
Pentamethylcyclopentadienyl Molybdenum(V) Complexes Derived from Iodoanilines: Synthesis, Structure, and ROP of ε-Caprolactone



Abstract
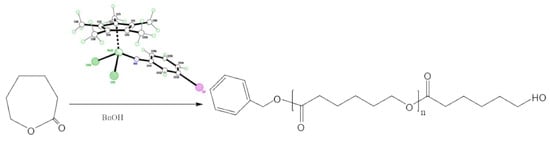
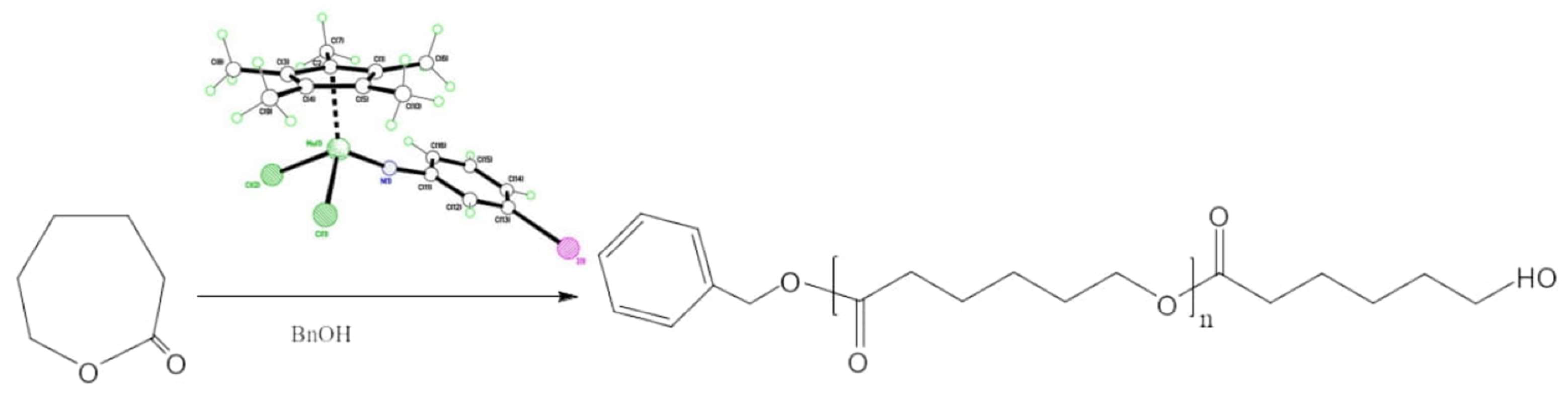
:
1. Introduction
2. Results
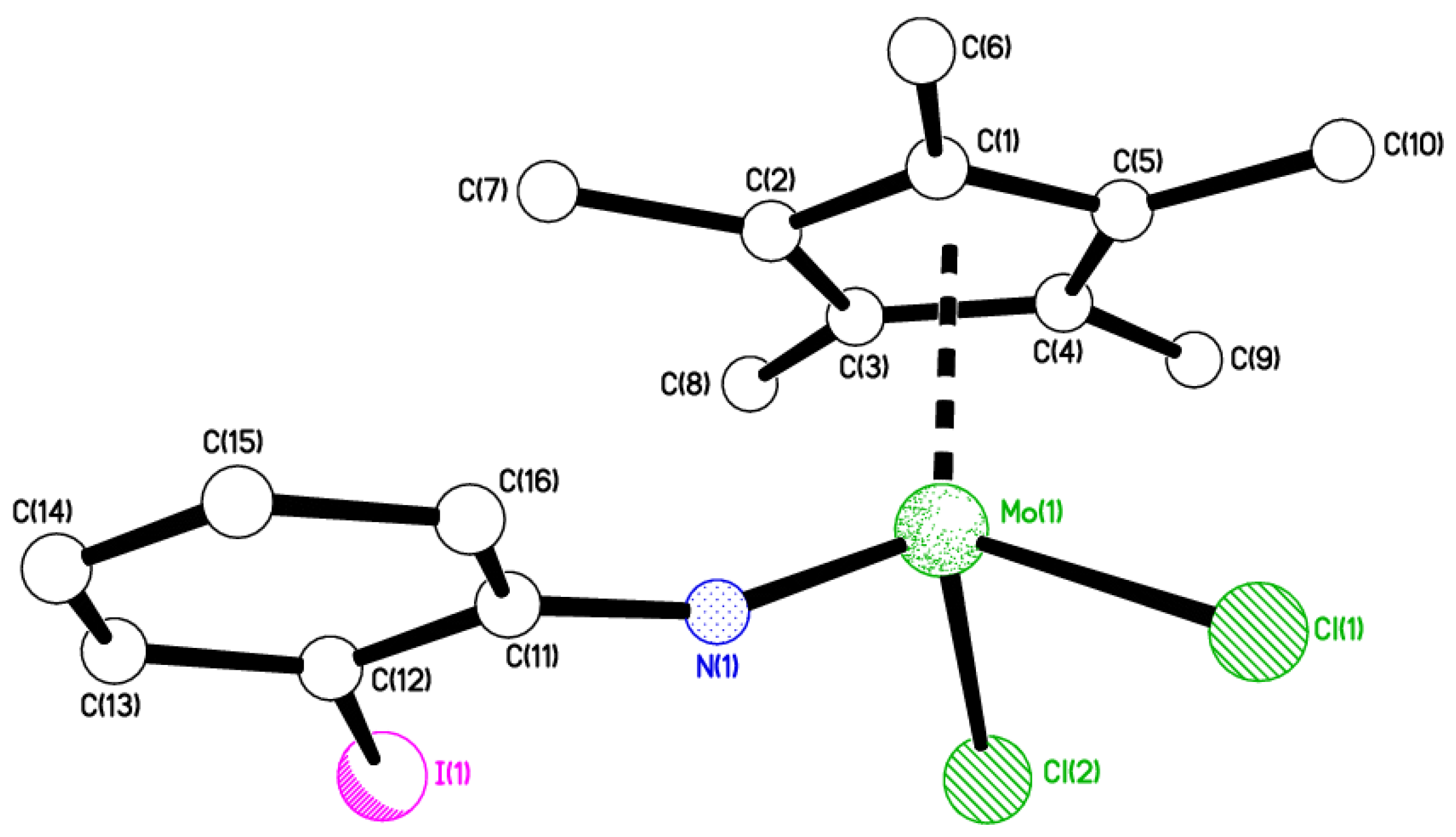
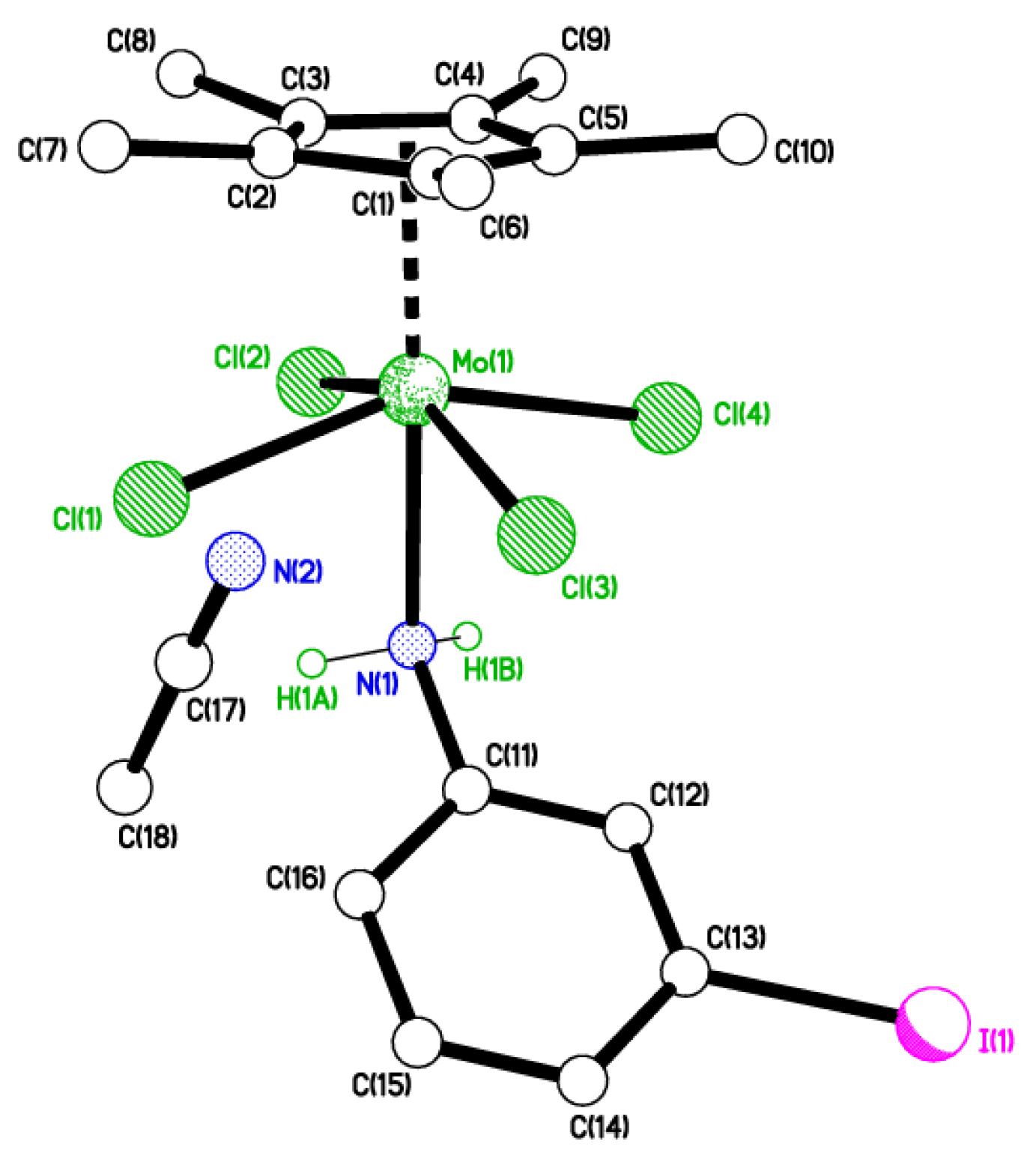
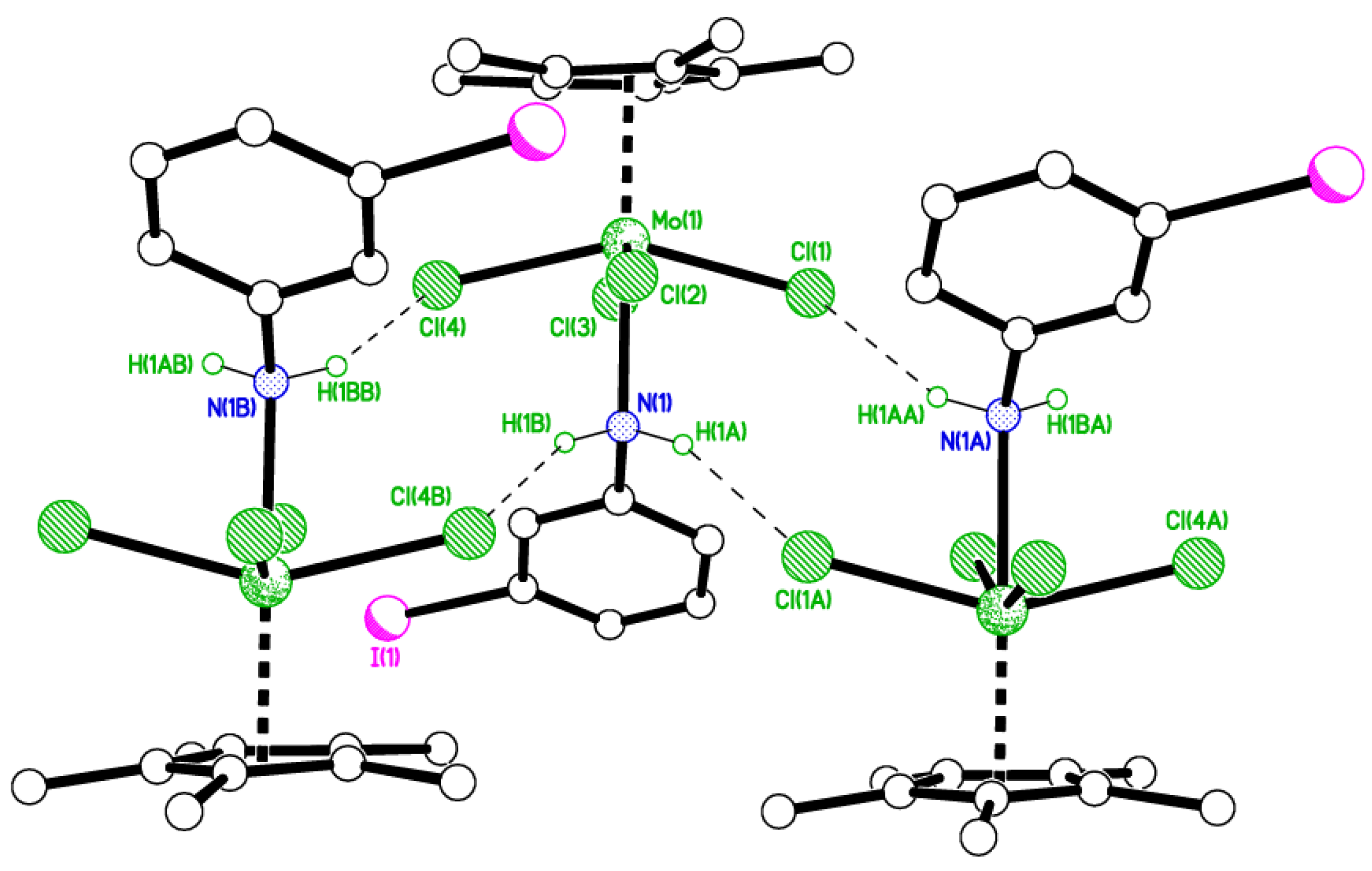
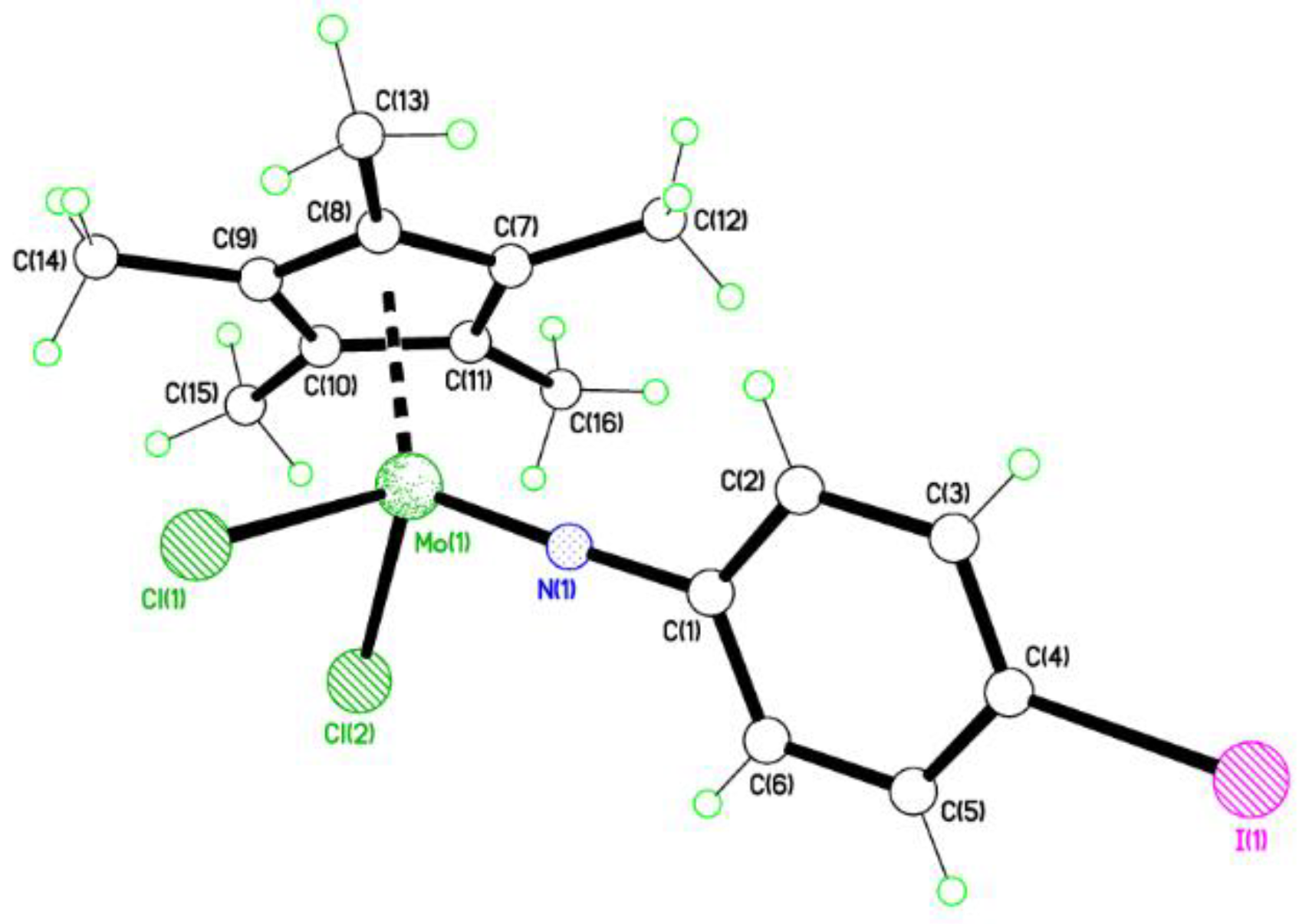
2.1. 2-Iodo Complexes
2.2. 3-Iodo Complexes
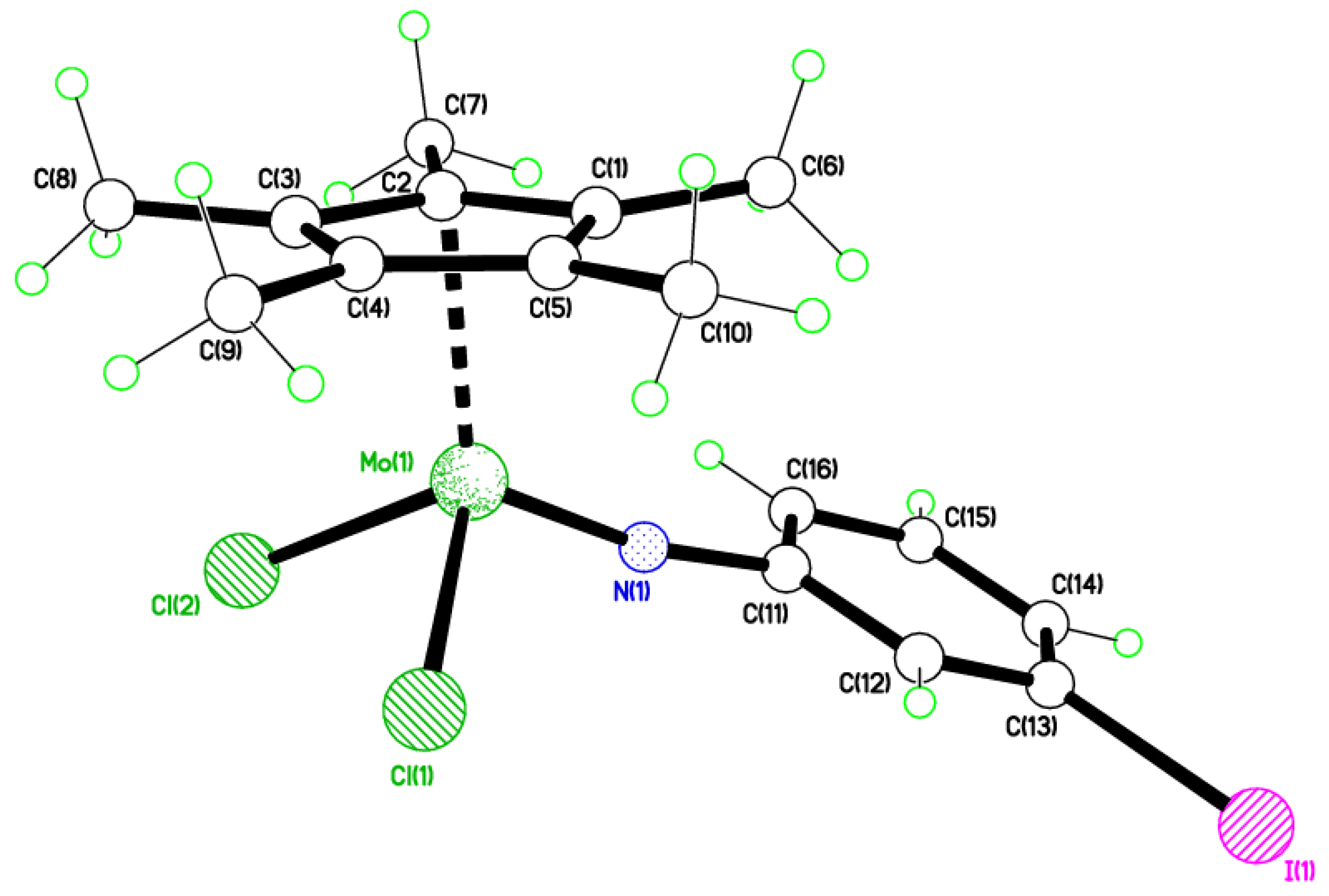
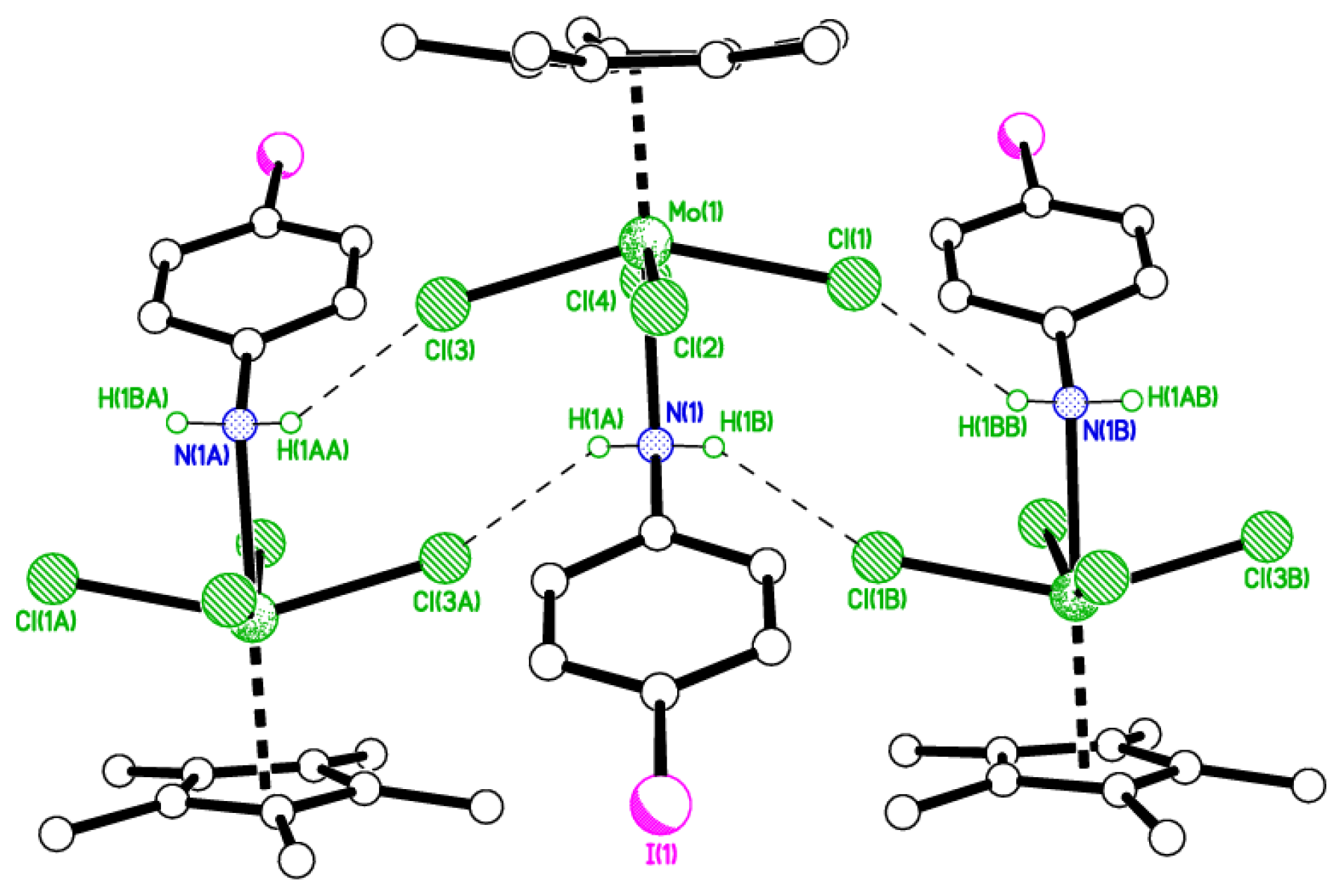
2.3. 4-Iodo Complexes
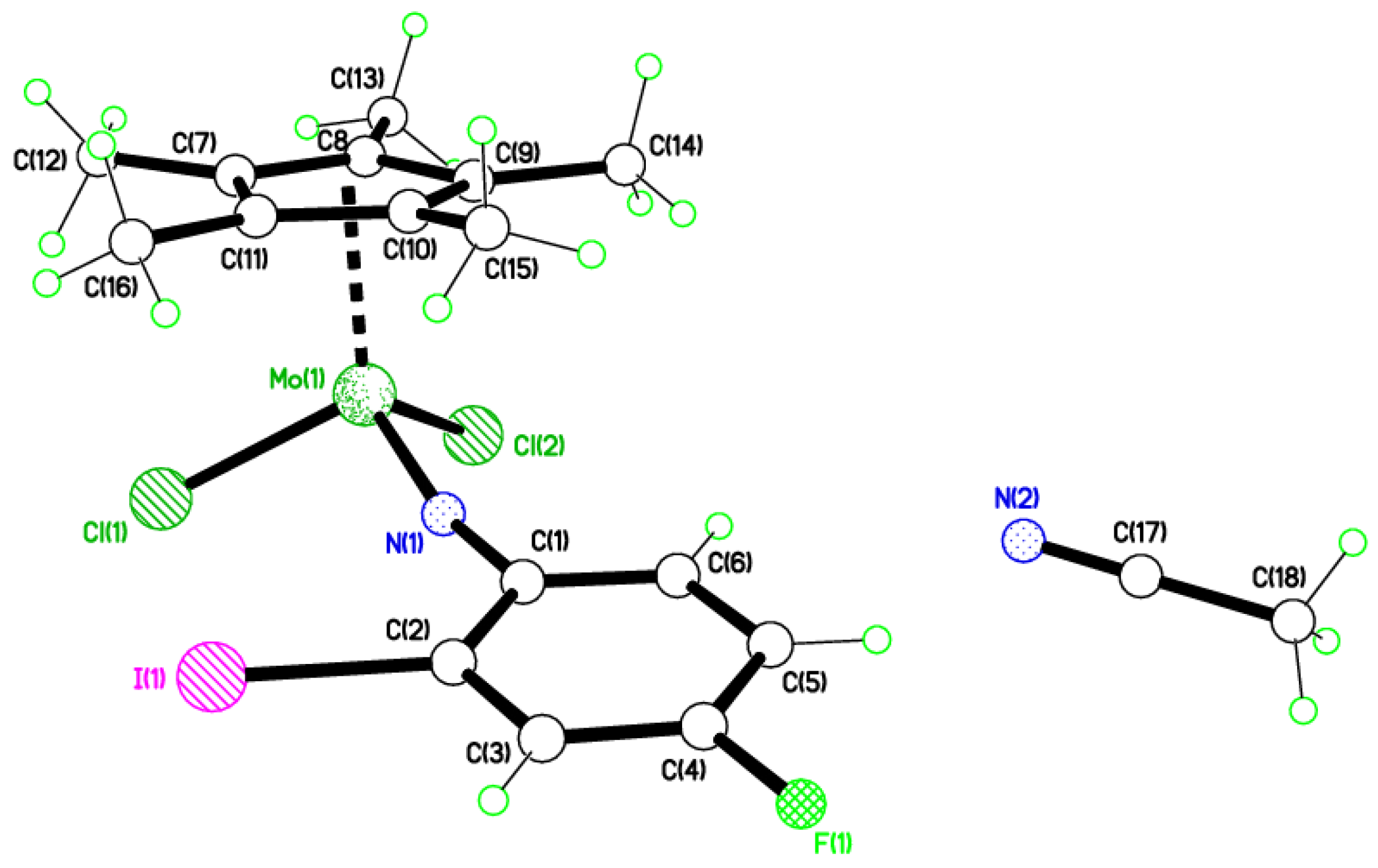
2.4. Use of 2-I,4-FC6H3NH2
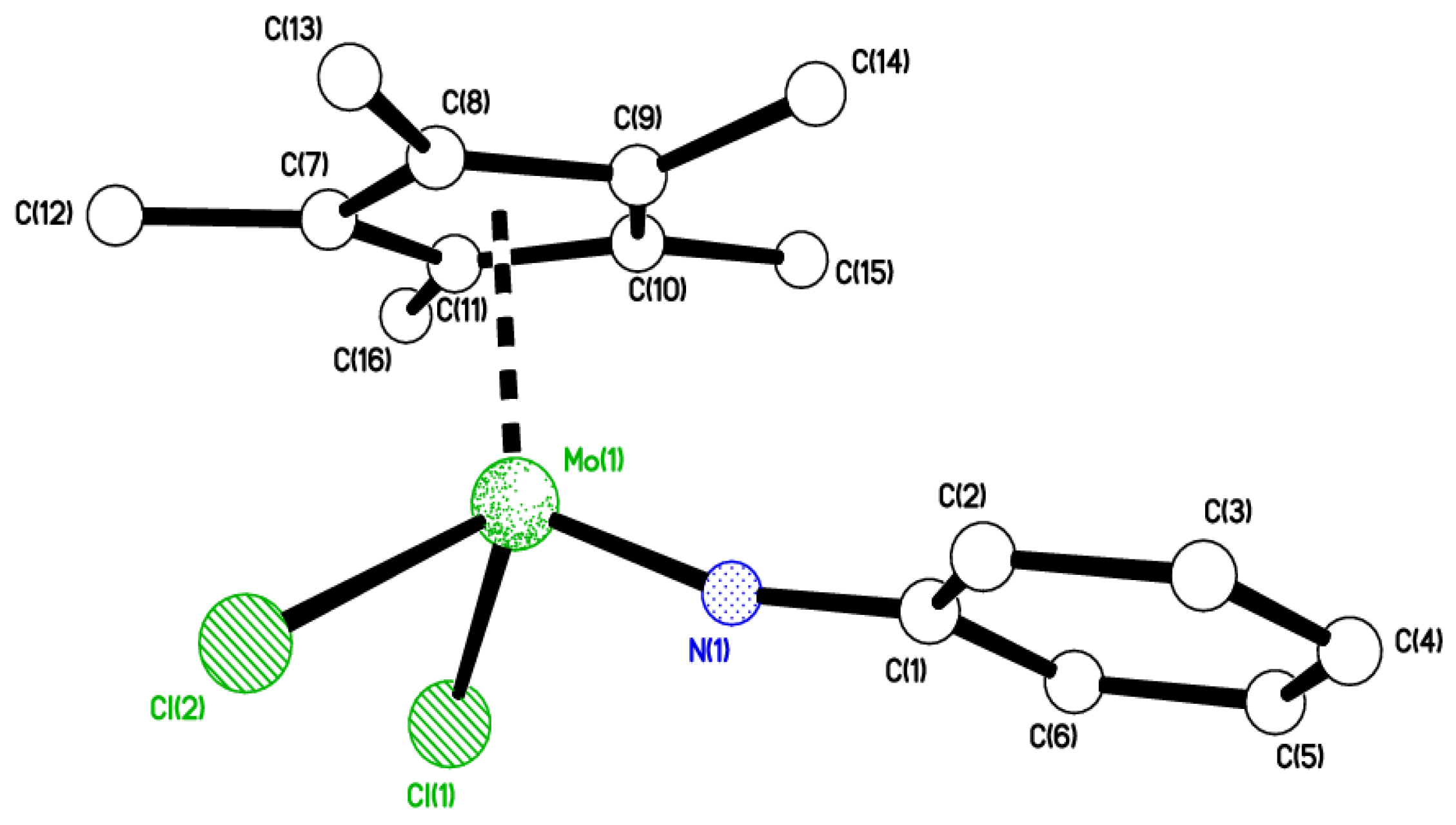
2.5. Use of Aniline
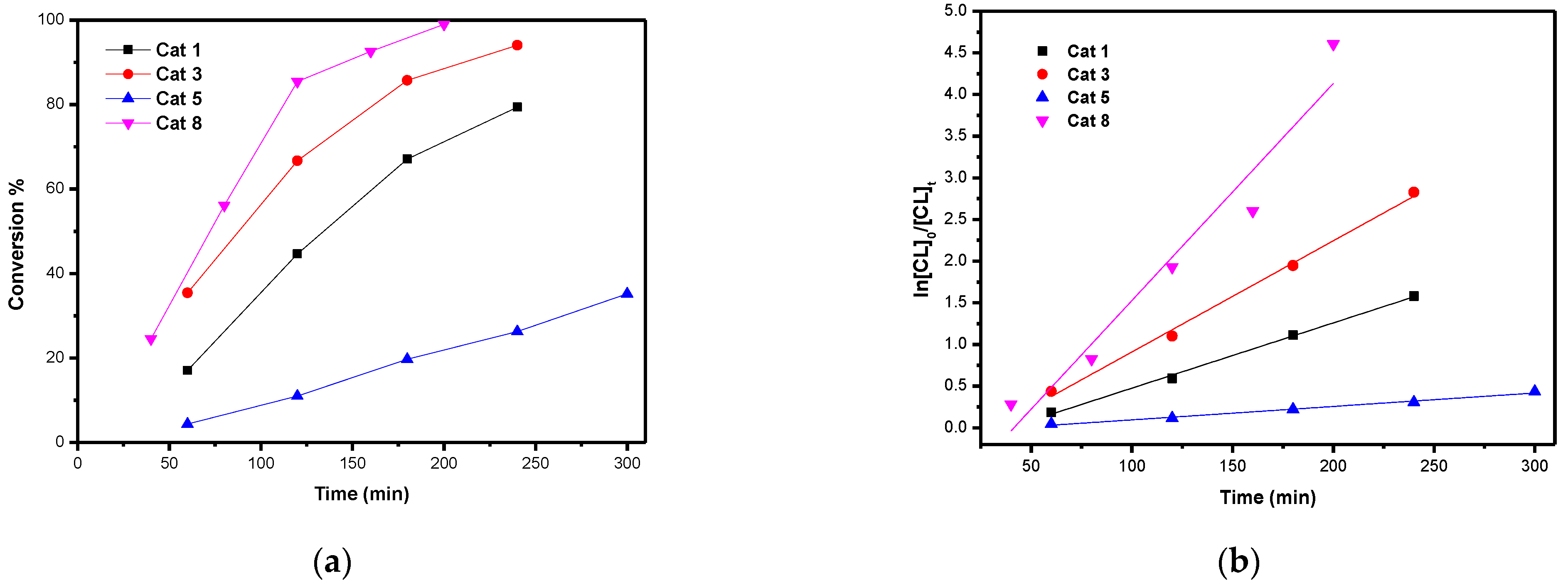
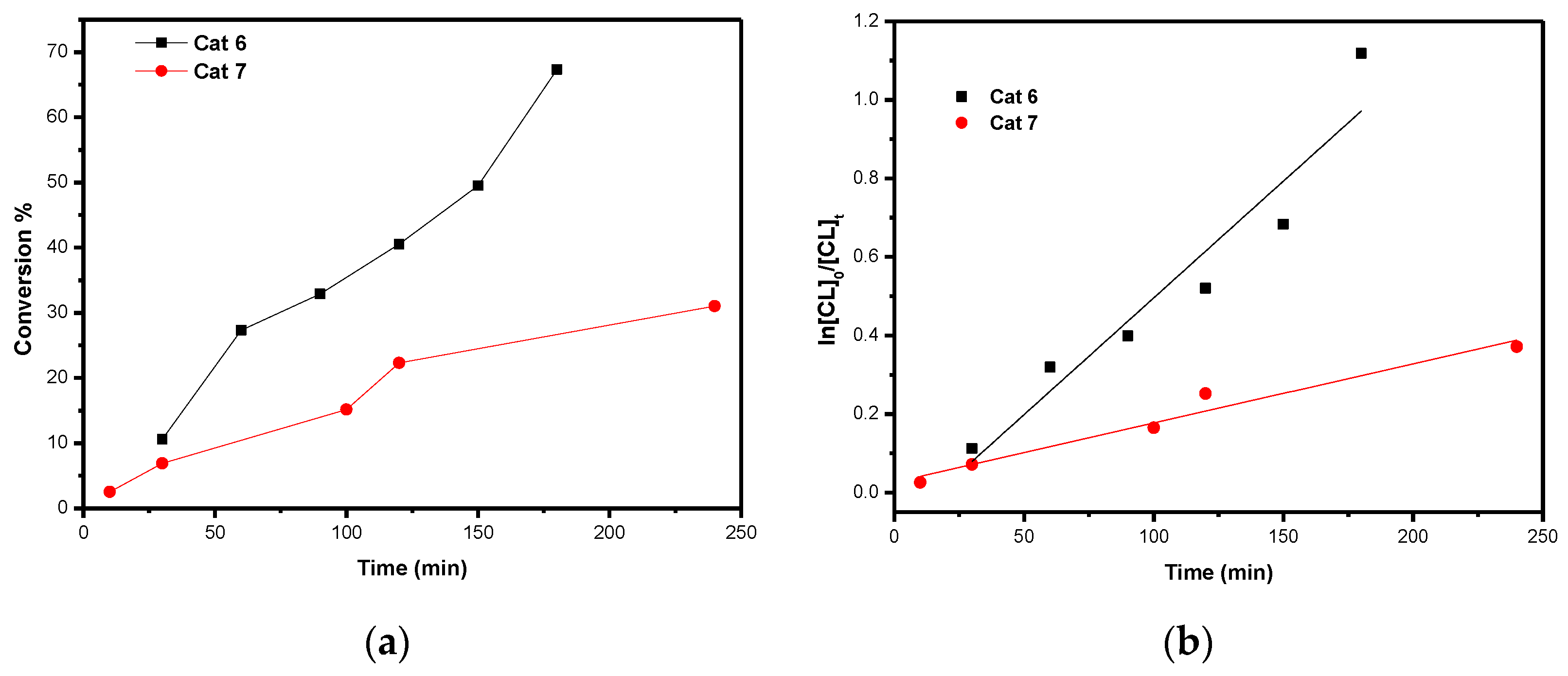
3. Ring Opening Polymerization (ROP) Studies
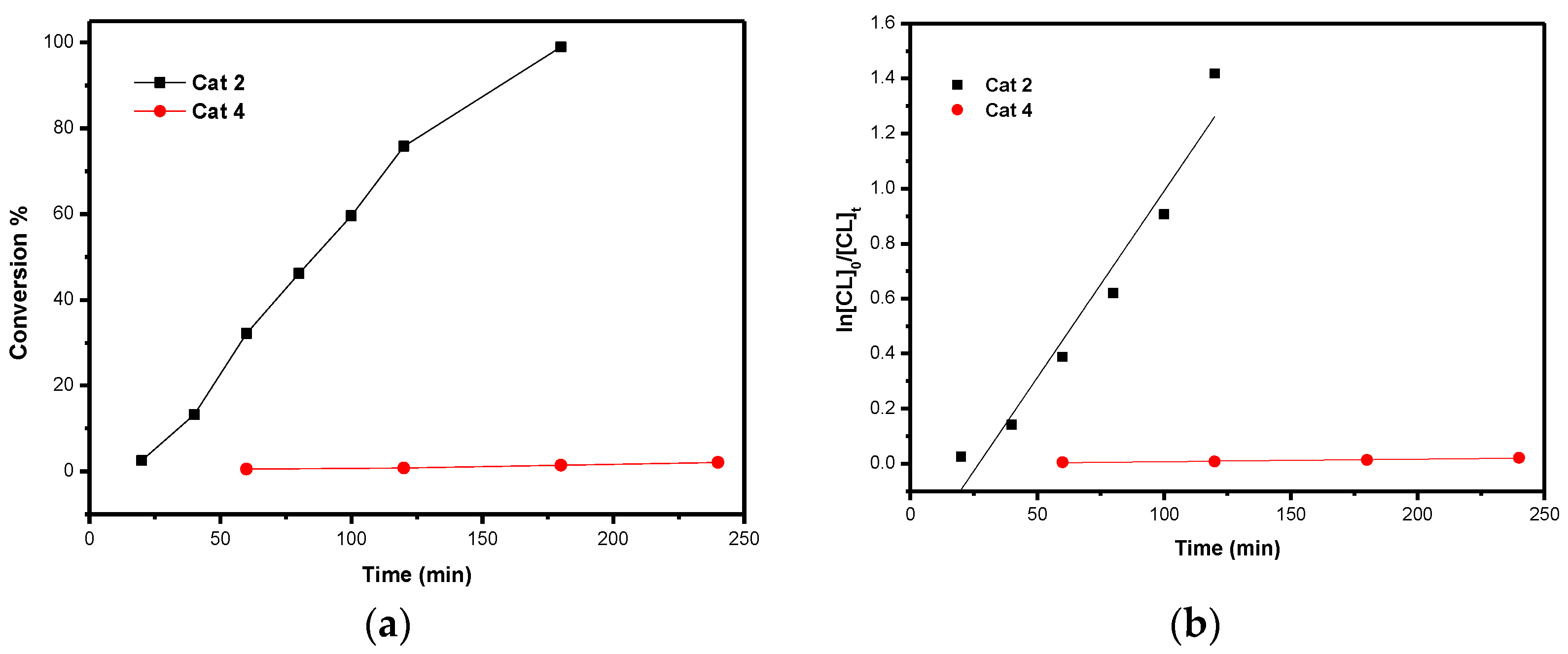
Kinetics
4. Materials and Methods
4.1. General
4.2. Synthesis of [Mo(η-C5Me5)Cl2(2-IC6H4N)] (1)
4.3. Synthesis of [Mo(η-C5Me5)Cl4(3-IC6H4NH2)]∙MeCN (2∙MeCN)
4.4. Synthesis of [Mo(η-C5Me5)Cl2(3-IC6H4N)] (3)
4.5. Synthesis of [Mo(η-C5Me5)Cl4(4-IC6H4NH2)] (4)
4.6. Synthesis of [Mo(η-C5Me5)Cl2(4-IC6H4N)] (5)
4.7. Synthesis of [Mo(η-C5Me5)Cl2(2-I,4-FC6H3N)]∙MeCN (6∙MeCN)
4.8. Synthesis of [Mo(η-C5Me5)Cl3(2-I,4-FC6H3N)] (7)
4.9. Synthesis of [Mo(η-C5Me5)Cl2(C6H5N)] (8)
4.10. ROP of ε-Caprolactone (ε-CL)
4.11. Polymerization Kinetics
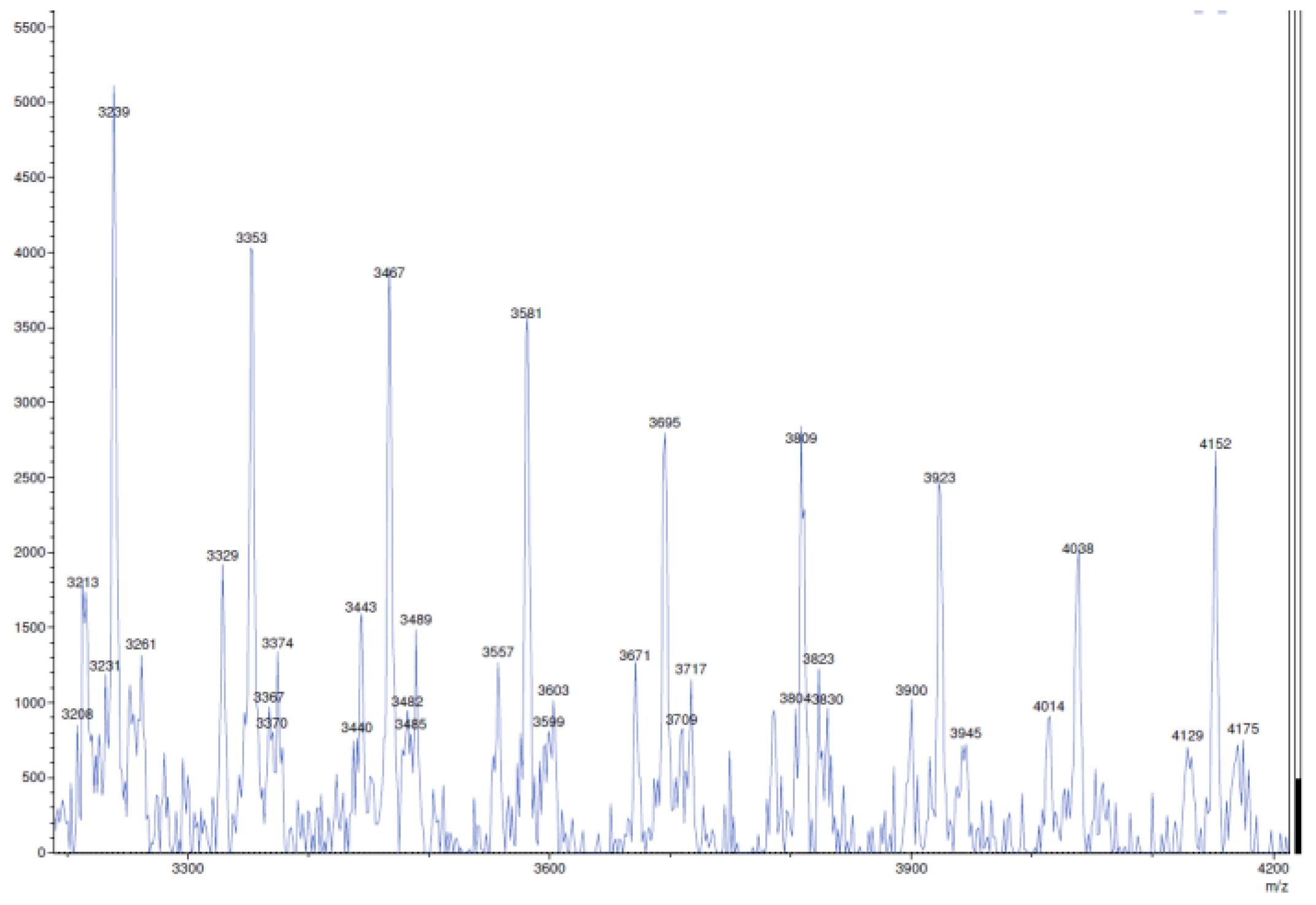
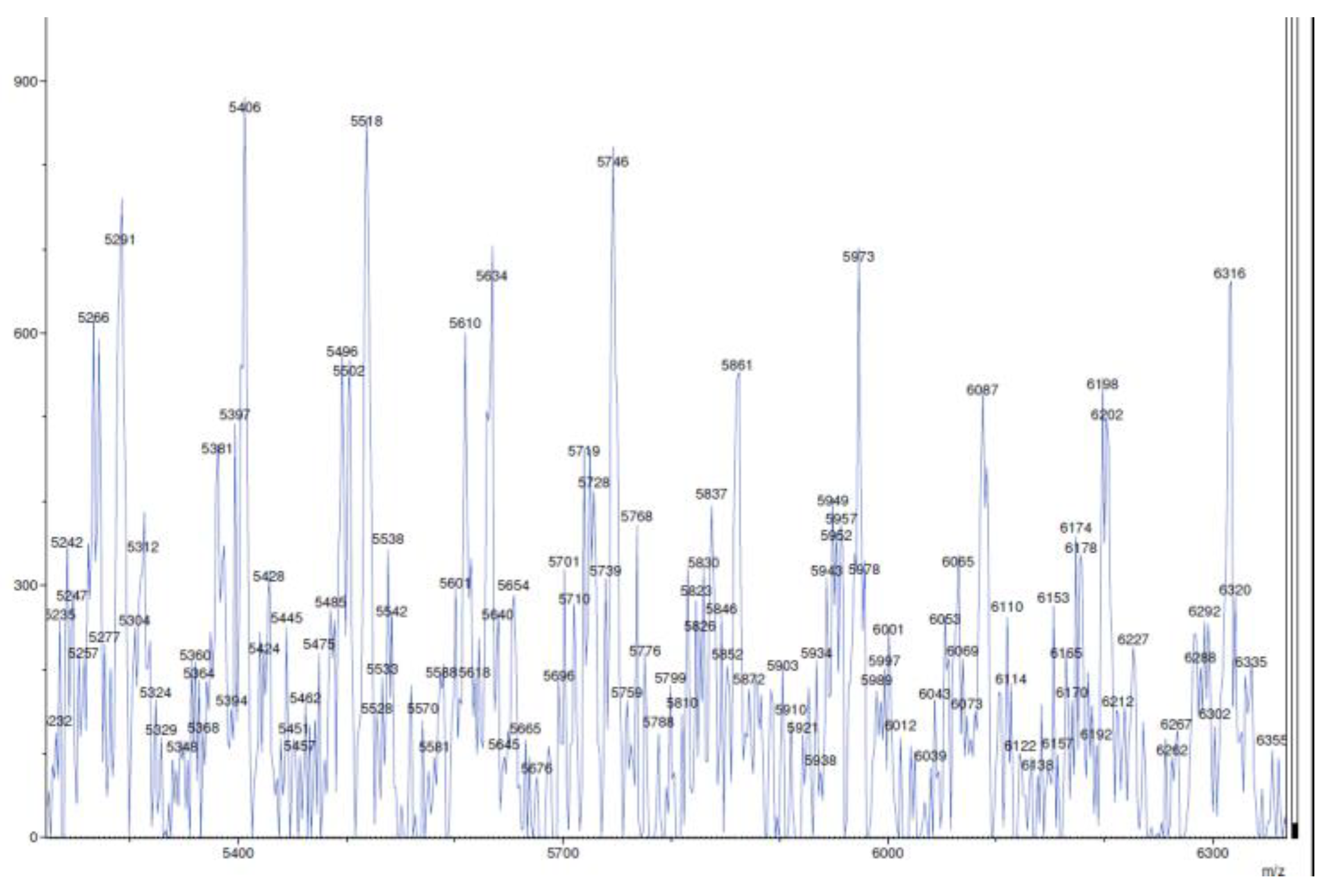
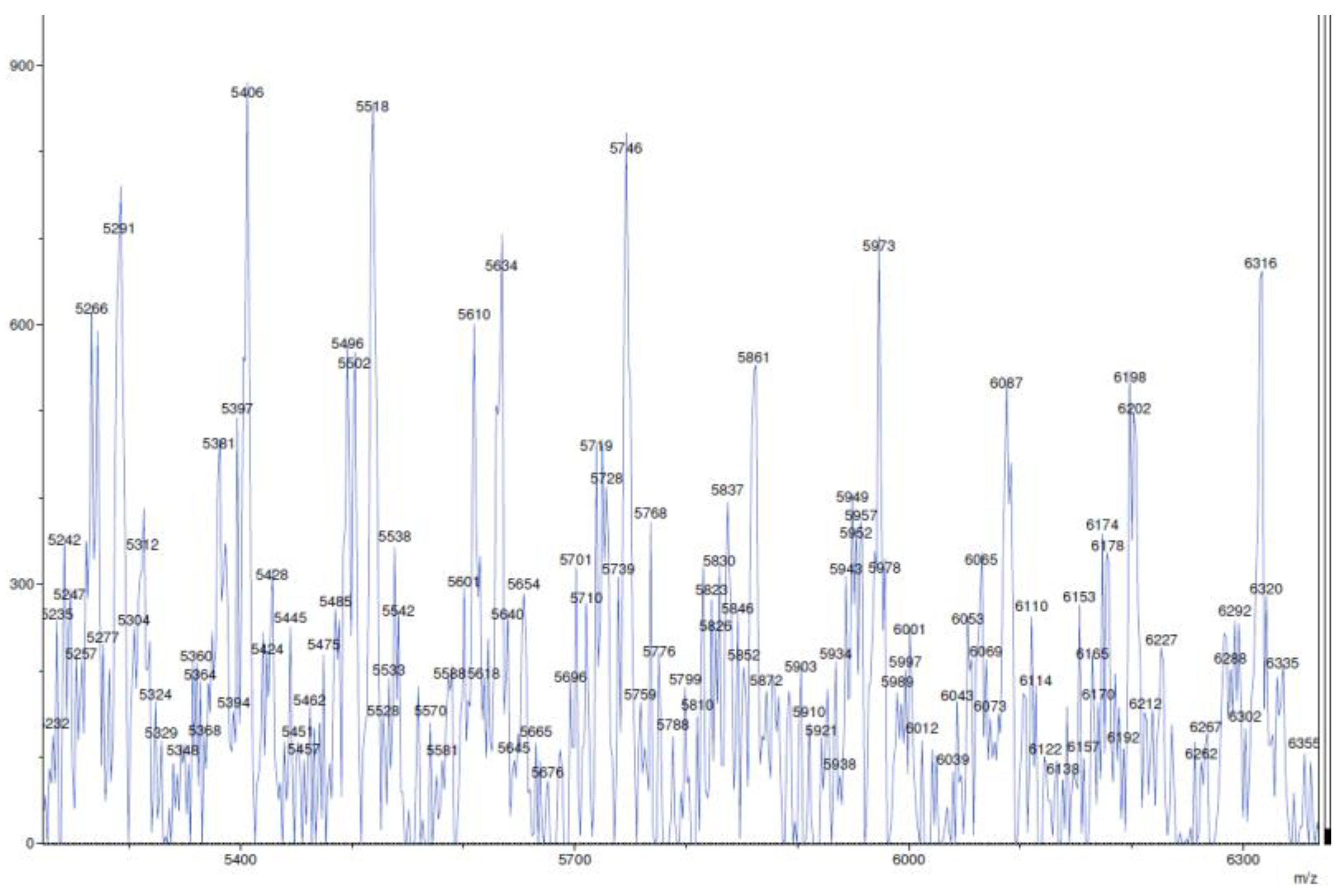
4.12. Polymer Samples Preparation for MALDI-TOF
4.13. Crystal Structure Determinations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schrock, R.R.; Hoveyda, A.H. Molybdenum and tungsten imido alkylidene complexes as efficient olefin-metathesis catalysts. Angew. Chem. Int. Ed. 2003, 42, 4592–4633. [Google Scholar] [CrossRef]
