Cobalt Based Catalysts on Alkali-Activated Zeolite Foams for N2O Decomposition

 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Results and Discussion
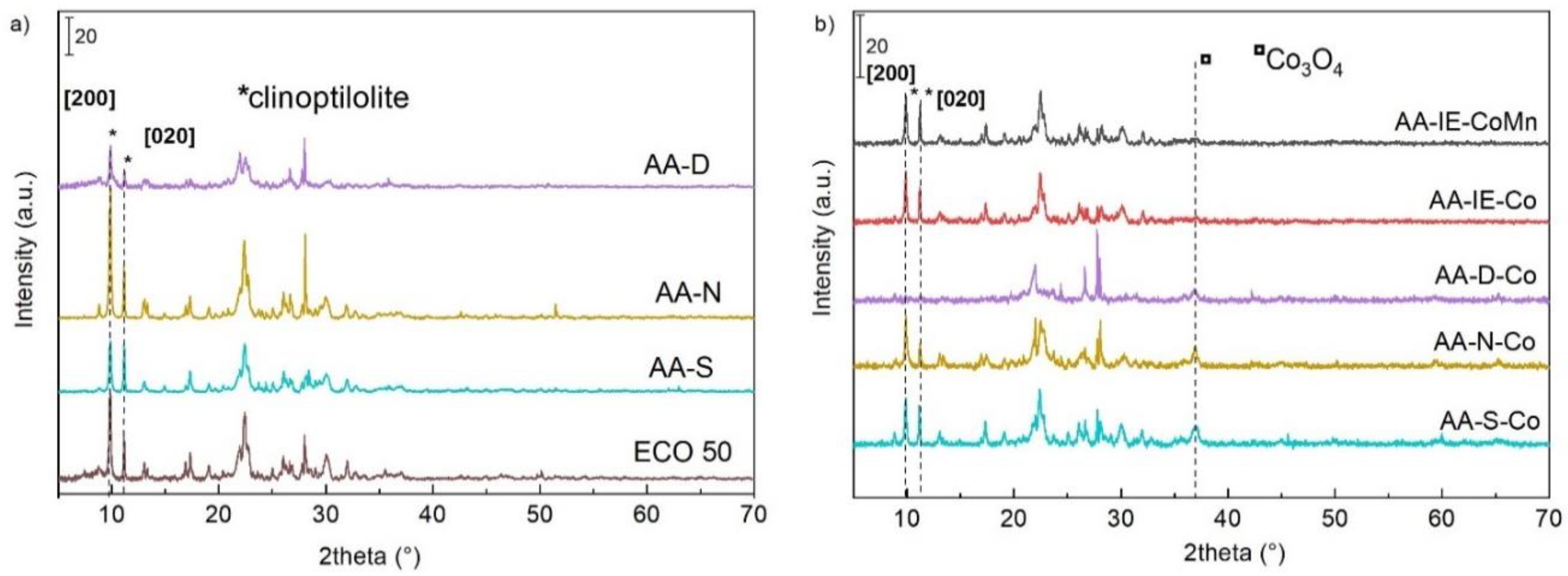
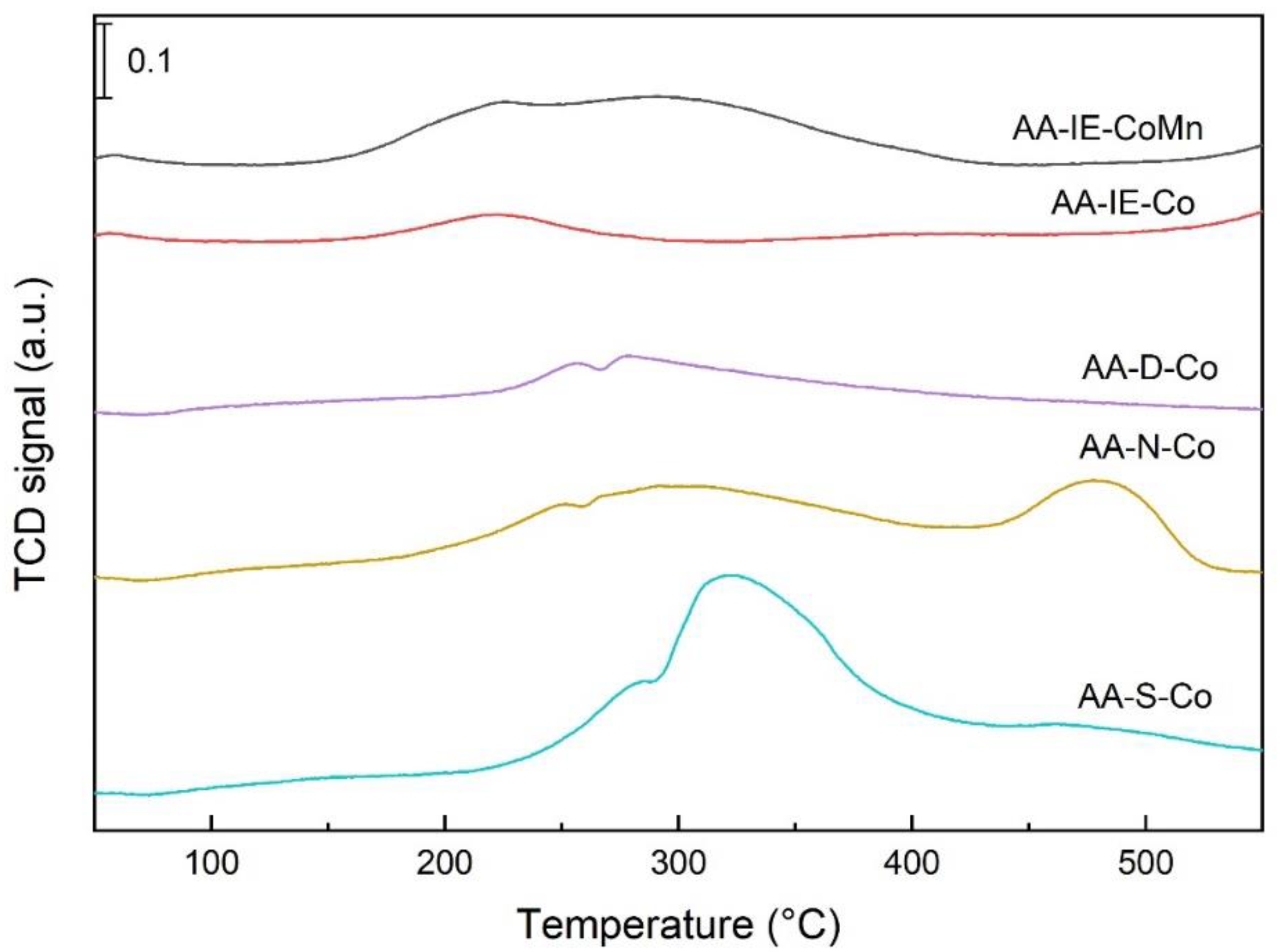
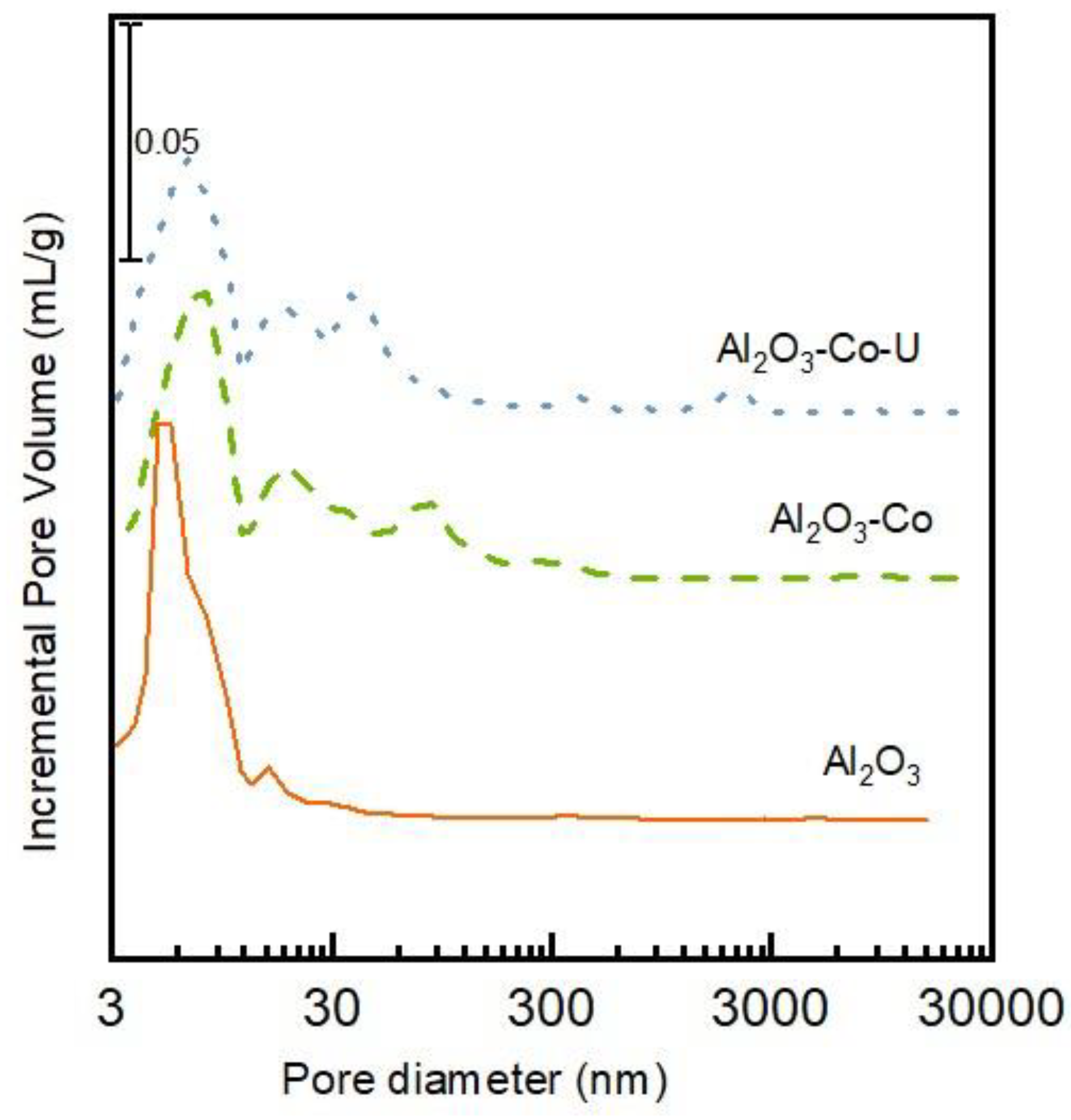
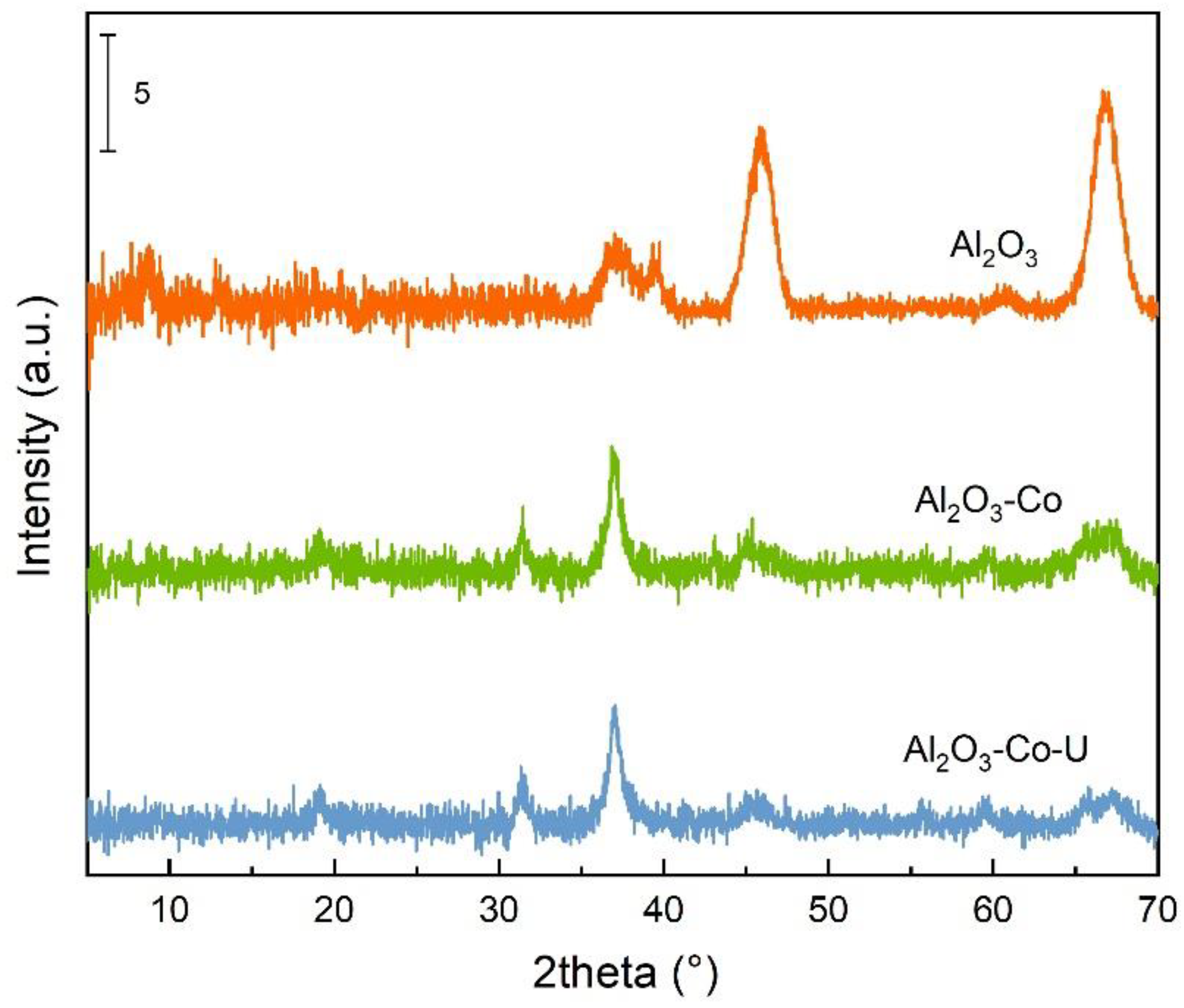
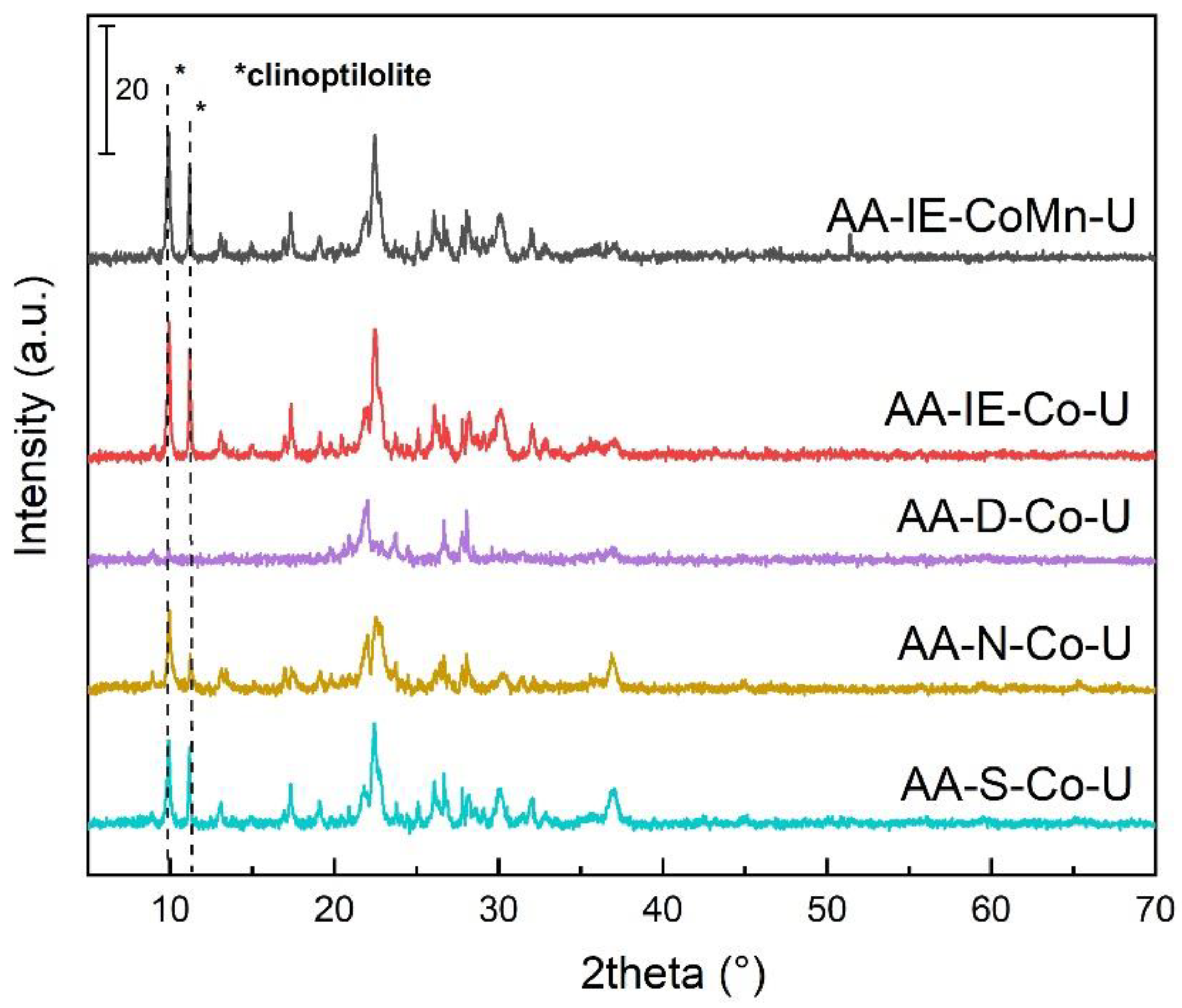
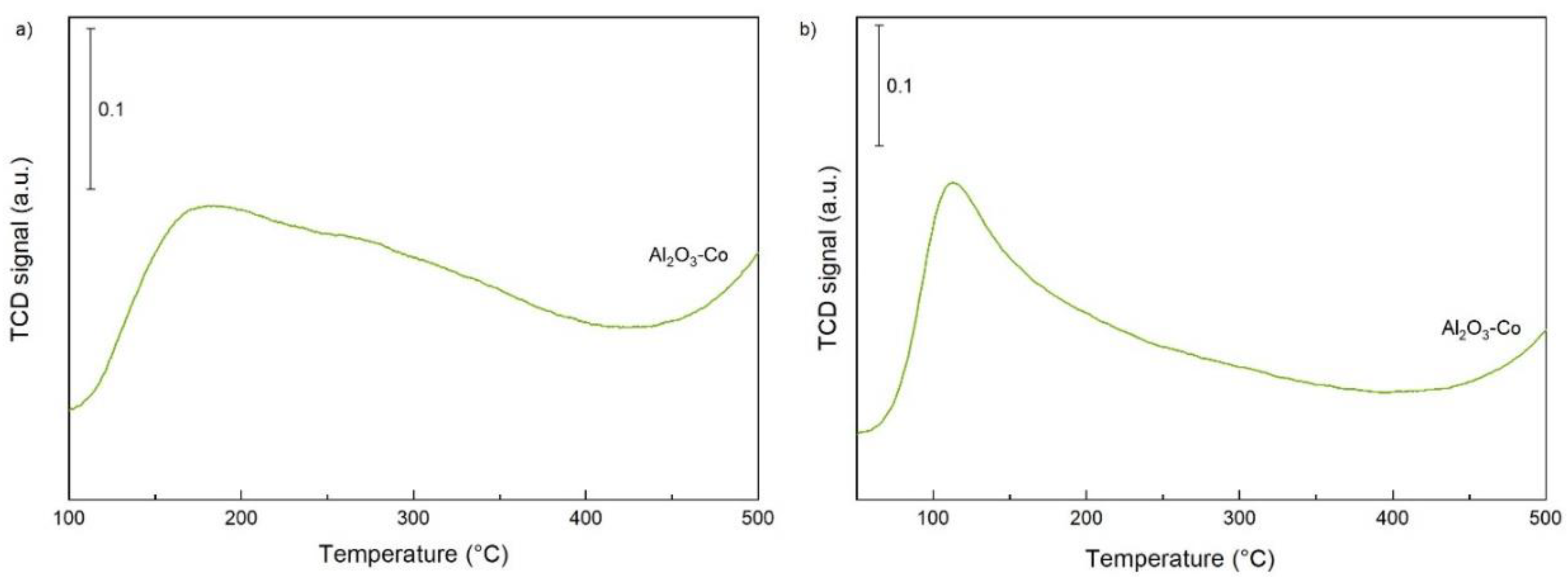
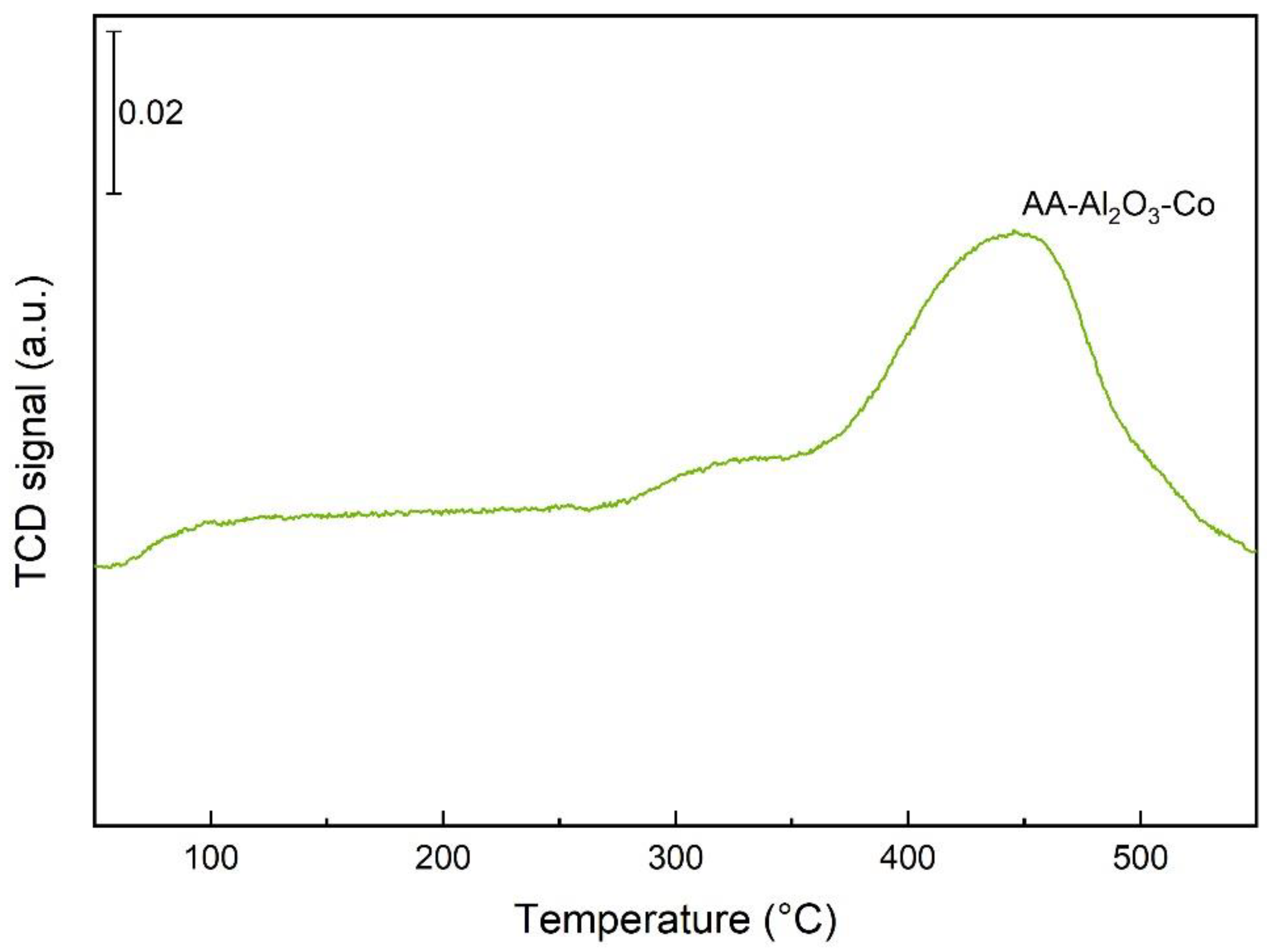
2.1. Characterisation of the Catalysts and Supports
