
Passivation of Co/Al2O3 Catalyst by Atomic Layer Deposition to Reduce Deactivation in the Fischer–Tropsch Synthesis

 , , ,
, , ,
Abstract
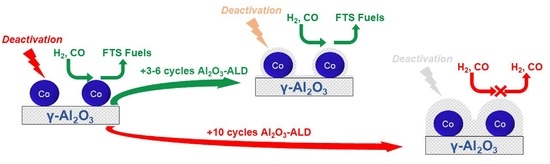
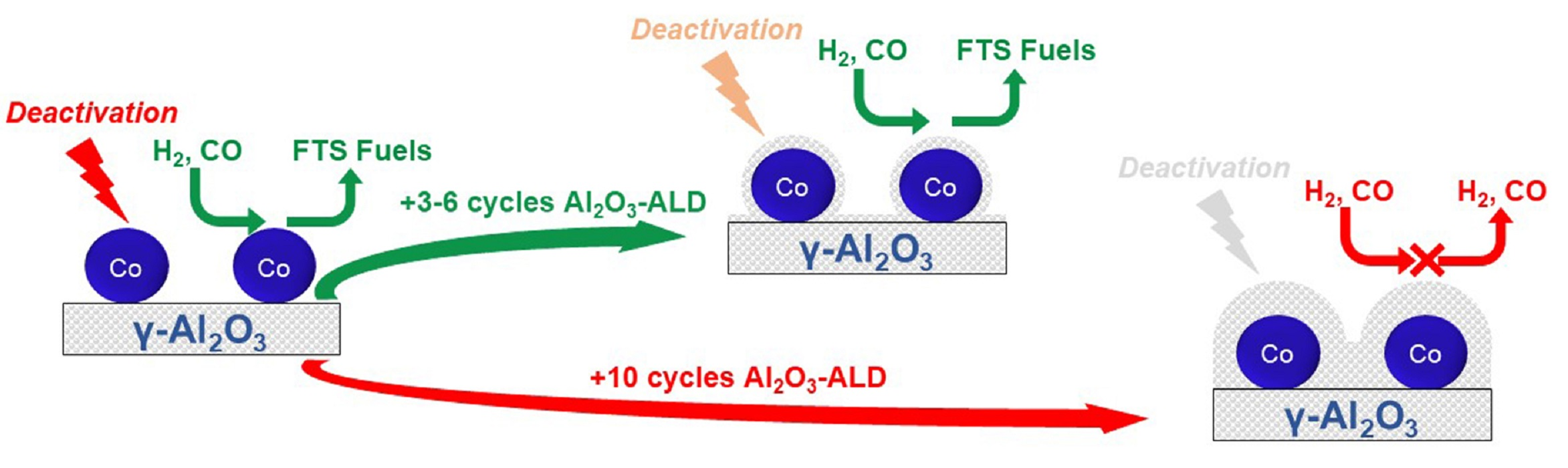
:
1. Introduction
2. Results and Discussion
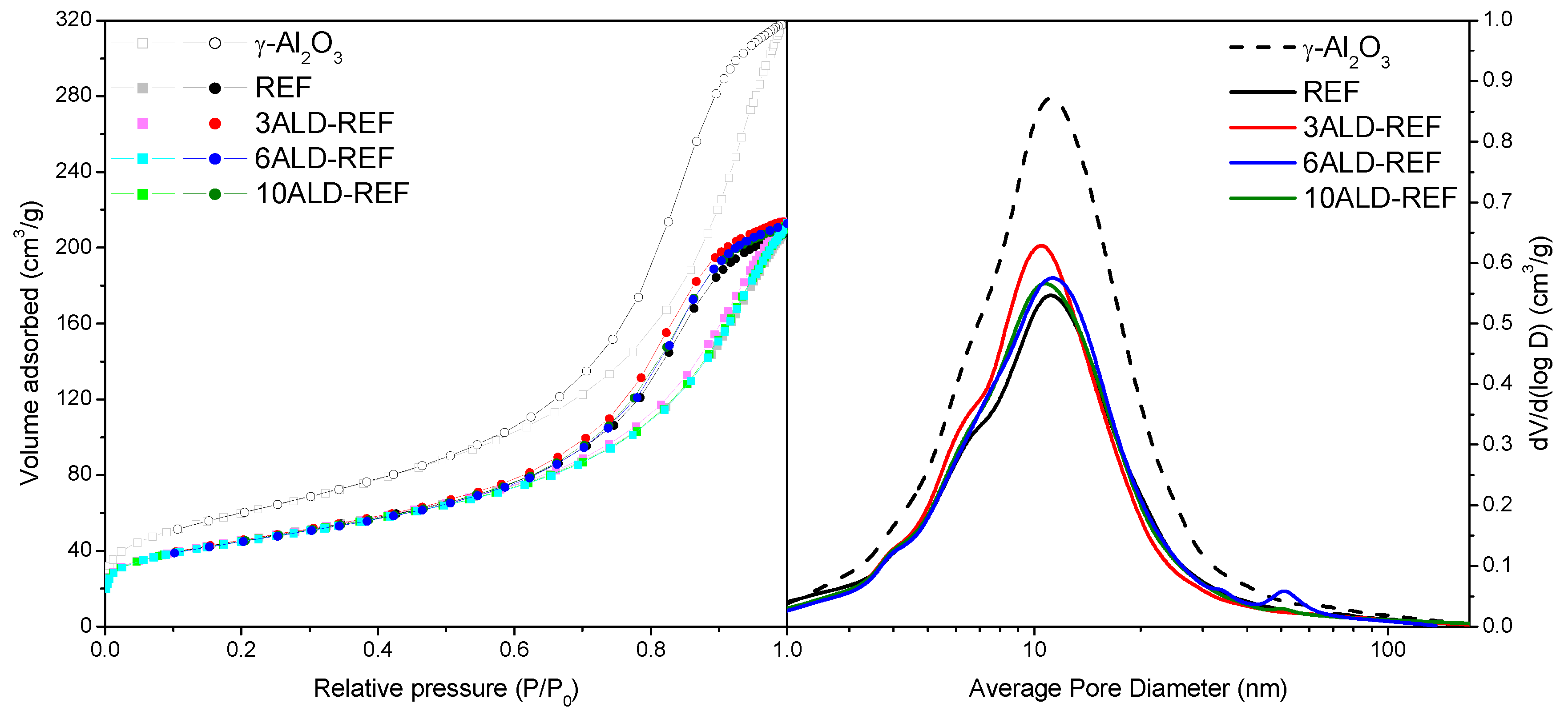
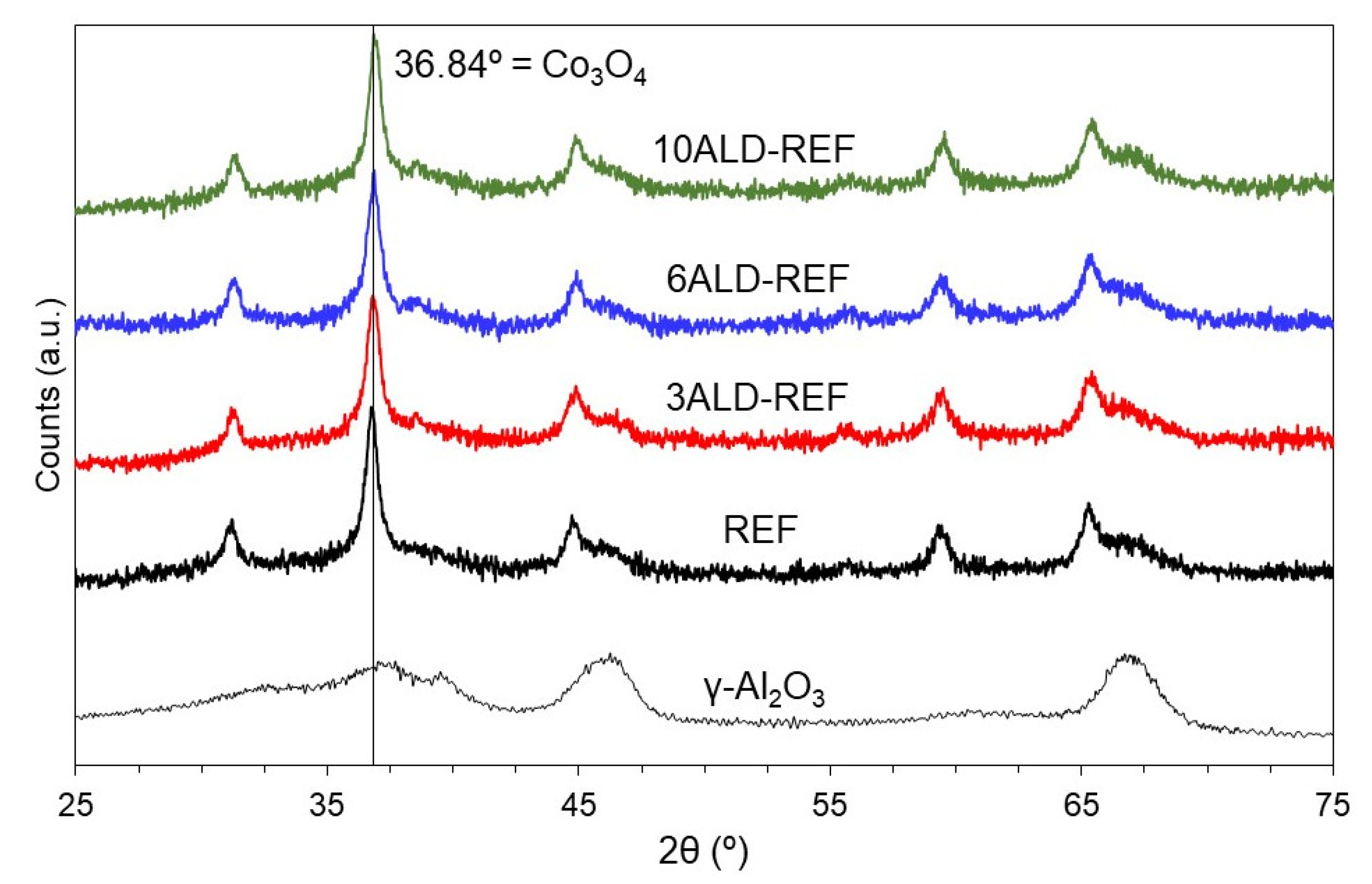
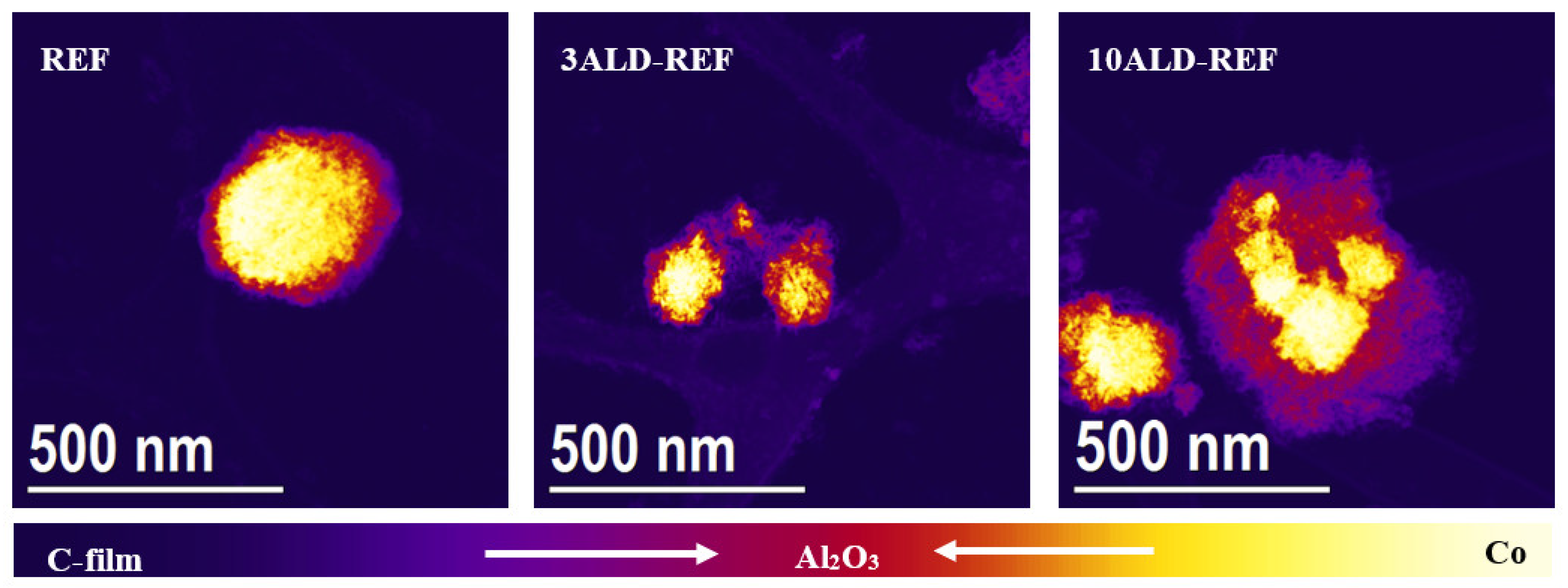
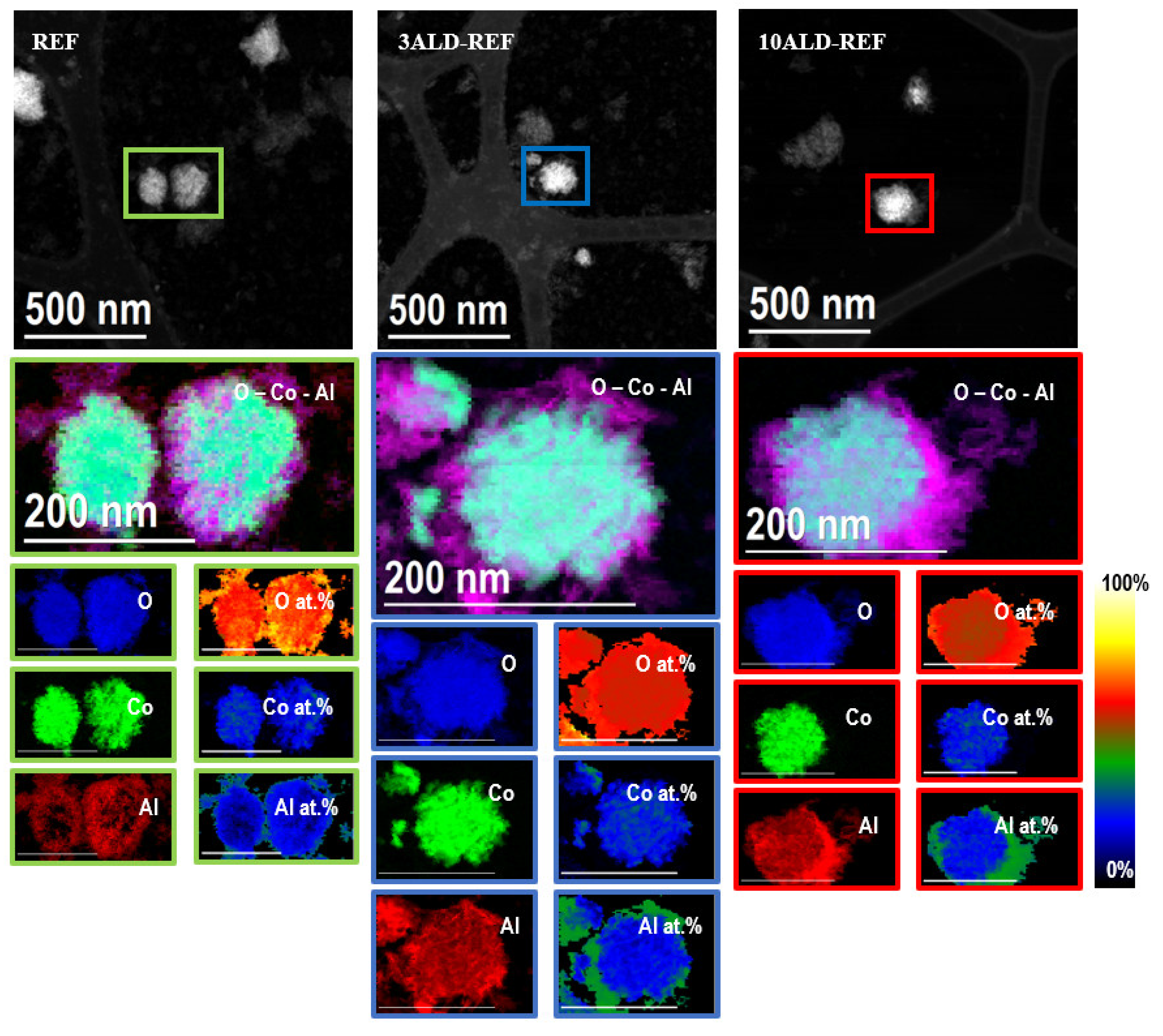
2.1. Catalyst Characterization
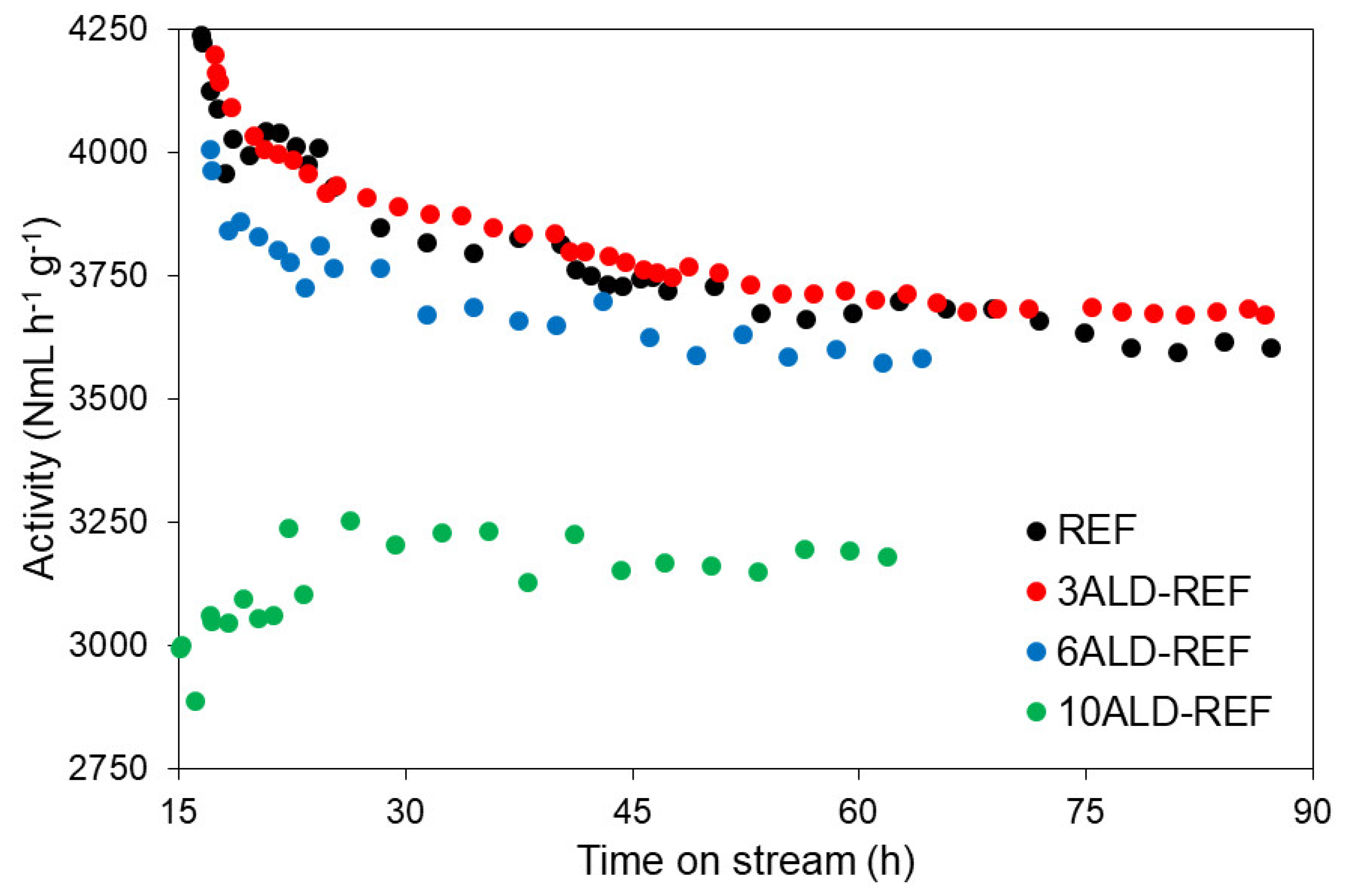
2.2. Catalytic Tests
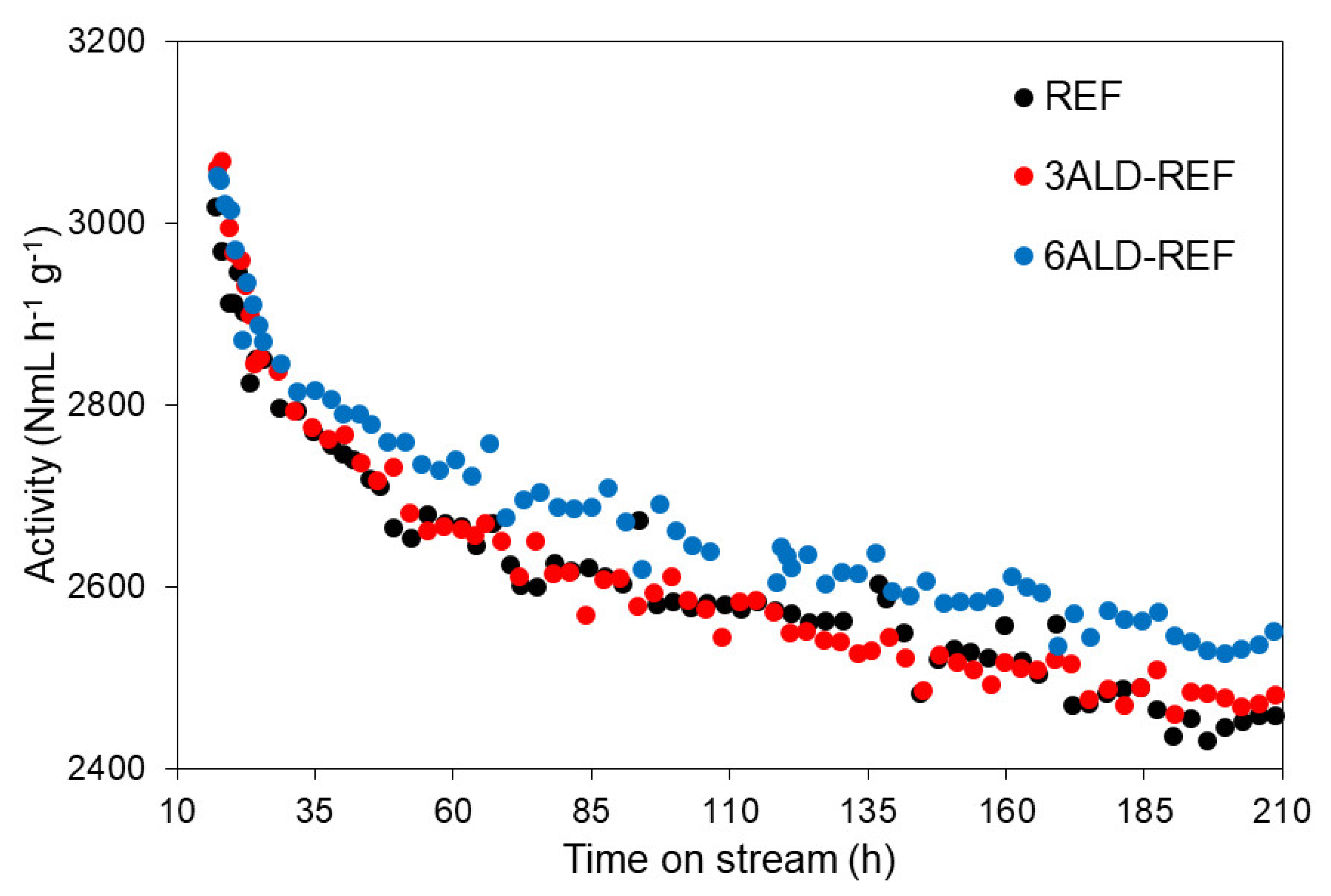
2.2.1. Catalytic Performance at Regular Conditions (XCO ≈ 50%)
2.2.2. Catalytic Performance at Accelerated Deactivation Conditions (XCO ≈ 60%)
3. Materials and Methods
3.1. ALD Passivation Procedure
3.2. Sample Characterization
3.3. Catalytic Tests
3.4. Evaluation of Catalytic Performance
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Panzone, C.; Philippe, R.; Chappaz, A.; Fongarland, P.; Bengaouer, A. Power-to-Liquid catalytic CO2 valorization into fuels and chemicals: Focus on the Fischer-Tropsch route. J. CO2 Util. 2020, 38, 314–347. [Google Scholar] [CrossRef]
- Pishahang, M.; Larring, Y.; Van Dijk, E.; Van Berkel, F.; Dahl, P.I.; Cobden, P.; McCann, M.; Bakken, E. Regenerative Copper-Alumina H2S Sorbent for Hot Gas Cleaning through Chemical Swing Adsorption. Ind. Eng. Chem. Res. 2016, 55, 1024–1032. [Google Scholar] [CrossRef]
