Ag-Based Catalysts in Heterogeneous Selective Oxidation of Alcohols: A Review

Abstract
:1. Introduction
2. Heterogeneous Gas-Phase Selective Oxidation
2.1. Monoalcohols
2.1.1. Methanol
2.1.2. Ethanol
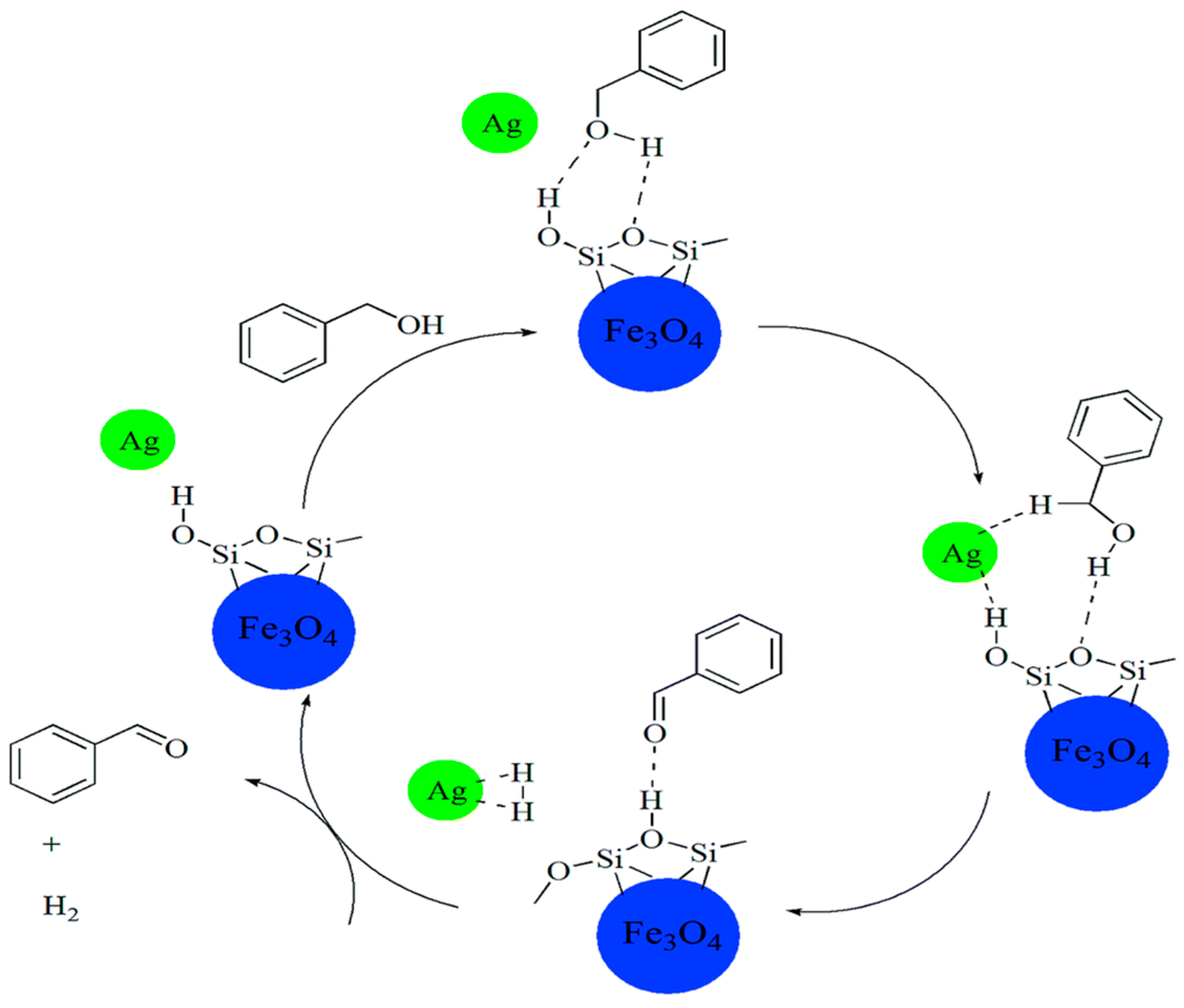
2.1.3. Benzyl Alcohol
2.1.4. Allyl Alcohol
2.2. Polyalcohols
2.2.1. Ethylene Glycol
2.2.2. Propylene Glycol
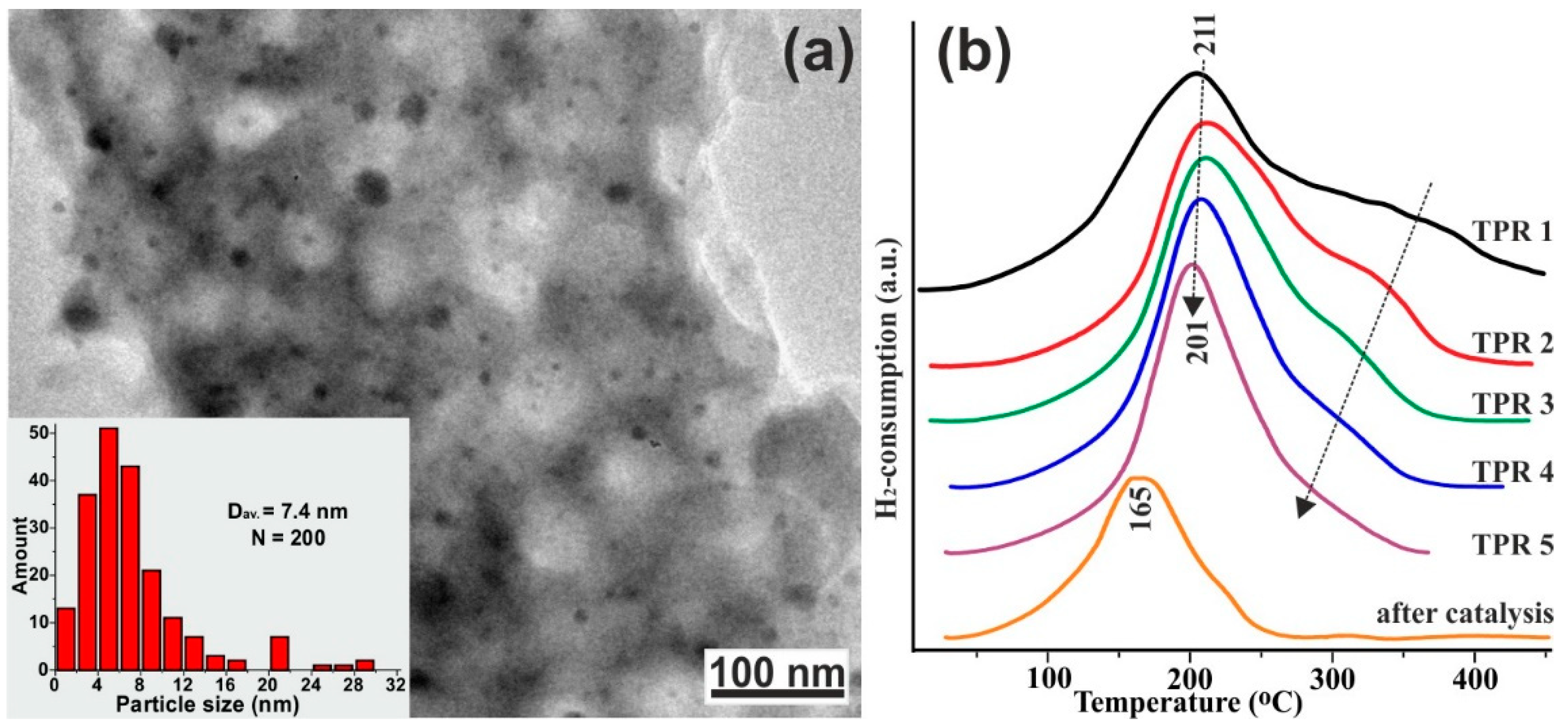
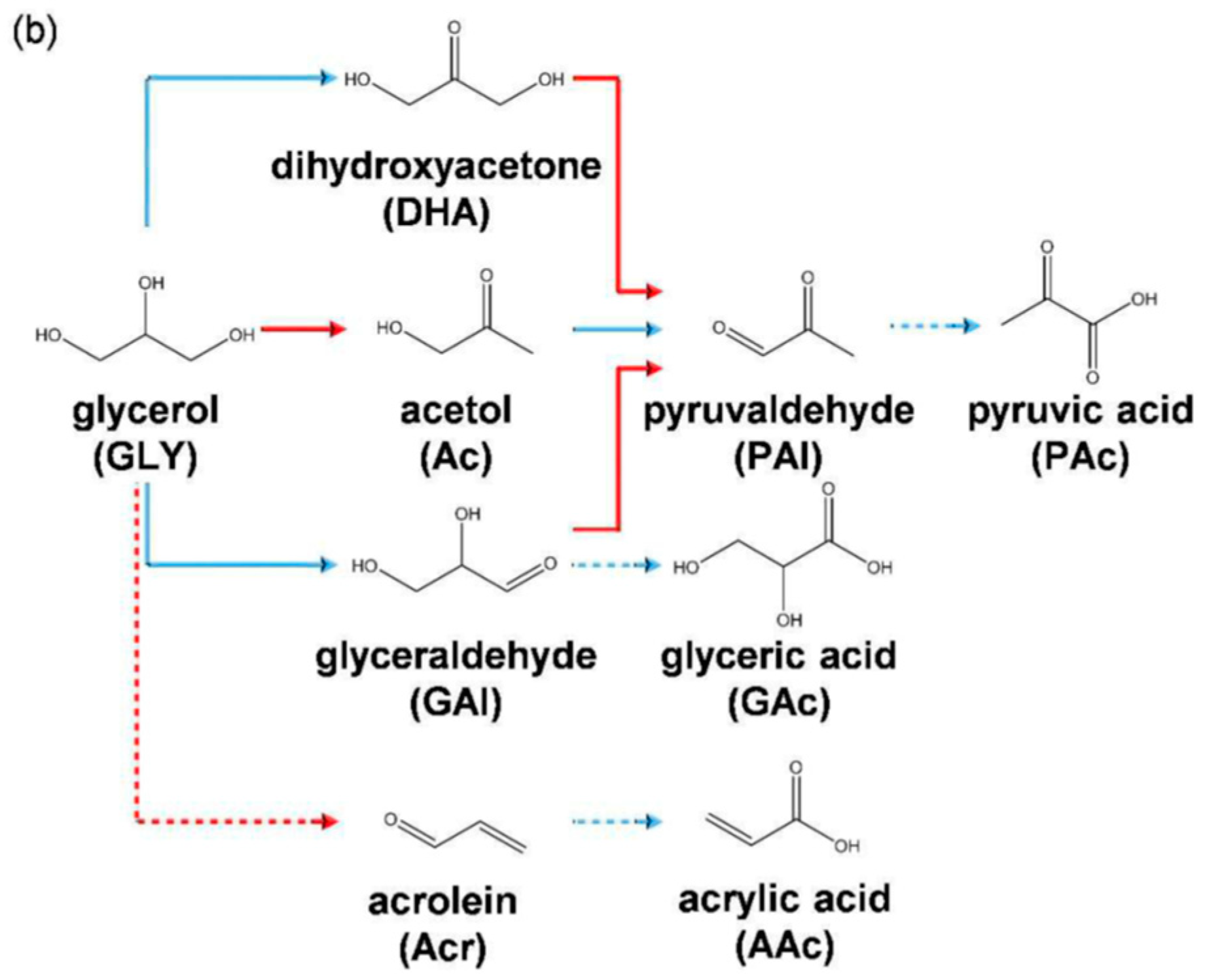
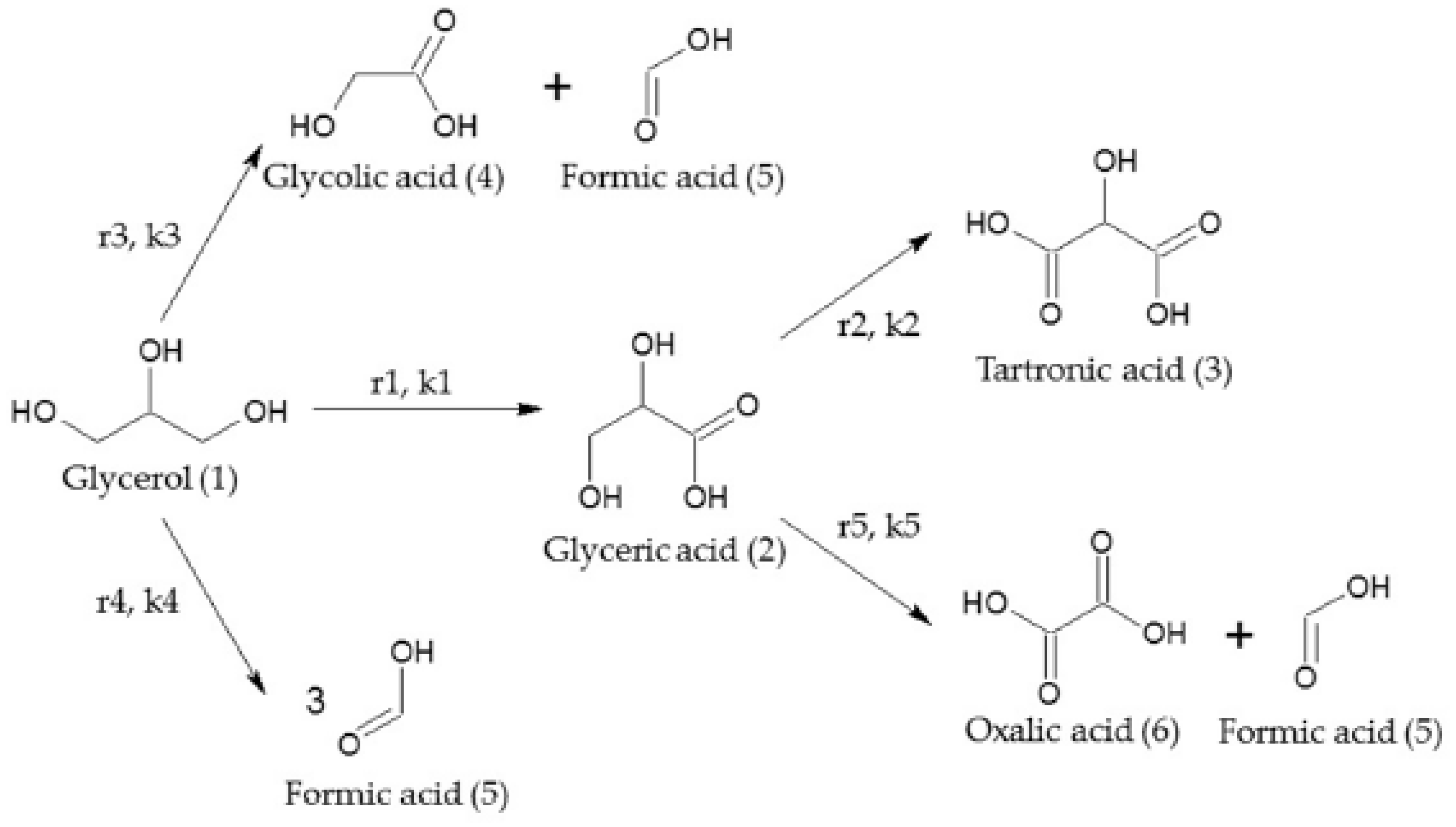
2.2.3. Glycerol
3. Heterogeneous Liquid-Phase Selective Oxidation
3.1. Benzyl Alcohol
3.2. Propylene Glycol
3.3. Glycerol
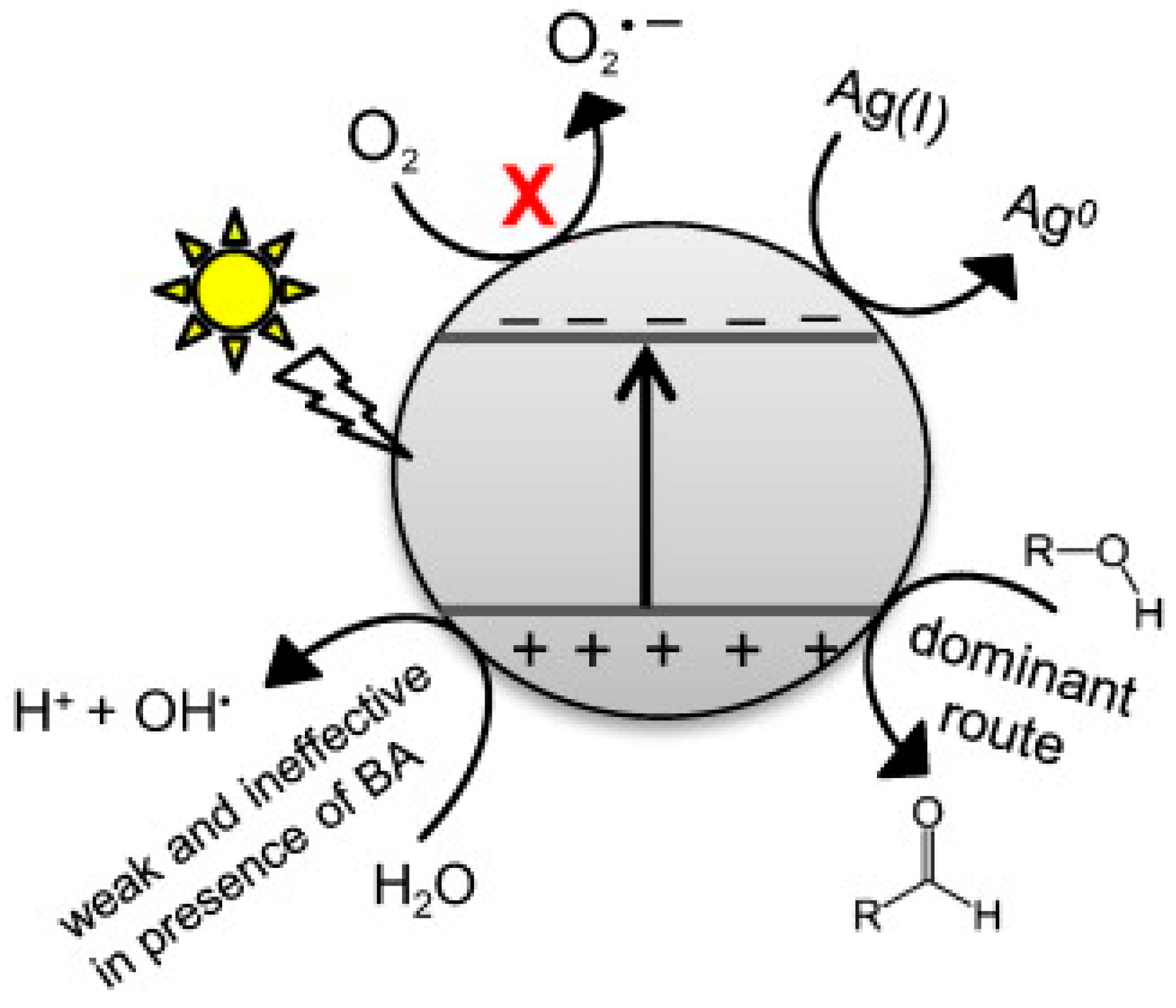
4. Selective Photooxidation of Alcohols
5. Conclusions and Outlook
- (1)
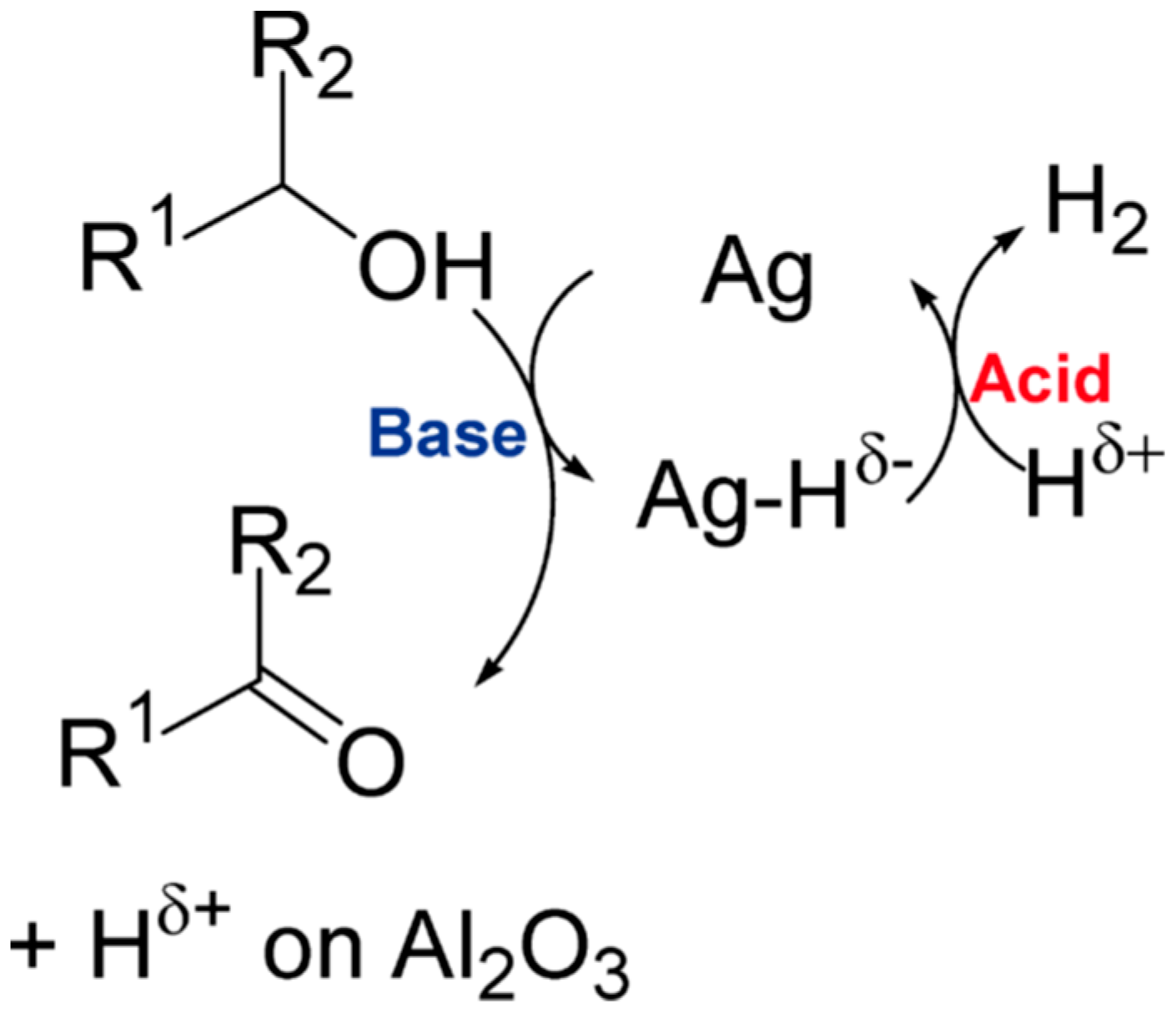
- the stabilization of the Ag-containing species (ultrasmall nanoparticles, clusters, ions) over the support surface due to balancing of its redox and acid-base properties,
- (2)
- the creating of the required “optimal” chemical surrounding for the metal site able to participate in the adsorption of the substrates, while the molecular oxygen is activated over the metal site,
- (3)
- the reducing of the process temperature, while keeping both high activity and selectivity of the catalysts, preventing the undesired processes (e.g., sintering, formation of carbon deposits, etc.),
- (4)
- the formation of surface oxygen species that promote the activation and transformation of the alcohol molecule, including the formation of O2−• species in photocatalytic applications.