- Gibson, V.C.; Redshaw, C.; Clegg, W.; Elsegood, M.R.J. Synthesis and characterization of molybdenum complexes bearing highly functionalized imido substituents. J. Chem. Soc. Dalton Trans. 1997, 18, 3207–3212. [Google Scholar] [CrossRef]
- Redshaw, C.; Wilkinson, G.; Sweet, T.K.N.; Hursthouse, M.B. Synthesis and X-ray structure of the organotungsten(V) o-phenylene diamido complex (η-C5Me5)WCl2{1,2-(HN)2C6H4}. Polyhedron 1993, 12, 2417–2420. [Google Scholar] [CrossRef]
- Redshaw, C.; Gibson, V.C.; Clegg, W.; Edwards, A.J.; Miles, B. Pentamethylcyclopentadienyl tungsten complexes containing imido, hydrazido and amino acid derived N-O chelate ligands. J. Chem. Soc. Dalton Trans. 1997, 18, 3343–3347. [Google Scholar] [CrossRef]
- Redshaw, C.; Wood, P.T.; Elsegood, M.R.J. Synthesis and structure of pentamethylcyclopentadienyl tungsten(V) complexes containing functionalized 6,12-epiiminodibenzo[b,f][1.5]diazocine ligands. Organometallics 2007, 26, 6501–6504. [Google Scholar] [CrossRef]
- Murray, R.C.; Blum, L.; Liu, A.H.; Schrock, R.R. Simple routes to mono(η5-pentamethylcyclopentadienyl) complexes of molybdenum(V) and tungsten(VI). Organometallics 1985, 4, 953–954. [Google Scholar] [CrossRef]
- Radius, U.; Sundermeyer, J. Organometall-Imide—Höhervalente Derivate der d-Metall-Säure, 2. Alkyl-, Aryl- und Cyclopentadienyl-substituierte Molybdän(VI)- und Wolfram(VI)-Imide. Chem. Ber. 1992, 125, 2183–2186. [Google Scholar] [CrossRef]
- Sundermeyer, J.; Radius, U.; Burschka, C. Organometall-Imide—Höhervalente Derivate der d-Metall-Säuren, 3. Synthese und Reaktionen von (Pentamethylcyclopentadienyl)(imido)-Komplexen des Molybdäns und Wolframs und eine effiziente Strategie zur Synthese der Organometallate NBu4[Cp*MO3] (M = Mo, W). Chem. Ber. 1992, 125, 2379–2384. [Google Scholar] [CrossRef]
- Abugideiri, F.; Brewer, G.A.; Desai, J.U.; Gordon, J.C.; Poli, R. (Pentamethylcyclopentadienyl)molybdenum(IV) chloride. Synthesis, Structure, and Properties. Inorg. Chem. 1994, 33, 3745–3751. [Google Scholar] [CrossRef]