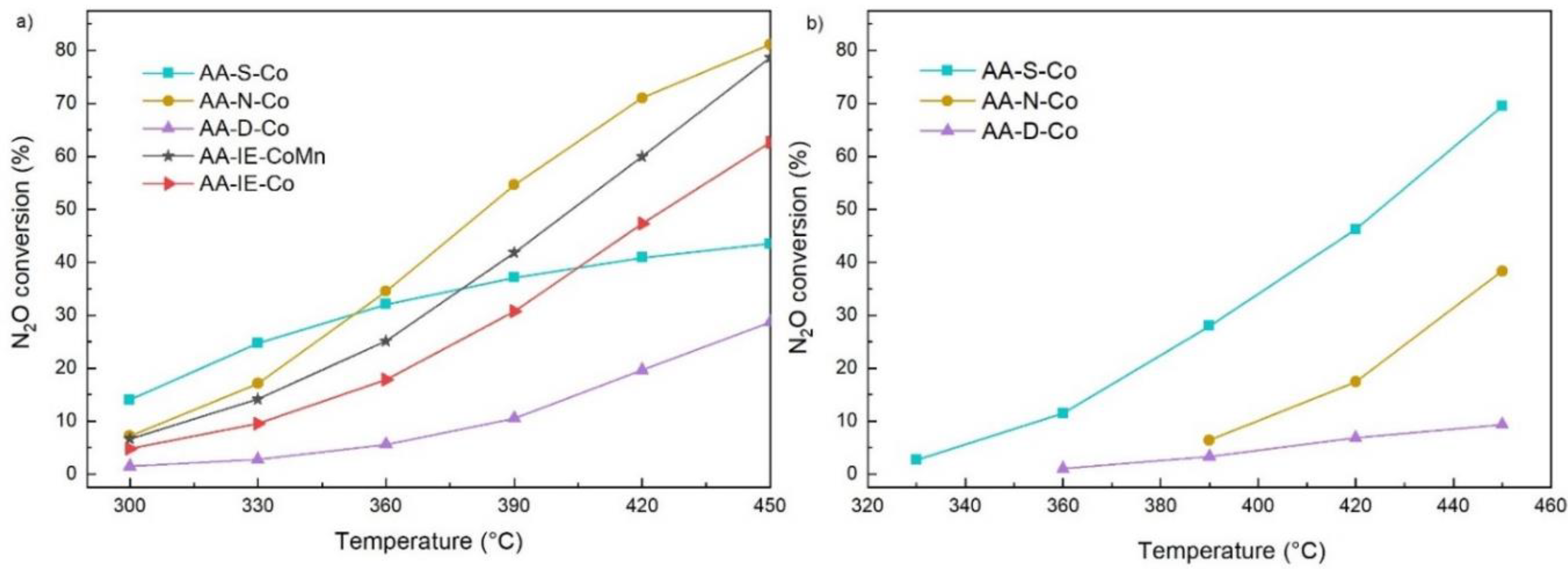
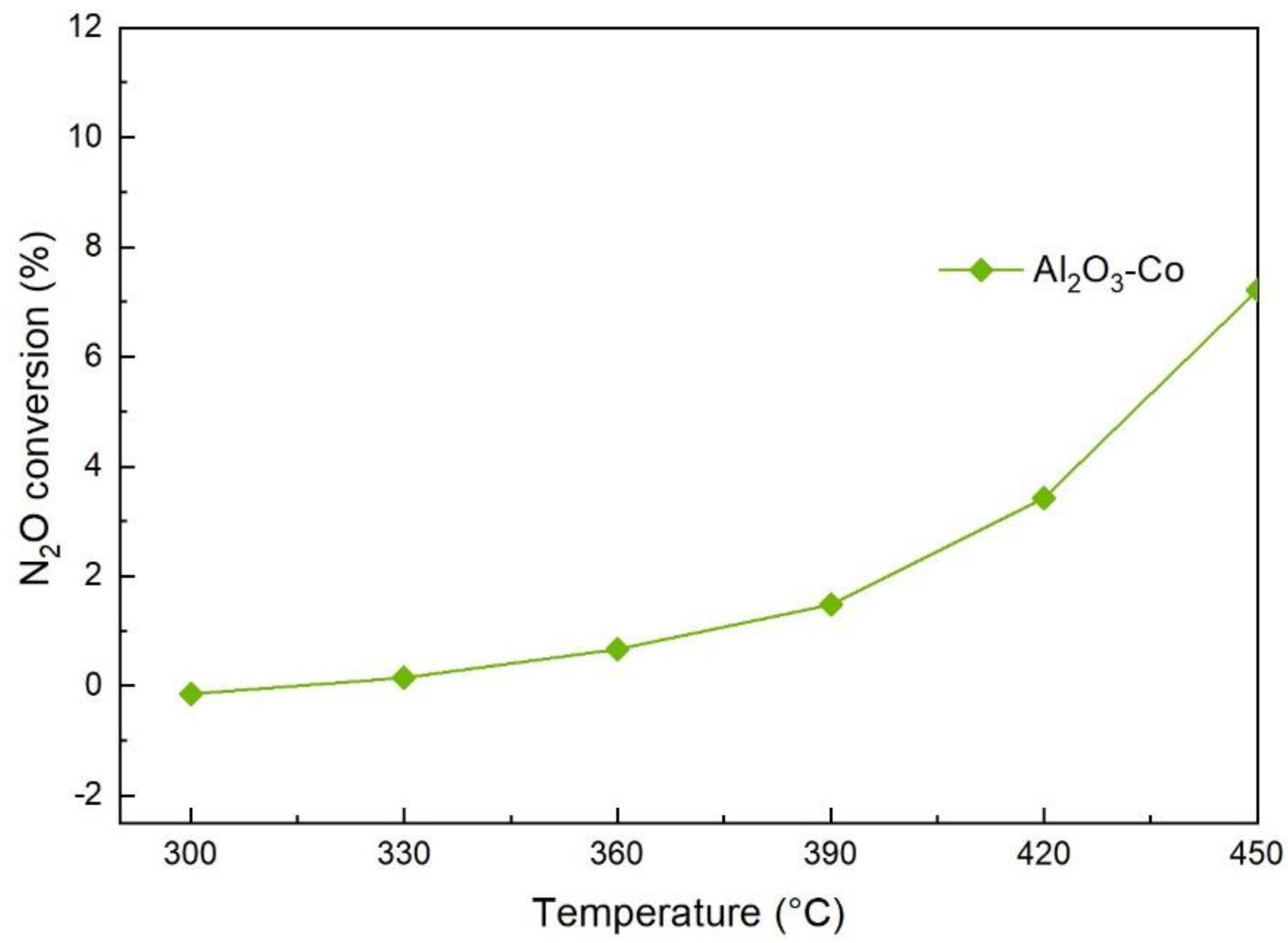
2.2. N2O Catalytic Decomposition
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Zeolite Foams
3.3. Post-Synthesis Modifications and Catalyst Synthesis
3.4. Characterisation of Supports and Prepared Catalysts
3.5. Catalytic Tests
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Chemical Composition (wt.%) | Si/Al Ratio (mol/mol) | Co/AM * Ratio | SSA ** (m2/g) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Si | Al | K | Na | Ca | Fe | Co | Mn | ||||
| Al2O3-Co | 0.1 | 49.0 | - | 0.3 | 0.1 | - | 5.2 | - | - | - | 212.9 |
| Sample | cSUM (μmol/g) | Tmax1 (°C) | cmax1 (μmol/g) | Pmax1 * (%) | Tmax2 (°C) | cmax2 (μmol/g) | Pmax2 * (%) | Tmax3 (°C) | cmax3 (μmol/g) | Pmax3 * (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Al2O3-Co | 296 | 173 | 178 | 60 | 283 | 118 | 40 | - | - | - |
| Sample | cSUM (μmol/g) | Tmax1 (°C) | cmax1 (μmol/g) | Pmax1 * (%) | Tmax2 (°C) | cmax2 (μmol/g) | Pmax2 * (%) | Tmax3 (°C) | cmax3 (μmol/g) | Pmax3 * (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Al2O3-Co | 145 | 109 | 31 | 21 | 144 | 89 | 61 | 263 | 25 | 18 |
| Sample | cSUM (μmol/g) | Tmax1 (°C) | cmax1 (μmol/g) | Pmax1 * (%) | Tmax2 (°C) | cmax2 (μmol/g) | Pmax2 * (%) | Tmax3 (°C) | cmax3 (μmol/g) | Pmax3 * (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Al2O3-Co | 109 | - | - | - | 435 | 85 | 80 | 479 | 24 | 20 |