- Tsakoumis, N.E.; Rønning, M.; Borg, Ø.; Rytter, E.; Holmen, A. Deactivation of cobalt based Fischer-Tropsch catalysts: A review. Catal. Today 2010, 154, 162–182. [Google Scholar] [CrossRef]
- Moodley, D.J.; van de Loosdrecht, J.; Saib, A.M.; Overett, M.J.; Datye, A.K.; Niemantsverdriet, J.W. Carbon deposition as a deactivation mechanism of cobalt-based Fischer–Tropsch synthesis catalysts under realistic conditions. Appl. Catal. A Gen. 2009, 354, 102–110. [Google Scholar] [CrossRef]
- Chen, W.; Kimpel, T.F.; Song, Y.; Chiang, F.K.; Zijlstra, B.; Pestman, R.; Wang, P.; Hensen, E.J.M. Influence of Carbon Deposits on the Cobalt-Catalyzed Fischer-Tropsch Reaction: Evidence of a Two-Site Reaction Model. ACS Catal. 2018, 8, 1580–1590. [Google Scholar] [CrossRef]
- Vázquez, F.V.; Koponen, J.; Ruuskanen, V.; Bajamundi, C.; Kosonen, A.; Simell, P.; Ahola, J.; Frilund, C.; Elfving, J.; Reinikainen, M.; et al. Power-to-X technology using renewable electricity and carbon dioxide from ambient air: SOLETAIR proof-of-concept and improved process concept. J. CO2 Util. 2018, 28, 235–246. [Google Scholar] [CrossRef]
- Rahmati, M.; Safdari, M.-S.; Fletcher, T.H.; Argyle, M.D.; Bartholomew, C.H. Chemical and Thermal Sintering of Supported Metals with Emphasis on Cobalt Catalysts During Fischer–Tropsch Synthesis. Chem. Rev. 2020, 120, 4455–4533. [Google Scholar] [CrossRef]