Acknowledgments
Funding
Conflicts of Interest
Abbreviations
| Ac | acetol |
| AcA | acetic acid |
| BA | benzoic acid |
| BAc | benzoic acid |
| BAld | benzaldehyde |
| BB | benzyl benzoate |
| DHA | dihydroxyacetone |
| EG | ethylene glycol |
| FA | formic acid |
| GLY | glycerol |
| GlyA | glyceric acid |
| GlyAl | glyceraldehydes |
| GlyAld | glycolic aldehyde |
| GlycA | glycolic acid |
| GO | glyoxal |
| GOA | glyoxalic acid |
| HA | hydroxyacetone |
| LA | lactic acid |
| LAld | lactaldehyde |
| MCF | mesostructured cellular foams |
| MF | methyl formate |
| MeGO | methyl glyoxal |
| NPs | nanoparticles |
| PG | propylene glycol |
| PVP | polyvinyl pyrrolidone |
| SS | stainless steel |
| ZFC | zeolite film coated copper grid |
References
- Kopylovich, M.N.; Ribeiro, A.P.C.; Alegria, E.C.B.A.; Martins, N.M.R.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Chapter Three—Catalytic Oxidation of Alcohols: Recent Advances. Adv. Organomet. Chem. 2015, 63, 91–174. [Google Scholar] [CrossRef]
- Mobley, J.K.; Crocker, M. Catalytic oxidation of alcohols to carbonyl compounds over hydrotalcite and hydrotalcite-supported catalysts. RSC Adv. 2015, 5, 65780–65797. [Google Scholar] [CrossRef]
- Ciriminna, R.; Pandarus, V.; Béland, F.; Xu, Y.-J.; Pagliaro, M. Heterogeneously Catalyzed Alcohol Oxidation for the Fine Chemical Industry. Org. Process. Res. Dev. 2015, 19, 1554–1558. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, C.; Li, H.; Zhao, X.; Song, L.; Li, X. An Overview of Selective Oxidation of Alcohols: Catalysts, Oxidants and Reaction Mechanisms. Catal. Surv. Asia 2016, 20, 13–22. [Google Scholar] [CrossRef]
- Sharma, A.; Kaur, H.; Shah, D. Selective Oxidation of Alcohols by Supported Gold Nanoparticles: Recent Advances. RSC Adv. 2016, 6, 28688–28727. [Google Scholar] [CrossRef]
- Hussain, M.A.; Joseph, N.; Kang, O.; Cho, Y.-H.; Um, B.-H.; Kim, J.W. Supported Metal Nanoparticles: Their Catalytic Applications to Selective Alcohol Oxidation. Appl. Chem. Eng. 2016, 27, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Vinod, C.P.; Wilson, K.; Lee, A.F. Recent advances in the heterogeneously catalysed aerobic selective oxidation of alcohols. J. Chem. Technol. Biotechnol. 2011, 86, 161–171. [Google Scholar] [CrossRef]
- Liu, F.; Wang, H.; Sapi, A.; Tatsumi, H.; Zherebetskyy, D.; Han, H.-L.; Carl, L.M.; Somorjai, G.A. Molecular orientations change reaction kinetics and mechanism: A review on catalytic alcohol oxidation in gas phase and liquid phase on size-controlled Pt nanoparticles. Catalysts 2018, 8, 226. [Google Scholar] [CrossRef]
- Parmeggiani, C.; Cardona, F. Transition metal based catalysts in the aerobic oxidation of alcohols. Green Chem. 2012, 14, 547–564. [Google Scholar] [CrossRef]
- Ochoa, J.V.; Cavani, F. Gas-phase oxidation of alcohols: Innovation in industrial technologies and recent developments. RSC Green Chem. 2015, 28, 203–230. [Google Scholar] [CrossRef]
- Dong, X.-Y.; Gao, Z.-W.; Yang, K.-F.; Zhang, W.-Q.; Xu, L.-W. Nanosilver as a new generation of silver catalysts in organic transformations for efficient synthesis of fine chemicals. Catal. Sci. Technol. 2015, 5, 2554–2574. [Google Scholar] [CrossRef]
- Wen, C.; Yin, A.; Dai, W.-L. Recent advances in silver-based heterogeneous catalysts for green chemistry processes. Appl. Catal. B 2014, 160–161, 730–741. [Google Scholar] [CrossRef]
- Davis, S.E.; Ide, M.S.; Davis, R.J. Selective oxidation of alcohols and aldehydes over supported metal nanoparticles. Green Chem. 2013, 15, 17–45. [Google Scholar] [CrossRef]
- Vodyankina, O.V.; Mamontov, G.V.; Dutov, V.V.; Kharlamova, T.S.; Salaev, M.A. Ag-containing nanomaterials in heterogeneous catalysis: Advances and Recent Trends. In Advanced Nanomaterials for Catalysis and Energy: Synthesis, Characterization and Applications, 1st ed.; Sadykov, V., Ed.; Elsevier: New York, NY, USA, 2019; pp. 145–175. [Google Scholar]
- Wisniewska, J.; Yang, C.-M.; Ziolek, M. Changes in bimetallic silver—Platinum catalysts during activation and oxidation of methanol and propene. Catal. Today 2018, in press. [Google Scholar] [CrossRef]
- Abdel Dayem, H.M.; Al-Shihry, S.S.; Hassan, S.A. Selective Methanol Oxidation to Hydrogen over Ag/ZnO Catalysts Doped with Mono- and Bi-Rare Earth Oxides. Ind. Eng. Chem. Res. 2014, 53, 19884–19894. [Google Scholar] [CrossRef]
- Ferrizz, R.M.; Wong, G.S.; Egami, T.; Vohs, J.M. Structure Sensitivity of the Reaction of Methanol on Ceria. Langmuir 2001, 17, 2464–2470. [Google Scholar] [CrossRef]
- Bahmanpour, A.M.; Hoadley, A.; Tanksale, A. Critical review and exergy analysis of formaldehyde production processes. Rev. Chem. Eng. 2014, 30, 583–604. [Google Scholar] [CrossRef]
- Brookes, C.; Bowker, M.; Wells, P.P. Catalysts for the Selective Oxidation of Methanol. Catalysts 2016, 6, 92. [Google Scholar] [CrossRef]
- Ghahraloud, H.; Farsi, M. Modeling and optimization of methanol oxidation over metal oxide catalyst in an industrial fixed bed reactor. J. Taiwan Inst. Chem. Eng. 2017, 81, 95–103. [Google Scholar] [CrossRef]
- Qian, M.; Liauw, M.A.; Emig, G. Formaldehyde synthesis from methanol over silver catalysts. Appl. Catal. A 2003, 238, 211–222. [Google Scholar] [CrossRef]
- Waterhouse, G.I.N.; Bowmaker, G.A.; Metson, J.B. Mechanism and active sites for the partial oxidation of methanol to formaldehyde over an electrolytic silver catalyst. Appl. Catal. A 2004, 265, 85–101. [Google Scholar] [CrossRef]
- Millar, G.J.; Collins, M. Industrial Production of Formaldehyde Using Polycrystalline Silver Catalyst. Ind. Eng. Chem. Res. 2017, 56, 9247–9265. [Google Scholar] [CrossRef]
- Pestryakov, A.N. Modification of silver catalysts for oxidation of methanol to formaldehyde. Catal. Today 1996, 28, 239–244. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Farrauto, R.J. Fundamentals of Industrial Catalytic Processes, 2nd ed.; John Wiley and Sons: Hoboken, NJ, USA, 2010; pp. 1–966. [Google Scholar]
- Millar, G.J.; Metson, J.B.; Bowmaker, G.A.; Cooney, R.P. Characterization of the Active Site for the Selective Oxidation of Methanol to Formaldehyde on Polycrystalline Silver Catalyst. J. Chem. Soc. Chem. Commun. 1994, 1717–1718. [Google Scholar] [CrossRef]
- Huang, W.; Sun, G.; Cao, T. Surface chemistry of group IB metals and related oxides. Chem. Soc. Rev. 2017, 46, 1977–2000. [Google Scholar] [CrossRef] [PubMed]
- Galvanin, F.; Cao, E.; Al-Rifai, N.; Dua, V.; Gavriilidis, A. Optimal design of experiments for the identification of kinetic models of methanol oxidation over silver catalyst. Chim. Oggi-Chem. Today 2015, 33, 51–56. [Google Scholar]
- Filho, A.C.P.; Filho, R.M. Hybrid training approach for artificial neural networks using genetic algorithms for rate of reaction estimation: Application to industrial methanol oxidation to formaldehyde on silver catalyst. Chem. Eng. J. 2010, 157, 501–508. [Google Scholar] [CrossRef]
- Aouat, Y.; Marom, G.; Avnir, D.; Gelman, V.; Shter, G.E.; Grader, G.S. Organically Doped Silver Nanoparticles Deposited on Titania Nanofibers: Enhanced Catalytic Methanol Oxidation. J. Phys. Chem. C 2013, 117, 22325–22330. [Google Scholar] [CrossRef]
- Binyamin, Y.; Shter, G.E.; Gelman, V.; Avnir, D.; Grader, G.S. Activated organically doped silver: Enhanced catalysis of methanol oxidation. Catal. Sci. Technol. 2011, 1, 1593–1599. [Google Scholar] [CrossRef]
- Halperin, V.; Shter, G.E.; Gelman, V.; Peselev, D.M.; Mann-Lahav, M.; Grader, G.S. Catalytic activity of electrospun Ag and Ag/carbon composite fibres in partial methanol oxidation. Catal. Sci. Technol. 2015, 5, 1153–1162. [Google Scholar] [CrossRef]
- Sobczak, I.; Dembowiak, E. The effect of AuAg-MCF and AuAg-NbMCF catalysts pretreatment on the gold-silver alloy formation and the catalytic behavior in selective methanol oxidation with oxygen. J. Mol. Catal. A 2015, 409, 137–148. [Google Scholar] [CrossRef]
- Wisniewska, J.; Ziolek, M. Formation of Pt-Ag alloy on different silicas-surface properties and catalytic activity in oxidation of methanol. RSC Adv. 2017, 7, 9534–9544. [Google Scholar] [CrossRef]
- Wisniewska, J.; Ziolek, M.; Artioli, N.; Daturi, M. The effect of niobium and tantalum on physicochemical and catalytic properties of silver and platinum catalysts based on MCF mesoporous cellular foams. J. Catal. 2016, 336, 58–75. [Google Scholar] [CrossRef]
- Aljama, H.; Yoo, J.S.; Nørskov, J.K.; Abild-Pedersen, F.; Studt, F. Methanol partial oxidation on Ag(111) from first principles. ChemCatChem. 2016, 8, 3621–3625. [Google Scholar] [CrossRef]
- Montoya, A.; Haynes, B.S. Methanol and Methoxide Decomposition on Silver. J. Phys. Chem. C 2007, 111, 9867–9876. [Google Scholar] [CrossRef]
- Montoya, A.; Haynes, B.S. DFT analysis of the reaction paths of formaldehyde decomposition on silver. J. Phys. Chem. A 2009, 113, 8125–8131. [Google Scholar] [CrossRef] [PubMed]
- Shirman, T.; Lattimer, J.; Luneau, M.; Shirman, E.; Reece, C.; Aizenberg, M.; Madix, R.J.; Aizenberg, J.; Friend, C.M. New Architectures for Designed Catalysts: Selective Oxidation using AgAu Nanoparticles on Colloid-Templated Silica. Chem. Eur. J. 2018, 24, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Nadeema, M.A.; Idriss, H. Photo-thermal reactions of ethanol over Ag/TiO2 catalysts. The role of silver plasmon resonance in the reaction kinetics. Chem. Commun. 2018, 54, 5197–5200. [Google Scholar] [CrossRef] [PubMed]
- Pomalaza, G.; Capron, M.; Ordomsky, V.; Dumeignil, F. Recent Break throughs in the Conversion of Ethanol to Butadiene. Catalysts 2016, 6, 203. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Ivanova, I.I.; Ordomsky, V.V.; Taarning, E. Design of a metal-promoted oxide catalyst for the selective synthesis of butadiene from ethanol. ChemSusChem 2014, 7, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.P.; da Costa Zonetti, P.; Appel, L.G. Chemicals from ethanol: The acetone synthesis from ethanol employing Ce0.75Zr0.25O2, ZrO2 and Cu/ZnO/Al2O3. Chem. Cent. J. 2017, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Aditiya, H.B.; Mahlia, T.M.I.; Chong, W.T.; Nur, H.; Sebayang, A.H. Second generation bioethanol production: A critical review. Renew. Sustain. Energy Rev. 2016, 66, 631–653. [Google Scholar] [CrossRef]
