-5 | Catalysts prepared by incipient wetness impregnation method. Feedstock: microcrystalline cellulose, xylan, pine wood and wood lignin. Catalyst/biomass ratio = 1, 3, 5, 7. Pyrolysis and catalytic upgrading temperature = 550–850 °C. | Both Mo2N/HZSM-5 and metal modified catalysts increased production of monocyclic aromatic hydrocarbons (MAHs) while decreasing the formation polycyclic aromatic hydrocarbons (PAHs), as compared to HZSM-5. Most effective in enhancing formation of MAHs and restraining the generation of PAHs was 10% Ce-Mo2N/HZSM-5. | [219] |
| Cu–Al-MCM-41 | Cu was introduced during the synthesis of Al-MCM-41. Feedstock: spruce wood and Miscanthus. Pyrolysis and catalytic upgrading temperature = 500 °C. | Catalyst produced more desirable compounds than the uncatalyzed sample, and the organic phase yield was increased close to the non-catalytic level. | [220] |
| Cu-Al, Fe-Al, and Zn-Al/MCM-41 | Catalysts prepared by ion-exchange. Feedstock: barkless spruce wood. Biomass/catalyst ratio = 1:5/0.7. Pyrolysis and catalytic upgrading temperature = 500 °C. | All catalysts, especially Fe–Al and Cu–Al-MCM-41 increased the yield of phenolic compounds. A low Si/Al ratio of MCM-41 was beneficial to improve the product yield and quality. | [221] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, L.; Bao, Z.; Xia, S.; Lu, Q.; Walters, K.B. Catalytic Pyrolysis of Biomass and Polymer Wastes. Catalysts 2018, 8, 659. https://doi.org/10.3390/catal8120659
AMA Style
Zhang L, Bao Z, Xia S, Lu Q, Walters KB. Catalytic Pyrolysis of Biomass and Polymer Wastes. Catalysts. 2018; 8(12):659. https://doi.org/10.3390/catal8120659
Chicago/Turabian StyleZhang, Laibao, Zhenghong Bao, Shunxiang Xia, Qiang Lu, and Keisha B. Walters. 2018. "Catalytic Pyrolysis of Biomass and Polymer Wastes" Catalysts 8, no. 12: 659. https://doi.org/10.3390/catal8120659
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.