- van de Loosdrecht, J.; Balzhinimaev, B.; Dalmon, J.-A.; Niemantsverdriet, J.W.; Tsybulya, S.V.; Saib, A.M.; van Berge, P.J.; Visagie, J.L. Cobalt Fischer-Tropsch synthesis: Deactivation by oxidation? Catal. Today 2007, 123, 293–302. [Google Scholar] [CrossRef]
- Wolf, M.; Fischer, N.; Claeys, M. Water-induced deactivation of cobalt-based Fischer–Tropsch catalysts. Nat. Catal. 2020, 3, 962–965. [Google Scholar] [CrossRef]
- Rytter, E.; Holmen, A. Deactivation and Regeneration of Commercial Type Fischer-Tropsch Co-Catalysts—A Mini-Review. Catalysts 2015, 5, 478–499. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Fang, X.; Xu, Y.; Liu, X. Investigation of the deactivation behavior of Co catalysts in Fischer–Tropsch synthesis using encapsulated Co nanoparticles with controlled SiO2 shell layer thickness. Catal. Sci. Technol. 2020, 10, 1182–1192. [Google Scholar] [CrossRef]
- Phaahlamohlaka, T.N.; Dlamini, M.W.; Mogodi, M.W.; Kumi, D.O.; Jewell, L.L.; Billing, D.G.; Coville, N.J. A sinter resistant Co Fischer-Tropsch catalyst promoted with Ru and supported on titania encapsulated by mesoporous silica. Appl. Catal. A Gen. 2018, 552, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.A.; Yang, N.; Bent, S.F. Nanoengineering Heterogeneous Catalysts by Atomic Layer Deposition. Annu. Rev. Chem. Biomol. Eng. 2017, 8, 41–62. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Elam, J.; Stair, P. Synthesis and stabilization of supported metal catalysts by atomic layer deposition. Acc. Chem. Res. 2013, 46. [Google Scholar] [CrossRef] [PubMed]