- Köhler, K.; Roesky, H.W.; Herzog, A.; Gornitzka, H.; Steiner, A.; Usón, I. Syntheses, Structures, and Reactivity of a Series of (Pentamethylcyclopentadienyl)molybdenum(V) and Tungsten(V) Imido Complexes. Inorg. Chem. 1996, 35, 1773–1777. [Google Scholar] [CrossRef]
- Pedraz, T.; Pellinghelli, M.A.; Royo, P.; Tiripicchio, A.; Vázquez de Miquel, A. Preparation of imido pentamethylcyclopentadienyl molybdenum(IV) complexes X-ray molecular structure of cis-[MoCp* Cl(μ-NtBu)]2·C6H6. J. Organomet. Chem. 1997, 534, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Sal, P.G.; Jiménez, I.; Martín, A.; Pedraz, T.; Royo, P.; Sellés, A.; Vázquez de Miguel, A. Synthesis of chloro and methyl imido cyclopentadienyl molybdenum and tungsten complexes. X-ray molecular structures of [WCp*Cl3(NtBu)], [MoCp*ClMe2(NtBu)] and [WCp*ClMe2(NtBu)]. Inorg. Chimia Acta 1998, 273, 270–278. [Google Scholar]
- Kubo, M.; Nakanishi, M.; Kimura, M. Process for the Preparation of Lactone Polyesters. Germany Patent No. DE2947978, 15 September 1981. [Google Scholar]
- Báez, J.E.; Martĭnez-Rosales, M.; Martĭnez-Richa, A. Ring-opening polymerization of lactones catalyzed by decamolybdate anion. Polymer 2003, 44, 6767–6772. [Google Scholar] [CrossRef]
- Báez, J.E.; Martĭnez-Richa, A. Synthesis and characterization of poly(ε-caprolactone) and copolyesters by catalysis with molybdenum compounds: Polymers with acid-functional asymmetric telechelic architecture. Polymer 2005, 46, 12118–12129. [Google Scholar] [CrossRef]
- Báez, J.E.; Marcos-Fernández, A.; Martĭnez-Richa, A. One-step route to α-hydroxyl-ω-(carboxylic acid) polylactones using catalysis by decamolybdate anion. Macromolecules 2005, 38, 1599–1608. [Google Scholar] [CrossRef]
- Mahha, Y.; Atlamsani, A.; Blais, J.-C.; Tessier, M.; Brégeault, J.M.; Salles, L. Oligomerization of ɛ-caprolactone and δ-valerolactone using heteropolyacid initiators and vanadium or molybdenum complexes. J. Mol. Catal. A Chem. 2005, 234, 63–73. [Google Scholar] [CrossRef]