- Rana, P.H.; Parikh, P.A. Bioethanol selective oxidation to acetaldehyde over Ag-CeO2: Role of metal-support interactions. New J. Chem. 2017, 41, 2636–2641. [Google Scholar] [CrossRef]
- Rana, P.H.; Parikh, P.A. Bioethanol valorization via its gas phase oxidation over Au &/or Ag supported on various oxides. J. Ind. Eng. Chem. 2017, 47, 228–235. [Google Scholar] [CrossRef]
- Guan, Y.; Hensen, E.J.M. Selective oxidation of ethanol to acetaldehyde by Au−Ir catalysts. J. Catal. 2013, 305, 135–145. [Google Scholar] [CrossRef]
- Sobolev, V.I.; Danilevich, E.V.; Koltunov, K.Y. Role of vanadium species in the selective oxidation of ethanol on V2O5/TiO2 catalysts. Kinet. Catal. 2013, 54, 730–734. [Google Scholar] [CrossRef]
- Silbaugh, T.L.; Devlaminck, P.; Sofranko, J.A.; Barteau, M.A. Selective oxidation of ethanol over Ag, Cu and Au nanoparticles supported on Li2O/γ-Al2O3. J. Catal. 2018, 364, 40–47. [Google Scholar] [CrossRef]
- Lippits, M.J.; Nieuwenhuys, B.E. Direct conversion of ethanol into ethylene oxide on gold-based catalysts: Effect of CeOx and Li2O addition on the selectivity. J. Catal. 2010, 274, 142–149. [Google Scholar] [CrossRef]
- Lippits, M.J.; Nieuwenhuys, B.E. Direct conversion of ethanol into ethylene oxide on copper and silver nanoparticles: Effect of addition of CeOx and Li2O. Catal. Today 2010, 154, 127–132. [Google Scholar] [CrossRef]
- Janlamool, J.; Jongsomjit, B. Oxidative dehydrogenation of ethanol over AgLi–Al2O3 catalysts containing different phases of alumina. Catal. Commun. 2015, 70, 49–52. [Google Scholar] [CrossRef]
- Shimizu, K.-I.; Satsuma, A. Silver Cluster Catalysts for Green Organic Synthesis. J. Jpn. Petrol. Inst. 2011, 54, 347–360. [Google Scholar] [CrossRef] [Green Version]
- Kolobova, E.; Pestryakov, A.; Mamontov, G.; Kotolevich, Y.; Bogdanchikova, N.; Farias, M.; Vosmerikov, A.; Vosmerikova, L.; Cortes Corberan, V. Low-temperature CO oxidation on Ag/ZSM-5 catalysts: Influence of Si/Al ratio and redox pretreatments on formation of silver active sites. Fuel 2017, 188, 121–131. [Google Scholar] [CrossRef]
- Xu, J.; Xu, X.-C.; Yang, X.-J.; Han, Y.-F. Silver/hydroxyapatite foam as a highly selective catalyst for acetaldehyde production via ethanol oxidation. Catal. Today 2016, 276, 19–27. [Google Scholar] [CrossRef]
- Blokhina, A.S.; Kurzina, I.A.; Sobolev, V.I.; Koltunov, K.Y.; Mamontov, G.V.; Vodyankina, O.V. Selective Oxidation of Alcohols over Si3N4 Supported Silver Catalysts. Kinet. Catal. 2012, 53, 477–481. [Google Scholar] [CrossRef]
- Vodyankina, O.V.; Blokhina, A.S.; Kurzina, I.A.; Sobolev, V.I.; Koltunov, K.Y.; Chukhlomina, L.N.; Dvilis, E.S. Selective oxidation of alcohols over Ag-containing Si3N4 catalysts. Catal. Today 2013, 203, 127–132. [Google Scholar] [CrossRef]
- Dutov, V.V.; Mamontov, G.V.; Sobolev, V.I.; Vodyankina, O.V. Silica-supported silver-containing OMS-2 catalysts for ethanol oxidative dehydrogenation. Catal. Today 2016, 278, 164–173. [Google Scholar] [CrossRef]
- Chen, J.; Li, J.; Liu, Q.; Huang, X.; Shen, W. Facile Synthesis of Ag-Hollandite Nanofibers and Their Catalytic Activity for Ethanol Selective Oxidation. Chin. J. Catal. 2007, 28, 1034–1036. [Google Scholar] [CrossRef]
- Chen, J.; Tang, X.; Liu, J.; Zhan, E.; Li, J.; Huang, X.; Shen, W. Synthesis and Characterization of Ag−Hollandite Nanofibers and Its Catalytic Application in Ethanol Oxidation. Chem. Mater. 2007, 19, 4292–4299. [Google Scholar] [CrossRef]
- Li, Z.; Xu, J.; Gu, X.; Wang, K.; Wang, W.; Zhang, X.; Zhang, Z.; Ding, Y. Selective Gas-Phase Oxidation of Alcohols over Nanoporous Silver. ChemCatChem 2013, 5, 1705–1708. [Google Scholar] [CrossRef]
- Liu, P.; Zhu, X.; Yang, S.; Li, T.; Hensen, E.J.M. On the metal–support synergy for selective gas-phase ethanol oxidation over MgCuCr2O4 supported metal nanoparticle catalysts. J. Catal. 2015, 331, 138–146. [Google Scholar] [CrossRef]
- Mamontov, G.V.; Grabchenko, M.V.; Sobolev, V.I.; Zaikovskii, V.I.; Vodyankina, O.V. Ethanol dehydrogenation over Ag-CeO2/SiO2 catalyst: Role of Ag-CeO2 interface. Appl. Catal. A 2016, 528, 161–167. [Google Scholar] [CrossRef]
- Guan, Y.; Hensen, E.J.M. Ethanol dehydrogenation by gold catalysts: The effect of the gold particle size and the presence of oxygen. Appl. Catal. A Gen. 2009, 361, 49–56. [Google Scholar] [CrossRef]
- Grabchenko, M.V.; Mamontov, G.V.; Zaikovskii, V.I.; La Parola, V.; Liotta, L.F.; Vodyankina, O.V. Design of Ag-CeO2/SiO2 catalyst for oxidative dehydrogenation of ethanol: Control of Ag–CeO2 interfacial interaction. Catal. Today 2018, in press. [Google Scholar] [CrossRef]
- Grabchenko, M.V.; Mamontov, G.V.; Zaikovskii, V.I.; Vodyankina, O.V. Effect of the metal—Support interaction in Ag/CeO2 catalysts on their activity in ethanol oxidation. Kinet. Catal. 2017, 58, 642–648. [Google Scholar] [CrossRef]
- Rodrigues, C.P.; Zonetti, P.C.; Silva, C.G.; Gaspar, A.B.; Appel, L.G. Chemicals from ethanol—The acetone one-pot synthesis. Appl. Catal. A 2013, 458, 111–118. [Google Scholar] [CrossRef]
- De Lima, A.F.F.; Zonetti, P.C.; Rodrigues, C.P.; Appel, L.G. The first step of the propylene generation from renewable raw material: Acetone from ethanol employing CeO2 doped by Ag. Catal. Today 2017, 279, 252–259. [Google Scholar] [CrossRef]
- Yamamoto, R.; Sawayama, Y.; Shibahara, H.; Ichihashi, Y.; Nishiyam, S.; Tsuruy, S. Promoted partial oxidation activity of supported Ag catalysts in the gas-phase catalytic oxidation of benzyl alcohol. J. Catal. 2005, 234, 308–317. [Google Scholar] [CrossRef]
- Jia, L.; Zhang, S.; Gu, F.; Ping, Y.; Guo, X.; Zhong, Z.; Su, F. Highly selective gas-phase oxidation of benzyl alcohol to benzaldehyde over silver-containing hexagonal mesoporous silica. Microporous Mesoporous Mater. 2012, 149, 158–165. [Google Scholar] [CrossRef]
- Ma, L.; Jia, L.; Guo, X.; Xiang, L. Catalytic activity of Ag/SBA-15 for low-temperature gas-phase selective oxidation of benzyl alcohol to benzaldehyde. Chin. J. Catal. 2014, 35, 108–119. [Google Scholar] [CrossRef]
- Mao, J.; Deng, M.; Xue, Q.; Chen, L.; Lu, Y. Thin-sheet Ag/Ni-fiber catalyst for gas-phase selective oxidation of benzyl alcohol with molecular oxygen. Catal. Commun. 2009, 10, 1376–1379. [Google Scholar] [CrossRef]
- Deng, M.; Zhao, G.; Xue, Q.; Chen, L.; Lu, Y. Microfibrous-structured silver catalyst for low-temperature gas-phase selective oxidation of benzyl alcohol. Appl. Catal. B 2010, 99, 222–228. [Google Scholar] [CrossRef]
- Zhao, L.; Kong, L.; Liu, C.; Wang, Y.; Dai, L. AgCu/SiC-powder: A highly stable and active catalyst for gas-phase selective oxidation of alcohols. Catal. Commun. 2017, 98, 1–4. [Google Scholar] [CrossRef]
- Liu, K.; Qin, T.; Sun, Y.; Hou, C.; Cao, X.; Jiang, S. Synergistic effect between Ag and Mn3O4 in the gas phase oxidation of alcohols. Catal. Commun. 2018, 113, 15–18. [Google Scholar] [CrossRef]
- Sawayama, Y.; Shibahara, H.; Ichihashi, Y.; Nishiyama, S.; Tsuruya, S. Promoting Effect and Role of Alkaline Earth Metal Added to Supported Ag Catalysts in the Gas-Phase Catalytic Oxidation of Benzyl Alcohol. Ind. Eng. Chem. Res. 2006, 45, 8837–8845. [Google Scholar] [CrossRef]
- Nguyen, T.T.N.; Huchede, M.; Blanco, E.; Morfin, F.; Rousset, J.L.; Massin, L.; Aouine, M.; Bellière-Baca, V.; Millet, J.M.M. An attempt to improve Ag-based catalysts for allyl alcohol oxidative dehydrogenation to acrolein. Appl. Catal. A 2018, 549, 170–178. [Google Scholar] [CrossRef]
- Yue, H.; Zhao, Y.; Ma, X.; Gong, J. Ethylene glycol: Properties, synthesis, and applications. Chem. Soc. Rev. 2012, 41, 4218–4244. [Google Scholar] [CrossRef] [PubMed]
- Vodyankina, O.V.; Kurina, L.N.; Boronin, A.I.; Salanov, A.N. Glyoxal Synthesis by Vapour-Phase Ethylene Glycol Oxidation on a Silver and Copper Catalysts. Stud. Surf. Sci. Catal. B 2000, 130, 1775–1780. [Google Scholar] [CrossRef]
- Xu, H.; Yin, G.; Deng, Z.; Yan, H.; Qin, F.; Shen, W. Method for Synthesizing Low-Impurity Content Glyoxal through Gas Phase Oxidation. Patent CN103,772,169A, 27 May 2015. [Google Scholar]
- Vodyankina, O.V.; Koscheev, S.V.; Yakushko, V.T.; Salanov, A.N.; Boronin, A.I.; Kurina, L.N. Physicochemical investigation of the copper and silver catalysts of the ethylene glycol oxidation. J. Mol. Catal. A Chem. 2000, 158, 381–387. [Google Scholar] [CrossRef]
- Gallezot, P.; Tretjak, S.; Christidis, Y.; Mattioda, G.; Schouteenten, A. Oxidative dehydrogenation of ethylene glycol into glyoxal: Effect of diethylphosphite on SiC-supported silver catalysts. J. Catal. 1993, 142, 729–734. [Google Scholar] [CrossRef]
- Deng, J.F.; Wang, J.; Xu, X. Oxidative dehydrogenation of glycol to glyoxal on a P-modified electrolytic silver catalyst. Catal. Lett. 1996, 36, 207–214. [Google Scholar] [CrossRef]
- Magaev, O.V.; Knyazev, A.S.; Vodyankina, O.V.; Dorofeeva, N.V.; Salanov, A.N.; Boronin, A.I. Active surface formation and catalytic activity of phosphorous-promoted electrolytic silver in the selective oxidation of ethylene glycol to glyoxal. Appl. Catal. A 2008, 344, 142–149. [Google Scholar] [CrossRef]
- Method for Continuous Industrial Production of Glyoxal. RU2,599,247 (C1), 2016.
- Salaev, M.A.; Krejker, A.A.; Magaev, O.V.; Malkov, V.S.; Knyazev, A.S.; Borisova, E.S.; Khanaev, V.M.; Vodyankina, O.V.; Kurina, L.N. Ethylene glycol oxidation over supported catalyst in tubular reactor. Chem. Eng. J. 2011, 172, 399–409. [Google Scholar] [CrossRef]
- Mamontov, G.V.; Magaev, O.V.; Knyazev, A.S.; Vodyankina, O.V. Influence of phosphate addition on activity of Ag and Cu catalysts for partial oxidation of alcohols. Catal. Today 2013, 203, 122–126. [Google Scholar] [CrossRef]
- Mamontov, G.V.; Knyazev, A.S.; Paukshtis, E.A.; Vodyankina, O.V. Adsorption and conversion of ethylene glycol on the surface of Ag-containing catalyst modified with phosphate. Kinet. Catal. 2013, 54, 735–743. [Google Scholar] [CrossRef]