- Gorey, T.J.; Dai, Y.; Anderson, S.L.; Lee, S.; Lee, S.; Seifert, S.; Winans, R.E. Selective growth of Al2O3 on size-selected platinum clusters by atomic layer deposition. Surf. Sci. 2020, 691, 121485. [Google Scholar] [CrossRef]
- Kwon, Y.J.; Ko, W.C.; Kang, S.; Kim, K.M.; Jeong, Y.K. Surface passivation of highly stable TiO2/V2O5 photocatalyst by atomic layer deposited-Al2O3. Appl. Surf. Sci. 2020, 507, 145128. [Google Scholar] [CrossRef]
- Baktash, E.; Littlewood, P.; Schomäcker, R.; Thomas, A.; Stair, P.C. Alumina coated nickel nanoparticles as a highly active catalyst for dry reforming of methane. Appl. Catal. B Environ. 2015, 179, 122–127. [Google Scholar] [CrossRef]
- Feng, H.; Lu, J.; Stair, P.C.; Elam, J.W. Alumina over-coating on Pd nanoparticle catalysts by atomic layer deposition: Enhanced stability and reactivity. Catal. Lett. 2011, 141, 512–517. [Google Scholar] [CrossRef]
- Lee, J.; Jackson, D.H.K.; Li, T.; Winans, R.E.; Dumesic, J.A.; Kuech, T.F.; Huber, G.W. Enhanced stability of cobalt catalysts by atomic layer deposition for aqueous-phase reactions. Energy Environ. Sci. 2014, 7, 1657–1660. [Google Scholar] [CrossRef]
- Moodley, D.J.; Saib, A.M.; Van De Loosdrecht, J.; Welker-Nieuwoudt, C.A.; Sigwebela, B.H.; Niemantsverdriet, J.W. The impact of cobalt aluminate formation on the deactivation of cobalt-based Fischer-Tropsch synthesis catalysts. Catal. Today 2011, 171, 192–200. [Google Scholar] [CrossRef]