- Reyes-López, S.Y.; Richa, A.M. The ring-opening polymerization of ε-caprolactone catalyzed by molybdenum trioxide: A kinetic approach study using NMR and DSC data. Macromol. Symp. 2013, 325–326, 21–37. [Google Scholar] [CrossRef]
- Maruta, Y.; Abiko, A. Bis(salicylaldehydato)dioxomolybdenum complexes: Catalysis for ring-opening polymerization. Polym. Bull. 2014, 71, 1433–1440. [Google Scholar] [CrossRef]
- Yang, W.; Zhao, Q.-K.; Redshaw, C.; Elsegood, M.R.J. Molybdenum complexes derived from the oxydianiline [(2-NH2C6H4)2O]: Synthesis, characterization and ε-caprolactone ROP capability. Dalton Trans. 2015, 44, 13133–13140. [Google Scholar] [CrossRef]
- Al-Khafaji, Y.; Prior, T.J.; Elsegood, M.R.J.; Redshaw, C. Molybdenum(VI) imido complexes derived from chelating phenols: Synthesis, characterization and ɛ-caprolactone ROP capability. Catalysts 2015, 5, 1928–1947. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Zhao, Y.; Prior, T.J.; Elsegood, M.R.J.; Wang, K.; Xing, T.; Redshaw, C. Mono-oxo molybdenum(VI) and tungsten(VI) complexes bearing chelating aryloxides: Synthesis, structure and ring opening polymerization of cyclic esters. Dalton Trans. 2019, 48, 1454–1466. [Google Scholar] [CrossRef] [Green Version]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulfield, D.; Engelage, E.; Mancheski, L.; Stoesser, J.; Huber, S.M. Crystal engineering with multipoint halogen bonding: Double two-point donors and acceptors at work. Chem. Eur. J. 2020, 26, 1567–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitrescu, D.; Shova, S.; Man, I.C.; Caira, M.R.; Popa, M.M.; Dumitrascu, F. 5-Iodo-1-arylpyrazoles as potential benchmarks for investigating the tuning of the halogen bonding. Crystals 2020, 10, 1149. [Google Scholar] [CrossRef]