- Knyazev, A.S.; Magaev, O.V.; Krejker, A.A.; Mamontov, G.V.; Knyazeva, E.M.; Dahnavi, E.M.; Vodyankina, O.V. Self-Organized nanostructured Ag/P2O5/SiO2 material as a catalyst for high-temperature selective oxidation of alcohols. Key Eng. Mater. 2015, 670, 126–132. [Google Scholar] [CrossRef]
- Mamontov, G.V.; Izaak, T.I.; Magaev, O.V.; Knyazev, A.S.; Vodyankina, O.V. Reversible Oxidation/Reduction of Silver Supported on Silica Aerogel: Influence of the Addition of Phosphate. Russ. J. Phys. Chem. A 2011, 85, 1540–1545. [Google Scholar] [CrossRef]
- Epiphanova, A.; Magaev, O.; Vodyankina, O. Formation and characterization of phosphate-modified silicate materials derived from sol-gel process. J. Sol-Gel Sci. Technol. 2012, 61, 509–517. [Google Scholar] [CrossRef]
- Dorofeeva, N.V.; Vodyankina, O.V.; Pavlova, O.S.; Mamontov, G.V. Synthesis of mixed zirconium-silver phosphates and formation of active catalyst surface for the ethylene glycol oxidation process. Stud. Surf. Sci. Catal. 2010, 175, 759–762. [Google Scholar] [CrossRef]
- Dorofeeva, N.V.; Vodyankina, O.V.; Sobolev, V.I.; Koltunov, K.Y.; Zaykovskii, V.I. Main routes of ethanol conversion under aerobic/anaerobic conditions over Ag-containing zirconium phosphate catalyst. Curr. Org. Synth. 2017, 14, 389–393. [Google Scholar] [CrossRef]
- Brik, Y.; Kacimi, M.; Bozon-Verduraz, F.; Ziyad, M. Characterization of active sites on AgHf2(PO4)3 inbutan-2-ol conversion. Microporous Mesoporous Mater. 2001, 43, 103–112. [Google Scholar] [CrossRef]
- Arsalane, S.; Ziyad, M.; Coudurier, G.; Védrine, J.C. Silver-cluster formation on AgZr2(PO4)3 and catalytic decomposition of butan-2-ol. J. Catal. 1996, 159, 162–169. [Google Scholar] [CrossRef]
- Salaev, M.A.; Poleshchuk, O.K.; Vodyankina, O.V. Ethylene glycol oxidation over Ag-containing catalysts: A theoretical study. J. Mol. Catal. A Chem. 2015, 396, 61–67. [Google Scholar] [CrossRef]
- Capote, A.J.; Madix, R.J. O-H and C-H Bond Activation in Ethylene Glycol by Atomic Oxygen on Ag(110): Heterometallacycle Formation and Selective Dehydrogenation to Glyoxal. J. Am. Chem. Soc. 1989, 111, 3570–3577. [Google Scholar] [CrossRef]
- Feng, Y.; Yin, H.; Wang, A.; Xue, W. Selectively catalytic oxidation of 1,2-propanediol to lactic, formic, and acetic acids over Ag nanoparticles under mild reaction conditions. J. Catal. 2015, 326, 26–37. [Google Scholar] [CrossRef]
- Salaev, M.A.; Poleshchuk, O.K.; Vodyankina, O.V. Propylene glycol oxidation over silver catalysts: A theoretical study. J. Mol. Catal. A Chem. 2016, 417, 36–42. [Google Scholar] [CrossRef]
- Xue, W.; Yin, H.; Lu, Z.; Feng, Y.; Wang, A.; Liu, S.; Shen, L.; Jia, X. Catalytic Oxidation of 1,2-Propanediol over Bimetallic Cu@Au Core/Shell Nanoparticles. Catal. Lett. 2016, 146, 1139–1152. [Google Scholar] [CrossRef]
- Griffin, M.B.; Rodriguez, A.A.; Montemore, M.M.; Monnier, J.R.; Williams, C.T.; Will Medlin, J. The selective oxidation of ethylene glycol and 1,2-propanediol on Au, Pd, and Au–Pd bimetallic catalysts. J. Catal. 2013, 307, 111–120. [Google Scholar] [CrossRef]
- Ebert, D.Y.; Dorofeeva, N.V.; Savel’eva, A.S.; Kharlamova, T.S.; Salaev, M.A.; Svetlichnyi, V.A.; Magaev, O.V.; Vodyankina, O.V. Silica-supported Fe-Mo-O catalysts for selective oxidation of propylene glycol. Catal. Today 2018, in press. [Google Scholar] [CrossRef]
- Ebert, D.Y.; Savel’eva, A.S.; Dorofeeva, N.V.; Vodyankina, O.V. FePO4/SiO2 Catalysts for Propylene Glycol Oxidation. Kinet. Catal. 2017, 58, 720–725. [Google Scholar] [CrossRef]
- Shen, J.; Shan, W.; Zhang, Y.; Du, J.; Xu, H.; Fan, K.; Shen, W.; Tang, Y. A novel catalyst with high activity for polyhydric alcohol oxidation: Nanosilver/zeolite film. Chem. Commun. 2004, 2880–2881. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Shan, W.; Zhang, Y.; Du, J.; Xu, H.; Fan, K.; Shen, W.; Tang, Y. Gas-phase selective oxidation of alcohols: In situ electrolytic nano-silver/zeolite film/copper grid catalyst. J. Catal. 2006, 237, 94–101. [Google Scholar] [CrossRef]
- Mao, J.; Deng, M.; Chen, L.; Liu, Y.; Lu, Y. Novel Microfibrous-Structured Silver Catalyst for High Efficiency Gas-Phase Oxidation of Alcohols. AIChE J. 2010, 56, 1545–1556. [Google Scholar] [CrossRef]
- Shen, J.; Du, J.-M.; Huang, J.-J.; Yang, X.-Y.; Shen, W.; Xu, H.-L.; Fan, K.-N. Gas-phase selective oxidation of 1, 2-propylene glycol over Ag/ZrO2 catalyst. Acta Chim. Sin. 2007, 65, 403–408. [Google Scholar]
- Yang, F.; Jing, X.; Huang, J.; Sun, D.; Li, Q. Microwave-Assisted Biosynthesis of Ag/ZrO2 Catalyst with Excellent Activity toward Selective Oxidation of 1,2-Propanediol. Ind. Eng. Chem. Res. 2015, 54, 5373–5380. [Google Scholar] [CrossRef]
- Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Della Pina, C. From Glycerol to Value-Added Products. Angew. Chem. Int. Ed. 2007, 46, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Dodekatos, G.; Schünemann, S.; Tüysüz, H. Recent Advances in Thermo-, Photo-, and Electrocatalytic Glycerol Oxidation. ACS Catal. 2018, 8, 6301–6333. [Google Scholar] [CrossRef]
- Lari, G.M.; Mondelli, C.; Pérez-Ramírez, J. Gas-Phase Oxidation of Glycerol to Dihydroxyacetone over Tailored Iron Zeolites. ACS Catal. 2015, 5, 1453–1461. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, M.; Han, Y. Selective oxidation of glycerol to formic acid in highly concentrated aqueous solutions with molecular oxygen using V-substituted phosphomolybdic acids. RSC Adv. 2014, 4, 35463–35466. [Google Scholar] [CrossRef]
- Lari, G.M.; García-Muelas, R.; Mondelli, C.; López, N.; Pérez-Ramírez, J. Glycerol oxidehydration to pyruvaldehyde over silver-based catalysts for improved lactic acid production. Green Chem. 2016, 18, 4682–4692. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Wallau, M.; Arends, I.W.C.E.; Schuchardt, U. Heterogeneous catalysts for liquid-phase oxidations: Philosophers’ stones or Trojan Horses? Acc. Chem. Res. 1998, 31, 485–493. [Google Scholar] [CrossRef]
- Cortes Corberan, V.; Gonzalez-Perez, M.E.; Martinez-Gonzalez, S.; Gomez-Aviles, A. Green oxidation of fatty alcohols: Challenges and opportunities. Appl. Catal. A 2014, 474, 211–223. [Google Scholar] [CrossRef]
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on platinum metal catalysts in aqueous solutions. Catal. Today 1994, 19, 247–283. [Google Scholar] [CrossRef]
- Kolobova, E.N.; Pestryakov, A.N.; Bogdanchikova, N.; Cortés Corberán, V. Silver catalysts for liquid-phase oxidation of alcohols in green chemistry: Challenges and outlook. Catal. Today 2018, in press. [Google Scholar] [CrossRef]
- Yang, Z.; Li, J.; Yang, X.; Xie, X.; Wu, Y. Gas-phase oxidation of alcohols over silver: The extension of catalytic cycles of oxidation of alcohols in liquid-phase. J. Mol. Catal. A 2005, 241, 15–22. [Google Scholar] [CrossRef]
- Beier, M.J.; Hansen, T.W.; Grunwaldt, J.-D. Selective liquid-phase oxidation of alcohols catalyzed by a silver-based catalyst promoted by the presence of ceria. J. Catal. 2009, 266, 320–330. [Google Scholar] [CrossRef]
- Olenin, A.Y.; Mingalev, P.G.; Lisichkin, G.V. Partial Catalytic Oxidation of Alcohols: Catalysts Based on Metals and Metal Coordination Compounds (a Review). Pet. Chem. 2018, 58, 577–592. [Google Scholar] [CrossRef]
- Park, Y.; Na, Y.; Pradhan, D.; Sohn, Y. Liquid-phase ethanol oxidation and gas-phase CO oxidation reactions over M doped (M = Ag, Au, Pd, and Ni) and MM’ codoped CeO2 nanoparticles. J. Chem. 2016, 2176576. [Google Scholar] [CrossRef]
- Liotta, L.F.; Venezia, A.M.; Deganello, G.; Longo, A.; Martorana, A.; Schay, Z.; Guczi, L. Liquid phase selective oxidation of benzyl alcohol over Pd-Ag catalysts supported on pumice. Catal. Today 2011, 66, 271–276. [Google Scholar] [CrossRef]
- Huang, X.; Wang, X.; Wang, X.; Wang, X.; Tan, M.; Ding, W.; Lu, X. P123-stabilized Au–Ag alloy nanoparticles for kinetics of aerobic oxidation of benzyl alcohol in aqueous solution. J. Catal. 2013, 301, 217–226. [Google Scholar] [CrossRef]
- Huang, X.; Wang, X.; Tan, M.; Zou, X.; Ding, W.; Lu, X. Selective oxidation of alcohols on P123-stabilized Au–Ag Alloy nanoparticles in aqueous solution with molecular oxygen. Appl. Catal. A 2013, 467, 407–413. [Google Scholar] [CrossRef]
- Zahed, B.; Hosseini-Monfared, H. A comparative study of silver-graphene oxide nanocomposites as a recyclable catalyst for the aerobic oxidation of benzyl alcohol: Support effect. Appl. Surf. Sci. 2015, 328, 536–547. [Google Scholar] [CrossRef]
- Adil, S.F.; Assal, M.E.; Khan, M.; Al-Warthan, A.; Rafiq, H.; Siddiqui, M. Nano silver-doped manganese oxide as catalyst for oxidation of benzyl alcohol and its derivatives: Synthesis, characterization, thermal study and evaluation of catalytic properties. Oxid. Commun. 2013, 36, 778–791. [Google Scholar]
- Alabbad, S.; Adil, S.F.; Assal, M.E.; Khan, M.; Alwarthan, A.; Siddiqui, M.R.H. Gold & silver nanoparticles supported on manganese oxide: Synthesis, characterization and catalytic studies for selective oxidation of benzyl alcohol. Arab. J. Chem. 2014, 7, 1192–1198. [Google Scholar] [CrossRef]
- Assal, M.E.; Shaik, M.R.; Kuniyil, M.; Khan, M.; Al-Warthan, A.; Alharthi, A.I.; Varala, R.; Siddiqui, M.R.H.; Adil, S.F. Ag2O nanoparticles/MnCO3, -MnO2 or –Mn2O3/highly reduced graphene oxide composites as an efficient and recyclable oxidation catalyst. Arab. J. Chem. 2018, in press. [Google Scholar] [CrossRef]
- Assal, M.E.; Shaik, M.R.; Kuniyil, M.; Khan, M.; Al-Warthan, A.; Siddiqui, M.R.H.; Adil, S.F. Ag2O nanoparticles-doped manganese immobilized on graphene nanocomposites for aeral oxidation of secondary alcohols. Metals 2018, 8, 468. [Google Scholar] [CrossRef]
- Mitsudome, T.; Mikami, Y.; Funai, H.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Oxidant-free alcohol dehydrogenation using a reusable hydrolactite-supported silver nanoparticle catalyst. Angew. Chem. Int. Ed. 2008, 120, 140–147. [Google Scholar] [CrossRef]
- Bayat, A.; Shakourian-Fard, M.; Ehyaei, N.; Hashemi, M.M. Silver nanoparticles supported on silica-coated ferrite as magnetically and reusable catalyst for oxidant-free alcohol dehydrogenation. RSC Adv. 2015, 5, 22503–22509. [Google Scholar] [CrossRef]