- Taheri Najafabadi, A.; Khodadadi, A.A.; Parnian, M.J.; Mortazavi, Y. Atomic layer deposited Co/γ-Al2O3 catalyst with enhanced cobalt dispersion and Fischer-Tropsch synthesis activity and selectivity. Appl. Catal. A Gen. 2016, 511, 31–46. [Google Scholar] [CrossRef]
- Eskelinen, P.; Fransila, S. Cobalt Catalyst Characterization and Modification by Atomic Layer Deposition for Fischer-Tropsch Synthesis; Aalto University: Espoo, Finland, 2019. [Google Scholar]
- Rouquerol, J.; Avnir, D.; Everett, D.H.; Fairbridge, C.; Haynes, M.; Pernicone, N.; Ramsay, J.D.F.; Sing, K.S.W.; Unger, K.K. Guidelines for the Characterization of Porous Solids. In Studies in Surface Science and Catalysis; Elsevier Science B.V.: Amsterdam, The Netherlands, 1994; Volume 87, pp. 1–9. [Google Scholar]
- Trépanier, M.; Tavasoli, A.; Dalai, A.K.; Abatzoglou, N. Co, Ru and K loadings effects on the activity and selectivity of carbon nanotubes supported cobalt catalyst in Fischer–Tropsch synthesis. Appl. Catal. A Gen. 2009, 353, 193–202. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the Development of Novel Cobalt Fischer−Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, J.; Ellis, P.R.; Humble, R.; Kelly, G.J.; McKenna, M.; West, J. Deactivation of alumina supported cobalt FT catalysts during testing in a Continuous-stirred tank reactor (CSTR). Appl. Catal. A Gen. 2018, 550, 28–37. [Google Scholar] [CrossRef]