- Kawakita, K.; Kakiuchi, Y.; Tsurugi, H.; Mashima, K.; Parker, B.F.; Arnold, J.; Tonks, I.A. Reactivity of terminal imido complexes of group 4—6 metals: Stoichiometric and catalytic reactions involving cycloaddition with unsaturated organic molecules. Coord. Chem. Rev. 2020, 407, 213118. [Google Scholar] [CrossRef]
- Nugent, W.A.; Haymore, B.L. Transition metal complexes containing organoimido (nr) and related ligands. Coord. Chem. Rev. 1980, 31, 123–175. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3–8. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Cat. | Temp (°C) | Conv. (%) b | Mn c | MnCalcd d | PDI e |
|---|---|---|---|---|---|---|
| 1 | 1 | 130 | 99 | 1590 | 56,499 | 1.35 |
| 2 | 1 | 70 | 99 | 4650 | 56,499 | 1.49 |
| 3 | 1 | 15 | 99 | 850 | 56,499 | 1.25 |
| 4 | 2 | 130 | 98.6 | 6840 | 56,271 | 1.33 |
| 5 | 2 | 70 | 99 | 5740 | 56,499 | 1.50 |
| 6 | 2 | 15 | 99 | 890 | 56,499 | 1.21 |
| 7 | 3 | 130 | >99 | 16,710 | 56,499 | 1.76 |
| 8 | 3 | 15 | 66.2 | 400 | 37,780 | 1.27 |
| 9 | 4 | 130 | 99 | 1015 | 56,499 | 1.62 |
| 10 | 4 | 70 | 99 | 2490 | 56,499 | 1.19 |
| 11 | 4 | 15 | 99 | 840 | 56,499 | 1.25 |
| 12 | 5 | 130 | 97.4 | 12,300 | 55,586 | 1.76 |
| 13 | 5 | 15 | 97.9 | 900 | 55,872 | 1.22 |
| 14 | 6 | 130 | 98.5 | 5700 | 56,214 | 1.46 |
| 15 | 6 | 15 | 96.3 | 840 | 54,958 | 1.09 |
| 16 | 7 | 130 | 98.7 | 21,470 | 56,328 | 2.10 |
| 17 | 7 | 15 | 96 | 850 | 54,787 | 1.08 |
| 18 | 8 | 130 | 99 | 7740 | 56,499 | 2.12 |
| 19 | 8 | 70 | 99 | 1560 | 56,499 | 1.30 |
| 20 | 8 | 20 | 94.9 | 470 | 54,159 | 1.19 |
| Entry | Cat. | Conv. (%) b | Mn c | MnCalcd d | PDI e |
|---|---|---|---|---|---|
| 1 | 1 | 94.7 | 12,490 | 54,045 | 2.41 |
| 2 | 2 | 97.6 | 3510 | 55,700 | 1.53 |
| 3 | 3 | >99 | 18,290 | 56,499 | 2.05 |
| 4 | 4 | 97.4 | 5820 | 55,586 | 1.37 |
| 5 | 5 | 98.5 | 26,780 | 56,214 | 2.00 |
| 6 | 6 | >99 | 19,580 | 56,499 | 1.76 |
| 7 | 7 | >99 | 12,450 | 56,499 | 1.22 |
| 8 | 8 | >99 | 10,990 | 56,499 | 2.35 |