- Qi, G.; Zhang, W.; Dai, Y. Selective catalysis for synthesis of benzaldehyde by magnetic Ag/Fe2O3. Asian J. Chem. 2014, 26, 6383–6386. [Google Scholar] [CrossRef]
- Bhuyan, B.; Paul, A.; Devi, M.; Dhar, S.S. A silver NP-dispersed water extract of fly ash as a green and efficient medium for oxidant-free dehydrogenation of benzyl alcohols. RSC Adv. 2018, 8, 1313–1319. [Google Scholar] [CrossRef] [Green Version]
- Yadav, G.D.; Yadav, A.K. Selective liquid phase oxidation of secondary alcohols into ketones by tert-butyl hydroperoxide on nano-fibrous Ag-OMS-2 catalyst. J. Mol. Catal. A Chem. 2013, 380, 70–77. [Google Scholar] [CrossRef]
- Shojaei, A.F.; Tabatabaeian, K.; Zanjanchi, M.A.; Moafi, H.F.; Modirpanah, N. Synthesis, characterization and study of catalytic activity of silver doped ZnO nanocomposite as an efficient catalyst for selective oxidation of benzyl alcohol. J. Chem. Sci. 2015, 127, 481–491. [Google Scholar] [CrossRef]
- Kolobova, E.; Kotolevich, Y.; Pakrieva, E.; Mamontov, G.; Farias, M.H.; Cortés Corberán, V.; Bogdanchikova, N.; Hemming, J.; Smeds, A.; Mäki-Arvela, P.; et al. Modified Ag/TiO2 systems: Promising catalysts for liquid-phase oxidation of alcohols. Fuel 2018, 234, 110–119. [Google Scholar] [CrossRef]
- Nagaraiju, P.; Balaraju, M.; Mohan Reddy, K.; Sai Prasad, P.S.; Lingaiah, N. Selective oxidation of allylic alcohols catalyzed by silver exchanged molybdovanado phosphoric acid catalyst in the presence of molecular oxygen. Catal. Commun. 2008, 9, 1389–1393. [Google Scholar] [CrossRef]
- Torbina, V.V.; Ivanchikova, I.D.; Kholdeeva, O.A.; Skobelev, I.Y.; Vodyankina, O.V. Propylene glycol oxidation with tert-butyl hydroperoxide over Cr-containing metal-organic frameworks MIL-101 and MIL-100. Catal. Today 2016, 278, 97–103. [Google Scholar] [CrossRef]
- Xue, W.; Feng, Y.; Yin, H.; Liu, S.; Wang, A.; Shen, L. Catalytic Oxidation of 1,2-Propanediol to Lactic Acid with O2 Under Atmospheric Pressure Over Pd–Ag Bimetallic Nanoparticles and Reaction Kinetics. J. Nanosci. Nanotechnol. 2016, 16, 9621–9633. [Google Scholar] [CrossRef]
- Feng, Y.; Xue, W.; Yin, H.; Meng, M.; Wang, A.; Liu, S. Selective oxidation of 1,2-propanediol to lactic acid catalyzed by hydroxyapatite-supported Pd and Pd–Ag nanoparticles. RSC Adv. 2015, 5, 106918–106929. [Google Scholar] [CrossRef]
- Albuquerque, E.M.; Borges, L.E.P.; Fraga, M.A. Lactic acid production from aqueous-phase selective oxidation of hydroxyacetone. J. Mol. Catal. A Chem. 2015, 400, 64–70. [Google Scholar] [CrossRef]
- Song, F. Zh.; Zhu, Q.L.; Yang, X.; Zhan, W.W.; Pachfule, P.; Tsumori, N.; Xu, Q. Metal–Organic Framework Templated Porous Carbon-Metal Oxide/Reduced Graphene Oxide as Superior Support of Bimetallic Nanoparticles for Efficient Hydrogen Generation from Formic Acid. Adv. Energy Mater. 2017, 1701416. [Google Scholar] [CrossRef]
- Liu, H.; Liu, X.; Yu, Y.; Yang, W.; Li, J.; Feng, M.; Li, H. Bifunctional Networked Ag/AgPd Core/Shell Nanowires for Highly Efficient Dehydrogenation of Formic Acid and Subsequent Reduction of Nitrate and Nitrite in Water. J. Mater. Chem. A 2018, 6, 4611–4616. [Google Scholar] [CrossRef]
- Bozell, J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Crottia, C.; Farnetti, E. Selective oxidation of glycerol catalyzed by iron complexes. J. Mol. Catal. A 2015, 396, 353–359. [Google Scholar] [CrossRef]
- Hirasawa, S.; Nakagawa, Y.; Tomishige, K. Selective oxidation of glycerol to dihydroxyacetone over Pd-Ag catalyst. Catal. Sci. Technol. 2012, 2, 1150–1152. [Google Scholar] [CrossRef]
- Hirasawa, S.; Watanabe, H.; Tokushi, K.; Nakagawa, Y.; Tomishige, K. Performance, structure and mechanism of Pd-Ag alloy catalyst for selective oxidation of glycerol to dihydroxyacetone. J. Catal. 2013, 300, 205–216. [Google Scholar] [CrossRef]
- Skrzyńska, E.; Zaid, S.; Girardon, J.-S.; Capron, M.; Dumeignil, F. Catalytic behavior of four different supported noble metals in the crude glycerol oxidation. Appl. Catal. A 2015, 499, 89–100. [Google Scholar] [CrossRef]
- Skrzyńska, E.; Zaid, S.; Addad, A.; Girardon, J.-S.; Capron, M.; Dumeignil, F. Performance of Ag/Al2O3 catalysts in the liquid phase oxidation of glycerol—Effect of preparation method and reaction conditions. Catal. Sci. Technol. 2016, 6, 3182–3196. [Google Scholar] [CrossRef]
- Skrzyńska, E.; Ftouni, J.; Girardon, J.-S.; Capron, M.; Jalowiecki-Duhamel, L.; Paul, J.-F.; Dumeignil, F. Quasi-homogeneous oxidation of glycerol by unsupported gold nanoparticles in the liquid phase. ChemSusChem 2012, 5, 2065–2078. [Google Scholar] [CrossRef] [PubMed]
- Zaid, E.; Skrzyńska, E.; Addad, A.; Nandi, S.; Jalowiecki-Duhamel, L.; Girardon, J.-S.; Capron, M.; Dumeignil, F. Development of Silver Based Catalysts Promoted by Noble Metal M (M = Au, Pd or Pt) for Glycerol Oxidation in Liquid Phase. Top Catal. 2017, 60, 1072–1081. [Google Scholar] [CrossRef]
- Besson, M.; Gallezot, P. Selective oxidation of alcohols and aldehydes on metal catalysts. Catal. Today 2000, 57, 127–141. [Google Scholar] [CrossRef]
- Diaz, J.A.; Skrzyńska, E.; Zaid, S.; Girardon, J.-S.; Capron, M.; Dumeignil, F.; Fongarland, P. Kinetic modeling of the glycerol oxidation in the liquid phase: Comparison of Pt, Au and Ag as active phases. J. Chem. Technol. Biotechnol. 2017, 92, 2267–2275. [Google Scholar] [CrossRef]
- Zope, B.N.; Hibbitts, D.D.; Neurock, M.; Davis, R.J. Reactivity of the gold/water interface during selective oxidation catalysis. Science 2007, 44, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.; Zhang, D.; Guan, H.; Huang, G.; Wang, X. Designation of highly efficient catalysts for one pot conversion of glycerol to lactic acid. Sci. Rep. 2016, 6, 29840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaskow, I.; Decyk, P.; Sobczak, I. The effect of copper and silver on the properties of Au-ZnO catalysts and its activity in glycerol oxidation. Appl. Surf. Sci. 2018, 444, 197–207. [Google Scholar] [CrossRef]
- Tamiolakis, I.; Lykakis, I.; Armatas, S. Mesoporous CdS-sensitized TiO2 nanoparticle assemblies with enhanced photocatalytic properties: Selective aerobic oxidation of benzyl alcohols. Catal. Today 2015, 250, 180–186. [Google Scholar] [CrossRef]
- Li, S.; Cai, J.; Wu, X.; Zheng, F. Sandwich-like TiO2@ZnO-based noble metal (Ag, Au, Pt, or Pd) for better photo-oxidation performance: Synergistic effect between noble metal and metal oxide phases. Appl. Surf. Sci. 2018, 443, 603–612. [Google Scholar] [CrossRef]
- Safaei, I.; Mohebbi, S.; Irani, M. Selective aerobic photocatalytic oxidation of benzyl alcohol over spherical structured WO3/TiO2 nanocomposite under visible light irradiation. J. Sol-Gel. Sci. Technol. 2018, 87, 170–182. [Google Scholar] [CrossRef]
- Li, S.; Cai, J.; Wu, X.; Liu, B.; Chen, Q.; Li, Y.; Zheng, F. TiO2@Pt@CeO2 nanocomposite as a bifunctional catalyst for enhancing photo-reduction of Cr(VI) and photo-oxidation of benzyl alcohol. J. Hazard. Mater. 2018, 346, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Magdziarz, A.; Colmenares, J.C.; Chernyayeva, O.; Lisovytskiy, D.; Grzonka, J.; Kurzydłowski, K.; Freindl, K.; Korecki, J. Insight into the synthesis procedure of Fe3+/TiO2-based photocatalyst applied in the selective photo-oxidation of benzyl alcohol under sun-imitating lamp. Ultrason. Sonochem. 2017, 38, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Amadelli, R.; Samiolo, L.; Maldotti, A.; Molinari, A.; Gazzoli, D. Selective Photooxidation and Photoreduction Processes at Surface-Modified by Grafted Vanadyl. Int. J. Photoenergy 2011, 1–10. [Google Scholar] [CrossRef]
- Yurdakal, S.; Tek, B.S.; Değirmenci, Ç.; Palmisano, G. Selective photocatalytic oxidation of aromatic alcohols in solar-irradiated aqueous suspensions of Pt, Au, Pd and Ag loaded TiO2 catalysts. Catal. Today 2017, 281, 53–59. [Google Scholar] [CrossRef]
- Si, J.; Liu, Y.; Chang, S.; Wu, D.; Tian, B.; Zhang, J. AgBr@TiO2/GO ternary composites with enhanced photocatalytic activity for oxidation of benzyl alcohol to benzaldehyde. Res. Chem. Intermed. 2016, 43, 2067–2080. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, P.; Bao, S.; Wang, Z.; Tian, B.; Zhang, J. Synthesis of sandwich-structured AgBr@Ag@TiO2 composite photocatalyst and study of its photocatalytic performance for the oxidation of benzyl alcohols to benzaldehydes. Chem. Eng. J. 2016, 306, 1151–1161. [Google Scholar] [CrossRef]
- Chen, Y.; Li, W.; Wang, J.; Yang, Q.; Hou, Q.; Ju, M. Gold nanoparticles-modified TiO2/SBA-15 nanocomposites as active plasmonic photocatalysts for the selective oxidation of aromatic alcohols. RSC Adv. 2016, 6, 70352–70363. [Google Scholar] [CrossRef]
- Feng, W.; Wu, G.; Li, L.; Guan, N. Solvent-free selective photocatalytic oxidation of benzyl alcohol over modified TiO2. Green Chem. 2011, 13, 3265–3272. [Google Scholar] [CrossRef]
- Yu, J.; Li, J.; Wei, H.; Zheng, J.; Su, H.; Wang, X. Hydrotalcite-supported gold catalysts for a selective aerobic oxidation of benzyl alcohol driven by visible light. J. Mol. Catal. A 2014, 395, 128–136. [Google Scholar] [CrossRef]
- Chassé, M.; Hallett-Tapley, G.L. Gold nanoparticle-functionalized niobium oxide perovskites as photocatalysts for visible light-induced aromatic alcohol oxidations. Can. J. Chem. 2018, 96, 664–671. [Google Scholar] [CrossRef]
- Zhang, X.; Ke, X.; Zhu, H. Zeolite-Supported Gold Nanoparticles for Selective Photooxidation of Aromatic Alcohols under Visible-Light Irradiation. Chem. Eur. J. 2012, 18, 8048–8056. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Li, X.; Zheng, R.; Chen, R.; Sun, X. Bimetallic (Au–Cu core)@(Ceria shell) nanotubes for photocatalytic oxidation of benzyl alcohol: Improved reactivity by Cu. J. Mater. Chem. A 2017, 5, 13382–13391. [Google Scholar] [CrossRef]