- Lögdberg, S.; Yang, J.; Lualdi, M.; Walmsley, J.C.; Järås, S.; Boutonnet, M.; Blekkan, E.A.; Rytter, E.; Holmen, A. Further insights into methane and higher hydrocarbons formation over cobalt-based catalysts with γ-Al2O3, α-Al2O3 and TiO2 as support materials. J. Catal. 2017, 352, 515–531. [Google Scholar] [CrossRef]
- de la Peña O’Shea, V.A.; Homs, N.; Fierro, J.L.G.; Ramírez de la Piscina, P. Structural changes and activation treatment in a Co/SiO2 catalyst for Fischer–Tropsch synthesis. Catal. Today 2006, 114, 422–427. [Google Scholar] [CrossRef]
- Argyle, M.D.; Frost, T.S.; Bartholomew, C.H. Cobalt Fischer–Tropsch Catalyst Deactivation Modeled Using Generalized Power Law Expressions. Top. Catal. 2014, 57, 415–429. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Sintering kinetics of supported metals: New perspectives from a unifying GPLE treatment. Appl. Catal. A Gen. 1993, 107, 1–57. [Google Scholar] [CrossRef]
- Tavasoli, A.; Malek Abbaslou, R.M.; Dalai, A.K. Deactivation behavior of ruthenium promoted Co/γ-Al2O3 catalysts in Fischer–Tropsch synthesis. Appl. Catal. A Gen. 2008, 346, 58–64. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
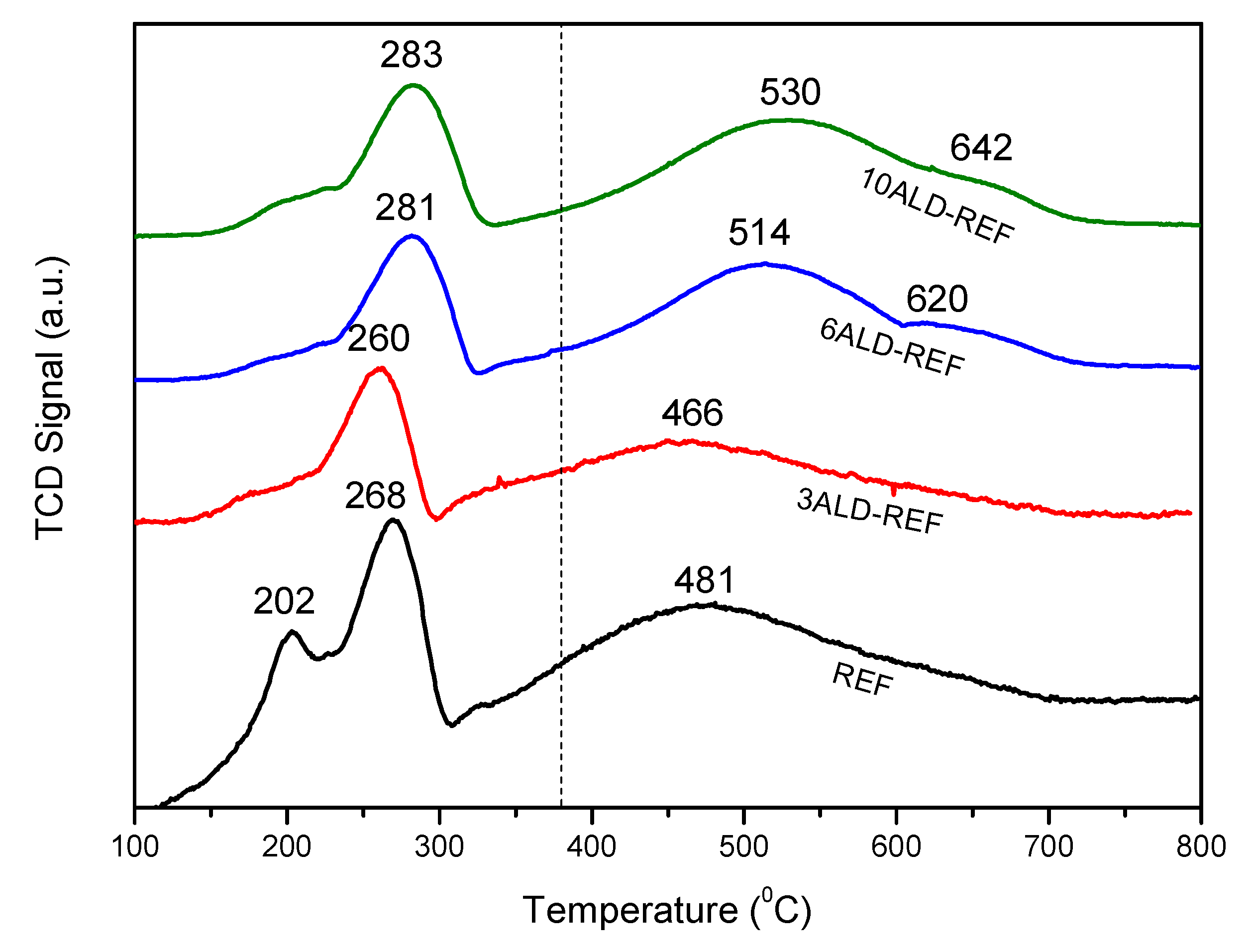
| Sample | BET Surface Area (m2/g) | Pore Volume (cm3/g) | TPR-Tmax (°C) a | Co Crystallite Size (nm) b | Extent of Co Reduction (%) c |
|---|---|---|---|---|---|
| γ-Al2O3 | 216 | 0.492 | - | - | - |