| Entry | Cat. | Conv. (%) c | Mn d | MnCalcd e | PDI f |
|---|---|---|---|---|---|
| 1 | 1 | 89.6 | 17,050 | 51,135 | 2.41 |
| 2 | 2 | 99 | 18,390 | 56,499 | 2.47 |
| 3 | 3 | 92.2 | 21,600 | 52,619 | 2.17 |
| 4 | 4 | 97.8 | 21,040 | 55,814 | 2.45 |
| 5 | 5 | >99 | 20,660 | 56,499 | 2.27 |
| 6 | 6 | >99 | 8640 | 56,499 | 1.71 |
| 7 | 7 | >99 | 7000 | 56,499 | 2.47 |
| 8 | 8 f | 99 | 710 | 56,499 | 1.38 |
| 9 | 8 | >99 | 7470 | 56,499 | 1.81 |
| Compound | 1 | 2·MeCN | 3 | 4 |
| Formula | C16H19Cl2NIMo | C16H21Cl4NIMo·(C2H3N) | C16H19Cl2NIMo | C16H21Cl4NIMo |
| Formula weight | 519.06 | 633.03 | 519.06 | 591.98 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | P21/c | P21/c |
| a (Å) | 13.959(7) | 8.6986(4) | 14.2972(2) | 11.8409(3) |
| b (Å) | 7.926(4) | 22.7727(7) | 17.0160(2) | 9.8584(2) |
| c (Å) | 16.248(8) | 11.3268(10) | 7.60500(12) | 17.9445(4) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 97.953(10) | 92.068(5) | 101.7956(15) | 105.356(3) |
| γ (°) | 90 | 90 | 90 | 90 |
| V (Å3) | 1780.4(15) | 2242.3(2) | 1811.08(4) | 2019.92(8) |
| Z | 4 | 4 | 4 | 4 |
| Temperature (K) | 120(2) | 100(2) | 100(2) | 100(2) |
| Wavelength (Å) | 0.6903 | 0.71073 | 0.71073 | 0.71073 |
| Calculated density (g.cm−3) | 1.936 | 1.875 | 1.904 | 1.947 |
| Absorption coefficient (mm−1) | 2.52 | 2.44 | 2.72 | 2.71 |
| Transmission factors (min./max.) | 0.770, 0.947 | 0.256, 1.000 | 0.595, 1.000 | 0.326, 1.000 |
| Crystal size (mm3) | 0.10 × 0.05 × 0.02 | 0.22 × 0.09 × 0.04 | 0.20 × 0.18× 0.04 | 0.12 × 0.04 × 0.02 |
| θ(max) (°) | 25.0 | 27.6 | 28.7 | 27.5 |
| Reflections measured | 7905 | 17958 | 36812 | 19181 |
| Unique reflections | 3172 | 8457 | 4670 | 4624 |
| Rint | 0.032 | 0.163 | 0.034 | 0.063 |
| Reflections with F2 > 2σ(F2) | 2863 | 7052 | 4519 | 3231 |
| Number of parameters | 195 | 242 | 196 | 221 |
| R1 [F2 > 2σ(F2)] | 0.048 | 0.094 | 0.016 | 0.036 |