- Qamar, M.; Elsayed, R.B.; Alhooshani, K.R.; Ahmed, M.I.; Bahnemann, D.W. Highly Efficient and Selective Oxidation of Aromatic Alcohols Photocatalyzed by Nanoporous Hierarchical Pt/Bi2WO6 in Organic Solvent-Free Environment. ACS Appl. Mater. Interfaces 2015, 7, 1257–1269. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, K.; Takemura, M.; Togo, H. Direct and Selective Benzylic Oxidation of Alkylarenes via C–H Abstraction Using Alkali Metal Bromides. Org. Lett. 2012, 14, 2414–2417. [Google Scholar] [CrossRef] [PubMed]
- Gazi, S.; Ananthakrishnan, R. Bromodimethylsulfonium bromide as a potential candidate for photocatalytic selective oxidation of benzylic alcohols using oxygen and visible light. RSC Adv. 2012, 2, 7781–7787. [Google Scholar] [CrossRef]
- Qamar, M.; Elsayed, R.B.; Alhooshani, K.R.; Ahmed, M.I.; Bahnemann, D.W. Chemoselective and highly efficient conversion of aromatic alcohols into aldehydes photo-catalyzed by Ag3PO4 in aqueous suspension under simulated sunlight. Catal. Commun. 2015, 58, 34–39. [Google Scholar] [CrossRef]
- DePuccio, D.P.; Landry, C.C. Photocatalytic oxidation of methanol using porous Au/WO3 and visible light. Catal. Sci. Technol. 2016, 6, 7512–7520. [Google Scholar] [CrossRef]
- Han, C.; Yang, X.; Gao, G.; Wang, J.; Lu, H.; Liu, J.; Tong, M.; Liang, X. Selective oxidation of methanol to methyl formate on catalysts of Au–Ag alloy nanoparticles supported on titania under UV irradiation. Green Chem. 2014, 16, 3603–3615. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, A.; Gao, G.; Han, D.; Han, C.; Wang, J.; Liu, J.; Tong, M. Photocatalytic oxidation of methanol to methyl formate in liquid phase over supported silver catalysts. Catal. Commun. 2014, 43, 192–196. [Google Scholar] [CrossRef]
- El-Roz, M.; Lakiss, L.; Telegeiev, I.; Lebedev, O.I.; Bazin, P.; Vicente, A.; Fernandez, C.; Valtchev, V. High-Visible-Light Photoactivity of Plasma-Promoted Vanadium Clusters on Nanozeolites for Partial Photooxidation of Methanol. ACS Appl. Mater. Interfaces 2017, 9, 17846–17855. [Google Scholar] [CrossRef] [PubMed]
- Lüken, A.; Muhler, M.; Strunk, J. On the role of gold nanoparticles in the selective photooxidation of 2-propanol over Au/TiO2. Phys. Chem. Chem. Phys. 2015, 17, 10391–10397. [Google Scholar] [CrossRef] [PubMed]
- Colmenares, J.C.; Magdziarz, A.; Chernyayeva, O.; Lisovytskiy, D.; Kurzydłowski, K.; Grzonka, J. Sonication-Assisted Low-Temperature Routes for the Synthesis of Supported Fe-TiO2 Econanomaterials: Partial Photooxidation of Glucose and Phenol Aqueous Degradation. ChemCatChem 2013, 5, 2270–2277. [Google Scholar] [CrossRef]
- Colmenares, J.C.; Magdziarz, A.; Bielejewska, A. High-value chemicals obtained from selective photo-oxidation of glucose in the presence of nanostructured titanium photocatalysts. Bioresour. Technol. 2011, 102, 11254–11257. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, E. Selective Photo-Oxidation of Cellobiose with TiO2-Supported Metal Nanoparticles. Master’s Thesis, Università di Bologna, Bologna, Italy, 2013. [Google Scholar]
- Colmenares, J.C.; Aramendía, M.A.; Marinas, A.; Marinas, J.M.; Urbano, F.J. Synthesis, characterization and photocatalytic activity of different metal-doped titania systems. Appl. Catal. A 2006, 306, 120–127. [Google Scholar] [CrossRef]
- Gupta, N.; Bansal, P.; Pal, B. Metal ion-TiO2 nanocomposites for the selective photooxidation of benzene to phenol and cycloalkanol to cycloalkanone. J. Exp. Nanosci. 2013, 10, 148–160. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, G.; Li, L.; Chen, H.; Huang, Q.; Du, X.; Tong, Z. Electrospun CeO2/Ag@carbon nanofiber hybrids for selective oxidation of alcohols. Powder Technol. 2017, 305, 597–601. [Google Scholar] [CrossRef]




























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst Composition | Product | Reaction Conditions | S, % | X, % | Ag, %wt. | SBET, m2/g | Mean Particle Size, nm | Ref. |
|---|---|---|---|---|---|---|---|---|
| CR@Ag/TiO2-nf (thermally activated Congo-red (CR) entrapped within silver deposited on TiO2 nanofibers) | formaldehyde | 550–200 °C, “methanol ballast process”, WHSV = 140–335 gmeth*gAg−1*h−1, methanol volume concentration = 0.5 g/L | 65 (550 °C) | 94 (550 °C) | – | – | Ag: Initial: 9.4; after oxidation: 44.9 | [30] |
| Ag/TiO2-nf | 60 (550 °C) | 74 (550 °C) | – | – | Initial: 5.2; after oxidation: 58.8 | |||
| Metallic Ag fibers | formaldehyde | temperature range: 550 to 150 °C, methanol ballast process, weight of Ag fiber mats = 45–70 g, CH3OH concentration = 500 mg/L, total pressure = 1.5 bar, feed gas mixture flow rate = 8.35 L/h, WHSV in the range of 60–90 h−1, | 82.4 | 94.2 | 100 | – | Uniform mats, containing fibres with 300–600 nm | [32] |
| Ag/C | 84.1 | 95.8 | 70 in the “green” Electrospun fibers | – | ~50 | |||
| AuAg(0.5)–NbMCF | formaldehyde | 0.02 g of the catalyst of the size diameter of 0.5 < d < 1 mm Supply rate of 40 cm3 min−1 | 99 | 42 | 0.5 | 393 | 6.7 | [33] |
| Catalyst | Ag, wt. % | Activity × 10−2, mol-Et/g-kat*h | TOF a, h−1 | Reaction Conditions | SAc, % | STY b, h−1 | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|
| GHSV, ml/g*h | T, oC | Et/O2 c | |||||||
| Ag/Fe-Si3N4 | 4.9 | 0.58 | 12.7 | 7200 | 283 | 1/9 | 96 | 4.9 | [57] |
| OMS-2 | - | 0.43 | - | 7200 | 170 | 1/9 | 98 | - | [58] |
| Ag/OMS-2-CP | 3.7 | 0.66 | 19.3 | 97 | 7.7 | [58] | |||
| Ag/OMS-2-Impr | 5 | 0.64 | 13.8 | 96 | 5.3 | [58] | |||
| Ag/OMS-2-CP | 3.7 | 1.41 | 41.3 | 36,000 | 200 | 58 | 9.8 | [58] | |
| Ag/OMS-2/SiO2-CP | 0.95 | 1.91 | 217.6 | 230 | 85 | 75.4 | [58] | ||
| OMS-2/SiO2 | - | 0.15 | - | 7200 | 170 | 98 | - | [58] | |
| Ag/OMS-2/SiO2-CI | 5.39 | 0.23 | 4.6 | 98 | 1.81 | [58] | |||
| Ag/OMS-2/SiO2-CP | 0.95 | 0.28 | 32.1 | 98 | 12.8 | [58] | |||
| Ag/OMS-2 | - | 1.25 | - | 36,000 | 190 | 1/2 | 98 | - | [59] |
| Ag/OMS-2 | - | 5.52 | - | 36,000 | 230 | 1/2 | 95 | 26 | [60] |
| rous Ag | 100 | 46.3 | 50 | 133,333 | 250 | 2/1 | 95 | 21 | [61] |
| Ag/MgCuCr2O4 | 0.89 | 1.23 | 149.2 | 100,000 | 225 | 1/3 | 99 | 65 | [62] |
| Catalyst Composition | Reaction Conditions | T, °C | S, % | Y, % | Ag Loading, %wt. | SBET, m2/g | Mean Particle Size, nm | Ref. |
|---|---|---|---|---|---|---|---|---|
| Ag(1.0)/SiO2 | continuous-flow fixed-bed reactor (Pyrex, 15 mm i.d. 0.2 g of catalyst total flow rate = 0.023 mol/min N2:O2:BA = 32:3:1 Temperature range: 227–377 K | 240 | Close to 100% | 49.9 | 1.0 | 190 | 76 (Ag) | [69] |
| CaO | 22.0 | 1.0 | – | – | ||||
| MgO | 14.0 | 1.0 | – | – | ||||
| SiO2 | 3.6 | 1.0 | – | – | ||||
| NaZSM-5 | 2.3 | 1.0 | – | – | ||||
| MCM-41 | 0.36 | 1.0 | – | – | ||||
| NaY | 0.27 | 1.0 | – | – | ||||
| Ag-HMS-25 | 0.5 g catalyst purified N2 (38 mL min−1), purified O2 (13 mL min−1), BA (0.10 mL min−1) total flow rate of 3.24 mmol min−1, O2/alcohol molar ratio = 0.6 | 575 | 96.0 | 96.0 | Si/Ag ratio = 25:1 | 605 | 23 | [70] |
| HMS | 724 | 85 | 9.0 | – | – | – | ||
| SBA-15 | catalyst (0.8 g) was sieved to 40–60 mesh powders and pretreated (O2:N2 volume ratio of 3:7, flow rate was 50 mL/min) at 550 °C for 2 h before testing purified N2 (35 mL/min), purified O2, WHSV = 4.7–12.5 h−1 vaporized at 220 °C | 240 | 60.5 | 1.21 | – | 676 | – | [71] |
| 4.1%Ag/SBA-15 | 85.2 | 63.7 | 4.1 | 541 | 5.5 | |||
| 5.3%Ag/SBA-15 | 96.9 | 91.0 | 5.3 | 546 | 6.4 | |||
| 8.1%Ag/SBA-15 | 96.0 | 90.4 | 8.1 | 476 | 10.2 | |||
| 16.5%Ag/SBA-15 | 91.5 | 88.4 | 16.5 | 411 | 17.4 | |||
| Ag/Ni-fiber | 0.3 g for the Ag/Ni-fiber, 1.0 g for both the electrolytic silver and Ag/α-Al2O3 Calcination temperature: 400–700 °C O2/ol = 0.6, WHSV = 25 h−1 for Ag/Ni-fiber, O2/ol = 0.6, T = 500 °C and WHSV = 8 h−1 for the electrolytic silver and Ag/α-Al2O3 | 380 | 90 | 80.1 | 10.2 | ~2.0 | – | [72] |
| Electrolytic silver | 79 | 44.2 | 100 | – | – | |||
| Ag/α-Al2O3 | 74 | 60.7 | – | – | – | |||
| Ag/Ni-fiber-M | WHSV = 20 h−1 Calcination temperature: 300–600 °C | 300 | 97.0 | 94.1 | 9.9 | – | ∼10 nm thick by ∼100 nm width | [73] |
| Ag/Ni-fiber | 380 | 87.0 | 80 | 9.7 | – | 200–300 | ||
| Ag2.5/SiC | reaction temperature = 280 °C O2/hydroxyl = 0.6 WHSV = 10 h−1 | 280 | 99.9 | 1.9 | – | 0.8 | 30–70 | [74] |
| Ag2.5Cu5/SiC | 99.6 | 98.9 | – | 0.20 | – | |||
| Ag2.5/SiC& Cu5/SiC | 98.9 | 62.1 | – | – | – | |||
| Ag2.5Fe5/SiC | 99.8 | 55.7 | – | – | – | |||
| SiC | 0.3 g (200–300 mesh) WHSV = 20 h−1 alcohol/O2/N2 is 1/0.6/2.4 | 280 | 97 | 2.9 | – | 0.15 | – | [75] |
| Ag1/SiC | 98 | 10.8 | 0.96 | – | – | |||
| Ag2/SiC | 99 | 14.9 | 1.88 | – | 36 | |||
| Ag3/SiC | 98 | 29.4 | 2.78 | – | – | |||
| Mn5/SiC | 98 | 7.8 | – | – | – | |||
| Ag1Mn5/SiC | 98 | 80.4 | 0.82 | – | – | |||
| Ag2Mn5/SiC | 98 | 92.1 | 1.93 | – | 34 | |||
| Ag3Mn5/SiC | 97 | 93.1 | 2.90 | – | – | |||
| Ag4Mn5/SiC | 98 | 93.1 | 3.65 | – | – | |||
| Ag5Mn5/SiC | 96 | 93.1 | 4.66 | – | – | |||
| Ag2/SiC& Mn5/SiC | 97 | 24.3 | – | – | – | |||
| Nanoporous Ag | 99 | 97 | – | – | – | |||
| Ag/SiO2 | continuous-flow fixed-bed reactor (Pyrex, 15 mm i.d.), 0.2 g of catalyst total flow rate = 0.023 mol/min N2:O2:BA = 32:3:1 Temperature range: 500–650 K | 240 | 97.4 | 1.5 | 1.0 | 160–260 | – | [76] |
| Ag/SiO2 + Ca/SiO2 | 96.5 | 4.6 | 1.0 | – | ||||
| Ca–Ag/SiO2 | 99.6 | 66.7 | 1.0 | – |
| Catalyst | ω(Ag), %(wt) | Oxidant | T, °C | t, h | Solvent | X, % | Selectivity, % | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|
| BAld | BA | BB | ||||||||
| Ag/pumice | 0.6 | O2 | 75 | CH3CN | 4.5 a | 100 | [122] | |||
| Ag/SiO2 + CeO2 | 10 | O2 | reflux | 2 | xylene | 98 | 95 | [119] | ||
| Ag/MnO2 | 1 | O2 | 100 | 2 | toluene | 100 | >99 | [126] | ||
| Ag2O-MnO2 | 1 b | O2 | 100 | 0.6 | toluene | 67.0 | >99 | [128] | ||
| Ag2O-MnO2/1%HRG | 1 b | O2 | 100 | 0.6 | toluene | 70.6 | >99 | [128] | ||
| Ag2O-MnO2/3%HRG | 1 b | O2 | 100 | 0.6 | toluene | 84.0 | >99 | [128] | ||
| Ag2O-MnO2/5%HRG | 1 b | O2 | 100 | 0.6 | toluene | 100.0 | >99 | [128] | ||
| Ag2O-MnO2/7%HRG | 1 b | O2 | 100 | 0.6 | toluene | 96.0 | >99 | [128] | ||
| Ag2O-MnO2/5%HRG c | 1 b | O2 | 100 | 0.6 | toluene | 95 | >99 | [128] | ||
| Ag2O-MnO2/5%HRG d | 1 b | O2 | 100 | 0.6 | toluene | 100 | >99 | [128] | ||
| Ag2O-MnO2/5%HRG e | 1 b | O2 | 100 | 0.6 | toluene | 44 | >99 | [128] | ||