| REF | 163 | 0.320 | 268/481 | 12.4 | 61.4 |
| 3ALD-REF | 163 | 0.331 | 260/466 | 11.6 | 56.5 |
| 6ALD-REF | 161 | 0.322 | 281/514 | 11.1 | 65.4 |
| 10ALD-REF | 161 | 0.327 | 283/530 | 12.1 | 11.1 |
| Sample | Time (h) | XCO (%) | Selectivity (%) | |||||
|---|---|---|---|---|---|---|---|---|
| CH4 | CO2 | C2–C4 a | C5+ | |||||
| REF | 16 | 43.5 | 15.9 | 0.70 | 4.78 | 78.6 | 1 | 1 |
| 87 | 35.9 | 14.9 | 0.59 | 11.6 | 73.0 | 1 | 1 | |
| 3ALD-REF | 17 | 43.0 | 17.2 | 0.76 | 5.2 | 76.9 | 0.990 | 0.967 |
| 87 | 36.7 | 15.2 | 0.57 | 10.9 | 73.3 | 1.018 | 1.026 | |
| 6ALD-REF | 17 | 40.1 | 15.3 | 0.74 | 7.6 | 76.3 | 0.945 | 0.895 |
| 64 | 34.8 | 14.0 | 0.66 | 11.5 | 73.9 | 0.972 b | 0.981 | |
| 10ALD-REF | 15 | 28.1 | 32.1 | 0.78 | 9.9 | 57.2 | 0.706 | 0.470 |
| 62 | 30.2 | 21.2 | 0.62 | 12.2 | 66.0 | 0.860 b | 0.760 | |
| Deactivation Order | 0 | 1 | 2 | |||
|---|---|---|---|---|---|---|
| Sample | ||||||
| REF | 2.38 × 10−4 | 0.8021 | 2.63 × 10−4 | 0.8154 | 2.92 × 10−4 | 0.8279 |
| 3ALD-REF | 2.02 × 10−4 | 0.8496 | 2.20 × 10−4 | 0.8619 | 2.41 × 10−4 | 0.8735 |
| 6ALD-REF | 1.73 × 10−4 | 0.7384 | 1.90 × 10−4 | 0.7528 | 2.07 × 10−4 | 0.7665 |
| Sample | Time (h) | XCO (%) | Selectivity (%) | |||||
|---|---|---|---|---|---|---|---|---|
| CH4 | CO2 | C2–C4 a | C5+ | |||||
| REF | 17 | 56.8 | 13.6 | 0.87 | 5.33 | 80.2 | 1 | 1 |
| 100 | 47.6 | 12.5 | 0.54 | 10.3 | 76.7 | 1 | 1 | |
| 200 | 44.5 | 12.6 | 0.53 | 10.7 | 76.2 | 1 | 1 | |