| wR2 (all data) | 0.135 | 0.226 | 0.040 | 0.089 |
| GOOF, S | 1.06 | 1.10 | 1.13 | 1.02 |
| Largest difference peak and hole (e Å−3) | 3.08 and −1.44 | 3.13 and −1.84 | 0.55 and −0.40 | 0.81 and −0.92 |
| Compound | 5 | 6 MeCN | 7 | 8 |
| Formula | C16H19Cl2NIMo | C18H21Cl2FN2IMo | C16H18Cl3FNIMo | C16H20Cl2NMo |
| Formula weight | 519.06 | 578.11 | 572.50 | 393.17 |
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | Pca21 | I2/a | Fdd2 | Pbca |
| a (Å) | 15.3004(5) | 14.45516(19) | 14.6309(3) | 12.93812(8) |
| b (Å) | 7.28895(19) | 8.64656(9) | 63.6134(13) | 14.34123(9) |
| c (Å) | 16.7395(4) | 33.2386(4) | 8.14550(17) | 17.82290(11) |
| α (°) β (°) γ (°) V (Å3) | 90 | 90 | 90 | 90 |
| 90 | 96.0961(12) | 90 | 90 | |
| 90 | 90 | 90 | 90 | |
| 1866.85(9) | 4130.91(9) | 7581.2(3) | 3307.01(4) | |
| Z Temperature (K) | 4 | 8 | 16 | 8 |
| 100(2) | 100(2) | 100(2) | 100(2) | |
| Wavelength (Å) Calculated density (g·cm−3) Absorption coefficient (mm−1) Transmission factors (min./max.) | 0.71073 | 0.71073 | 0.71073 | 1.54178 |
| 1.847 | 1.859 | 2.006 | 1.579 | |
| 2.64 | 2.40 | 2.75 | 9.36 | |
| 0.722, 1.000 | 0.775, 1.000 | 0.960, 1.000 | 0.537, 0.893 | |
| Crystal size (mm3) θ(max) (°) | 0.20 × 0.18 × 0.12 | 0.24 × 0.16 × 0.08 | 0.24 × 0.18 × 0.03 | 0.09 × 0.04 × 0.02 |
| 28.7 | 28.7 | 33.2 | 70.5 | |
| Reflections measured | 19105 | 36899 | 59080 | 113441 |
| Unique reflections | 4744 | 5345 | 6772 | 3153 |
| Rint | 0.037 | 0.016 | 0.061 | 0.043 |
| Reflections with F2 > 2σ(F2) | 4704 | 5113 | 6613 | 3095 |
| Number of parameters | 196 | 232 | 213 | 273 |
| R1 [F2 > 2σ(F2)] | 0.024 | 0.016 | 0.042 | 0.038 |
| wR2 (all data) | 0.062 | 0.037 | 0.095 | 0.084 |
| GOOF, S | 1.06 | 1.10 | 1.12 | 1.32 |
| Largest difference peak and hole (e Å−3) | 1.08 and −0.96 | 0.47 and −0.45 | 2.69 and −1.49 | 0.46 and −0.81 |
| Compound | 2-I NH3Cl | 3-I NH3Cl | ||
| Formula | C6H7NI·Cl | C6H7NI·Cl | ||