| Ag5%Au/MnO2 c | 5 | O2 | 100 | 1.5 | toluene | 69.51 | >99 | [127] | ||
| Ag5%Au/MnO2 d | 5 | O2 | 100 | 1.5 | toluene | 100 | >99 | [127] | ||
| Ag5%Au/MnO2 e | 5 | O2 | 100 | 1.5 | toluene | 19.90 | >99 | [127] | ||
| Ag/GOSH | 9.58 | O2 | 80 | 24 | MeCN | 7 | 19 | 36 | 45 | [125] |
| Ag/GOSH+NHPI | 9.58 | O2 | 80 | 24 | MeCN | 61 | 58 | 13 | 29 | [125] |
| Ag/GO+NHPI | 5.41 | O2 | 80 | 24 | MeCN | 33 | 55 | 18 | 27 | [125] |
| Ag/rGO+NHPI | 15.14 | O2 | 80 | 24 | MeCN | 12 | 8 | 25 | 67 | [125] |
| Ag/ZnO | 3 | TBHP | reflux | 0.25 | CH3CN | 90 | [135] | |||
| Ag/ZnO | 3 | TBHP | reflux | 0.25 | C2H5OH | 40 | [135] | |||
| Ag/ZnO | 3 | TBHP | reflux | 0.25 | CH2Cl2 | 30 | [135] | |||
| Ag/ZnO | 3 | H2O2 | reflux | 1 | CH3CN | 30 | [135] | |||
| 0.005% Ag/HT | 0.005 | - f | 130 | 16 | p-xylene | >99 | >99 | [130] | ||
| Fe3O4@SiO2-Ag | 3.2 | - f | reflux | 24 | toluene | 98 | 99 | [131] | ||
| Ag/Fe2O3 | 2(mol) | H2O2 | 80 | 12 | ~69 | ~90 | [132] | |||
| Ag@WEFA | - | -f | 4 | - | 96 g | [133] | ||||
| Au | O2 | 30 | 2 | P123-H2O | 50 | 21 | 50 | 28 | [124] | |
| Au0.99Ag0.01 | O2 | 30 | 2 | P123-H2O | 65 | 16 | 57 | 27 | [124] | |
| Au0.98Ag0.02 | O2 | 30 | 2 | P123-H2O | 74 | 10 | 65 | 25 | [124] | |
| Au0.95Ag0.05 | O2 | 30 | 2 | P123-H2O | 82 | 9 | 67 | 24 | [124] | |
| Au0.90Ag0.10 | O2 | 30 | 2 | P123-H2O | 77 | 10 | 65 | 25 | [124] | |
| Au0.85Ag0.15 | O2 | 30 | 2 | P123-H2O | 63 | 12 | 59 | 29 | [124] | |
| Catalyst | P(O2), bar | T, °C | t, h | NaOH/PG | X, % | Selectivity, % | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| LA | HA | FA | AcA | |||||||
| AgCA | 10 | 120 | 4 | 2 | 100 | 6.3 | - | 30.5 | 63.2 | [98] |
| AgSDBS | 10 | 120 | 4 | 2 | 81.9 | 48.1 | - | 17.6 | 35.3 | [98] |
| AgTween | 10 | 120 | 4 | 2 | 65.6 | 62.0 | - | 13.8 | 24.2 | [98] |
| AgDS | 10 | 120 | 4 | 2 | 58.8 | 27.4 | - | 23.8 | 48.8 | [98] |
| AgPVP | 10 | 120 | 4 | 2 | 42.6 | 22.1 | - | 22.1 | 42.5 | [98] |
| AgTween | 10 | 80 | 4 | 2 | 28.0 | 41.0 | - | 20 | 39 | [98] |
| Ag2/HAP | 10 | 100 | 2 | 2 | 15.7 | 57.2 | - | 14.4 | 28.4 | [140] |
| Ag1.8Pd0.2/HAP | 10 | 100 | 2 | 2 | 22.6 | 69.4 | - | 9.8 | 20.8 | [140] |
| Ag1.5Pd0.5/HAP | 10 | 100 | 2 | 2 | 64.3 | 85.0 | - | 4.5 | 10.5 | [140] |
| Ag1Pd1/HAP | 10 | 100 | 2 | 2 | 86.3 | 88.8 | - | 3.1 | 8.1 | [140] |
| Ag1Pd1/HAP | 10 | 80 | 2 | 2 | 62.3 | 91.0 | - | 3.1 | 5.9 | [140] |
| Ag | 1 | 85 | 4 | 2 | 8.6 | 73.2 | 7.1 | 5.7 | 14.4 | [139] |
| Ag0.95Pd0.05 | 1 | 85 | 4 | 2 | 31.2 | 87.4 | 5.8 | 2.2 | 4.6 | [139] |
| Ag0.85Pd0.15 | 1 | 85 | 4 | 2 | 61.8 | 93.3 | 3.1 | 1.32 | 2.3 | [139] |
| Ag0.7Pd0.3 | 1 | 85 | 4 | 2 | 88.2 | 78.4 | 0 | 6.4 | 15.2 | [139] |
| Ag0.5Pd0.5 | 1 | 85 | 4 | 2 | 99.8 | 76.0 | 0 | 8.6 | 15.5 | [139] |
| Ag0.85Pd0.15 | 1 | 95 | 4 | 2 | 73.2 | 96.2 | 0 | 1.2 | 2.6 | [139] |
| Catalyst | P(O2), Bar | T, °C | NaOH/GLY | t, h | X, % | Selectivity, % | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DHA | GlyAl | GlyA | GlycA | FA | LA | |||||||
| Pd/C+Ag/C | 3 | 80 | 0 | 4 | 1.8 | 70.7 | 18.8 | 7.1 | 2.2 | - | - | [146] |
| AgPd/C | 3 | 80 | 0 | 4 | 6.7 | 74.6 | 14.6 | 7.2 | 1.4 | - | - | [146] |
| Ag/C | 3 | 80 | 0 | 24 | 0.3 | 84.0 | 5.0 | 11.0 | <0.1 | - | - | [146] |
| AgPd/C | 3 | 80 | 0 | 24 | 24.5 | 79.1 | 3.6 | 11.2 | 5.8 | - | - | [146] |
| Pretreated 1Ag2Pd/C | 3 | 80 | 0 | 24 | 10.9 | 77.7 | 8.6 | 9.6 | 2.7 | - | - | [146] |
| Pretreated 1Ag1Pd/C | 3 | 80 | 0 | 24 | 20.0 | 82.2 | 4.6 | 8.0 | 3.4 | - | - | [146] |
| Pretreated 2Ag1Pd/C | 3 | 80 | 0 | 24 | 16.4 | 85 | 4.6 | 5.4 | 2.2 | - | - | [146] |
| 1Ag2Pd/SiO2 | 3 | 80 | 0 | 4 | 7.0 | 76.4 | 13.5 | 6.9 | 1.4 | - | - | [147] |
| 1Ag1Pd/SiO2 | 3 | 80 | 0 | 4 | 4.9 | 86.6 | 8.0 | 2.7 | 1.2 | - | - | [147] |
| 2Ag1Pd/SiO2 | 3 | 80 | 0 | 4 | 4.0 | 91.6 | 4.8 | 1.6 | 1.7 | - | - | [147] |
| 1Ag1Pd/TiO2 | 3 | 80 | 0 | 4 | 1.7 | 90.0 | 5.9 | 2.9 | 0.7 | - | - | [147] |
| 1Ag1Pd/Al2O3 | 3 | 80 | 0 | 4 | 6.8 | 86.4 | 7.3 | 3.4 | 0.6 | - | - | [147] |
| 1Ag1Pd/ZrO2 | 3 | 80 | 0 | 4 | 3.2 | 91.3 | 6.2 | 2.1 | 0.2 | - | - | [147] |
| 1Ag1Pd/CeO2 | 3 | 80 | 0 | 4 | 0.3 | 82.3 | 4.1 | 5.9 | 3.4 | - | - | [147] |
| Au/Al2O3 | 5 | 60 | 4 | - | - | - | - | 59.8 a 60.4 b | 19.7 a 20.7 b | 10.2 a 12.5 b | - | [148] |
| Pd/Al2O3 | 5 | 60 | 4 | - | - | - | - | 91.7 a 85.8 b | 2.8 a 2.6 b | 0 a 1.0 b | - | [148] |
| Pt/Al2O3 | 5 | 60 | 4 | - | - | - | - | 75.6 a 74.0 b | 10.7 a 9.9 b | 11.0 a 8.1 b | - | [148] |
| Ag/Al2O3 | 5 | 60 | 4 | - | - | - | - | 27.8 a 27.2 b | 35.9 a 44.8 b | 35.3 a 28.0 b | - | [148] |
| Ag/Al2O3 c | 5 | 60 | 4 | - | - | - | - | 49.5 a | 27.0 a | 17.6 a | - | [148] |
| Ag/Al2O3 calcination | 5 | 100 | 1 | 2 | 42.6 | - | - | 13.0 | 51.4 | 33.5 | - | [149] |
| 0.7% Ag/Al2O3 HCHO-MeOH | 5 | 100 | 1 | 2 | 19.9 | - | - | 24.9 | 39.4 | 34.2 | - | [149] |
| 1.1% Ag/Al2O3 HCHO-MeOH | 5 | 100 | 1 | 2 | 48.3 | - | - | 9.7 | 51.6 | 32.4 | - | [149] |
| 2.3% Ag/Al2O3 HCHO-MeOH | 5 | 100 | 1 | 2 | 48.6 | - | - | 9.4 | 54.1 | 32.6 | - | [149] |
| 3.6% Ag/Al2O3 HCHO-MeOH | 5 | 100 | 1 | 2 | 48.8 | - | - | 7.6 | 56.0 | 33.3 | - | [149] |
| Ag/Al2O3 N2H4-H2O | 5 | 100 | 1 | 2 | 32.8 | - | - | 15.8 | 44.8 | 32.3 | - | [149] |
| Ag/Al2O3 N2H4-MeOH | 5 | 100 | 1 | 2 | 48.5 | - | - | 10.0 | 50.6 | 31.7 | - | [149] |
| Ag/Al2O3 NaBH4-H2O | 5 | 100 | 1 | 2 | 41.2 | - | - | 14.4 | 44.3 | 30.8 | - | [149] |
| Ag/Al2O3 NaBH4-MeOH | 5 | 100 | 1 | 2 | 47.6 | - | - | 10.7 | 50.6 | 32.0 | - | [149] |
| 95Ag5Au/CeO2 | 5 | 60 | 4 | 5 | 43.8 | - | - | 23.3 | 46.2 | 25.2 | - | [151] |
| 95Ag5Pt/CeO2 | 5 | 60 | 4 | 5 | 54.2 | - | - | 18.9 | 51.0 | 27.4 | - | [151] |
| 95Ag5Pd/CeO2 | 5 | 60 | 4 | 5 | 37.1 | - | - | 25.8 | 44.9 | 24.5 | - | [151] |
| 90Ag10Pt/CeO2 | 5 | 60 | 4 | 5 | 43.1 | - | - | 19.2 | 50.7 | 27.9 | - | [151] |
| 50Ag50Pt/CeO2 | 5 | 60 | 4 | 5 | 20.0 | - | - | 48.3 | 28.4 | 20.6 | - | [151] |
| H3PMo12O40 | 5 | 60 | 0 | 5 | 83 | 3 | 3 | 4 | - | - | 72 | [155] |
| Ag3PMo12O40 | 5 | 60 | 0 | 5 | 89 | 6 | 2 | 3 | - | - | 81 | [155] |
| Ag2HPMo12O40 | 5 | 60 | 0 | 5 | 87 | 5 | 3 | 3 | - | - | 78 | [155] |
| Ag1H2PMo12O40 | 5 | 60 | 0 | 5 | 85 | 5 | 3 | 4 | - | - | 75 | [155] |
| 2%Au/ZnO | 6 | 60 | 2 | 5 | 50 | - | - | 61 | 21 | 10 | - | [156] |
| 2%Au2%Ag/ZnO | 6 | 60 | 2 | 5 | 10 | - | - | 77 | 8 | 7 | - | [156] |
| 2%Au1%Cu/ZnO | 6 | 60 | 2 | 5 | 95 | - | - | 59 | 17 | 12 | - | [156] |
| Substrate | Products | Lamp Properties | Catalyst | Preparation Technique | Efficiency | Ref. |
|---|---|---|---|---|---|---|
| BA | BAld BAc | 300 W Xe arc lamp | TiO2@Ag@ZnO TiO2@Au@ZnO TiO2@Pt@ZnO TiO2@Pd@ZnO | Sol-gel (support) Simple preparation (metal NPs) | TiO2@Ag@ZnO 86.2% conversion 51.1% BAld selectivity 33% BAc selectivity | [158] |
| BA | BAld BAc | 300 W Xe arc lamp λmax ≥ 420 nm cut-off filter | AgBr@TiO2 AgBr@Ag@TiO2 | Double-jet precipitation (AgBr@TiO2) Photodeposition (Ag) | O2 atmosphere AgBr@Ag@TiO2-0.325 In acetonitrile: 90% conversion, 95% selectivity In water: 38% conversion, 47% selectivity | [165] |
| BA | BAld | 300 W Xe arc lamp λmax ≥ 420 nm cut-off filter | AgBr@TiO2/GO | Hummers method (GO support) Hydrothermal method (core-shell structured catalyst) | 78% yield of BAld for AgBr@TiO2/GO | [164] |
| 4-methoxybenzyl alcohol (MBA) | 4-methoxybenzyl aldehyde | 1500 W solar simulator (Xe lamp) | Ag/TiO2, Pt/TiO2, Au/TiO2, Pd/TiO2 | Photodeposition | Aldehyde selectivity: pH = 1 100% for most catalysts pH = 7 30% for Pd-TiO2-0.5%-100 30% for Pt-TiO2-0.5%-100 15% for Ag-TiO2-0.5%-100 13% for Ag-TiO2-1%-100 pH = 13 32% for Au-TiO2-0.5%-100 33% for Pt-TiO2-0.5%-100 12% for Ag-TiO2-0.5%-100 21.5% for Ag-TiO2-1%-100 | [164] |
| BA 4-MBA Cinnamyl alcohol | BAld | Sunlight simulator | Ag3PO4 | Precipitation | BAld: >85% conversion >99% selectivity ~85% yield p-Anisaldehyde >85% conversion >99% selectivity ~85% yield Cinnamyl alcohol ~90% conversion >90% selectivity ~81% yield | [175] |
| Cyclohexanol Cycloheptanol | Cyclohexanone Cycloheptanone &others | 125 W Hg lamp 10.4 mW/cm2 | 0.5–20% wt. Ag/TiO2 &others | Chemical reduction by NaBH4 | 75 mmol of product vs. 375 mmol for TiO2 P25 | [185] |
| Methanol | Methyl formate Formaldehyde CO2 | 500 W Hg lamp | Au/TiO2 Ag/TiO2 AuAgTiO2 | Chemical reduction by NaBH4 | AuAgTiO2 82% conversion 84% selectivity | [177] |
| Methanol | Methyl formate | 500 W Hg lamp | Ag/TiO2 Ag/SiO2 | Chemical reduction by NaBH4 | ~22 mmol∙g−1∙h−1 for 3% Ag/TiO2 ~22 mmol∙g−1∙h−1 for 3% Ag/SiO2 | [178] |
| Cellobiose | Glucose | Mod. LZC-4, Luzchem Research Inc. ON, CAN 14 UVB (84 lx) or Vis (19,900 lx) lamps | Ag/TiO2, Au/TiO2, AgAu/TiO2 | Wetness impregnation; Colloidal methods | 36% conversion, 2% glucose selectivity vs. 57% conversion, 3% glucose selectivity for TiO2 P25 | [183] |
| BA | BAld BAc Benzylbenzoate CO2 &others | 250 W Hg lamp λmax = 365 nm | Ag/TiO2 Au/TiO2 Pt/TiO2 Pd/TiO2 Rh/TiO2 Ir/TiO2 | Photodeposition | 81% BAld selectivity for Ag/TiO2 92% BAld selectivity for Ir/TiO2 | [167] |
| 2-propanol | Acetone CO2 | λmax = 365 nm 3.25 W/cm2 | Ag0.01Ti0.99O2 | Sol-gel + Magnetic stirring or ultrasound treatment | 38–40% conversion 97% acetone selectivity vs. sol-gel TiO2: 34–38% conversion 97% acetone selectivity vs. TiO2 P25: 46% conversion 97% acetone selectivity | [184] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torbina, V.V.; Vodyankin, A.A.; Ten, S.; Mamontov, G.V.; Salaev, M.A.; Sobolev, V.I.; Vodyankina, O.V. Ag-Based Catalysts in Heterogeneous Selective Oxidation of Alcohols: A Review. Catalysts 2018, 8, 447. https://doi.org/10.3390/catal8100447
Torbina VV, Vodyankin AA, Ten S, Mamontov GV, Salaev MA, Sobolev VI, Vodyankina OV. Ag-Based Catalysts in Heterogeneous Selective Oxidation of Alcohols: A Review. Catalysts. 2018; 8(10):447. https://doi.org/10.3390/catal8100447
Chicago/Turabian StyleTorbina, Viktoriia V., Andrei A. Vodyankin, Sergey Ten, Grigory V. Mamontov, Mikhail A. Salaev, Vladimir I. Sobolev, and Olga V. Vodyankina. 2018. "Ag-Based Catalysts in Heterogeneous Selective Oxidation of Alcohols: A Review" Catalysts 8, no. 10: 447. https://doi.org/10.3390/catal8100447