| 3ALD-REF | 17 | 56.7 | 14.9 | 0.96 | 5.64 | 78.5 | 1.014 | 0.977 |
| 100 | 47.7 | 12.4 | 0.56 | 10.0 | 77.0 | 1.011 | 1.001 | |
| 200 | 44.8 | 12.3 | 0.51 | 10.3 | 76.9 | 1.013 | 1.016 | |
| 6ALD-REF | 17 | 56.8 | 16.3 | 1.08 | 6.72 | 75.9 | 1.012 | 0.946 |
| 100 | 48.4 | 14.1 | 0.70 | 10.8 | 74.4 | 1.029 | 0.986 | |
| 200 | 45.7 | 13.4 | 0.58 | 10.7 | 75.3 | 1.034 | 1.015 | |
| Reference Sample | Number of ALD Cycles | Nomenclature |
|---|---|---|
| 16Co/γ-Al2O3 (Johnson Matthey) | 0 | REF |
| 3 | 3ALD-REF | |
| 6 | 6ALD-REF | |
| 10 | 10ALD-REF |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-López, J.A.; Guilera, J.; Biset-Peiró, M.; Enache, D.; Kelly, G.; Andreu, T. Passivation of Co/Al2O3 Catalyst by Atomic Layer Deposition to Reduce Deactivation in the Fischer–Tropsch Synthesis. Catalysts 2021, 11, 732. https://doi.org/10.3390/catal11060732
Díaz-López JA, Guilera J, Biset-Peiró M, Enache D, Kelly G, Andreu T. Passivation of Co/Al2O3 Catalyst by Atomic Layer Deposition to Reduce Deactivation in the Fischer–Tropsch Synthesis. Catalysts. 2021; 11(6):732. https://doi.org/10.3390/catal11060732
Chicago/Turabian StyleDíaz-López, José Antonio, Jordi Guilera, Martí Biset-Peiró, Dan Enache, Gordon Kelly, and Teresa Andreu. 2021. "Passivation of Co/Al2O3 Catalyst by Atomic Layer Deposition to Reduce Deactivation in the Fischer–Tropsch Synthesis" Catalysts 11, no. 6: 732. https://doi.org/10.3390/catal11060732