| Formula weight | 255.48 | 255.48 | ||
| Crystal system | Monoclinic | Monoclinic | ||
| Space group | P21/c | P21/n | ||
| a (Å) | 9.24697(19) | 4.51643(17) | ||
| b (Å) | 9.29797(18) | 5.99872(17) | ||
| c (Å) | 9.3626(2) | 30.1104(11) | ||
| α (°) β (°) γ (°) V (Å3) | 90 | 90 | ||
| 100.672(2) | 92.257(4) | |||
| 90 | 90 | |||
| 791.05(3) | 815.14(5) | |||
| Z Temperature (K) | 4 | 4 | ||
| 100(2) | 100(2) | |||
| Wavelength (Å) Calculated density (g.cm−3) Absorption coefficient (mm−1) Transmission factors (min./max.) | 0.71073 | 0.71073 | ||
| 2.145 | 2.082 | |||
| 4.30 | 4.17 | |||
| 0.328, 1.000 | 0.588, 1.000 | |||
| Crystal size (mm3) θ(max) (°) | 0.18 × 0.09 × 0.09 | 0.16 × 0.12 × 0.01 | ||
| 28.7 | 28.7 | |||
| Reflections measured | 15911 | 13319 | ||
| Unique reflections | 2041 | 2077 | ||
| Rint | 0.025 | 0.050 | ||
| Reflections with F2 > 2σ(F2) | 1984 | 1840 | ||
| Number of parameters | 95 | 91 | ||
| R1 [F2 > 2σ(F2)] | 0.012 | 0.029 | ||
| wR2 (all data) | 0.029 | 0.068 | ||
| GOOF, S | 1.11 | 1.09 | ||
| Largest difference peak and hole (e Å−3) | 0.42 and −0.39 | 2.79 and −0.91 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, T.; Elsegood, M.R.J.; Dale, S.H.; Redshaw, C. Pentamethylcyclopentadienyl Molybdenum(V) Complexes Derived from Iodoanilines: Synthesis, Structure, and ROP of ε-Caprolactone. Catalysts 2021, 11, 1554. https://doi.org/10.3390/catal11121554
Xing T, Elsegood MRJ, Dale SH, Redshaw C. Pentamethylcyclopentadienyl Molybdenum(V) Complexes Derived from Iodoanilines: Synthesis, Structure, and ROP of ε-Caprolactone. Catalysts. 2021; 11(12):1554. https://doi.org/10.3390/catal11121554
Chicago/Turabian StyleXing, Tian, Mark R. J. Elsegood, Sophie H. Dale, and Carl Redshaw. 2021. "Pentamethylcyclopentadienyl Molybdenum(V) Complexes Derived from Iodoanilines: Synthesis, Structure, and ROP of ε-Caprolactone" Catalysts 11, no. 12: 1554. https://doi.org/10.3390/catal11121554