References
- Choya, A.; De Rivas, B.; Gutiérrez-Ortiz, J.I.; Velasco, J.R.G.; López-Fonseca, R. Synthesis, Characterization and Kinetic Behavior of Supported Cobalt Catalysts for Oxidative after-Treatment of Methane Lean Mixtures. Materials 2019, 12, 3174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.-K.; Wang, C.-B.; Chiu, H.-C.; Chien, S.-H. In situ FTIR Study of Cobalt Oxides for the Oxidation of Carbon Monoxide. Catal. Lett. 2003, 86, 63–68. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Wang, L.; Zhang, C.; He, H. Catalytic oxidation of formaldehyde over manganese oxides with different crystal structures. Catal. Sci. Technol. 2015, 5, 2305–2313. [Google Scholar] [CrossRef]
- Hrachovcová, K.; Tišler, Z.; Svobodová, E.; Šafář, J. Modified Alkali Activated Zeolite Foams with Improved Textural and Mechanical Properties. Minerals 2020, 10, 483. [Google Scholar] [CrossRef]
- Atkins, P.W.; Jones, L.L. Chemistry: Molecules, Matter, and Change, 3rd ed.; W. H. Freeman and Company: New York, NY, USA, 1997. [Google Scholar]
- Hidalgo-Herrador, J.M.; Tišler, Z.; Vráblík, A.; Velvarská, R.; Lederer, J. Acid-modified phonolite and foamed zeolite as supports for NiW catalysts for deoxygenation of waste rendering fat. React. Kinet. Mech. Catal. 2019, 126, 773–793. [Google Scholar] [CrossRef]
- Tišler, Z.; Velvarská, R.; Skuhrovcová, L.; Pelíšková, L.; Akhmetzyanova, U. Key Role of Precursor Nature in Phase Composition of Supported Molybdenum Carbides and Nitrides. Materials 2019, 12, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tišler, Z.; Horacek, J.; Safar, J.; Velvarska, R.; Peliskova, L.; Kocik, J.; Gherib, Y.; Marklova, K.; Bulánek, R.; Kubička, D. Clinoptilolite foams prepared by alkali activation of natural zeolite and their post-synthesis modifications. Microporous Mesoporous Mater. 2019, 282, 169–178. [Google Scholar] [CrossRef]
- Maniak, G.; Stelmachowski, P.; Kotarba, A.; Sojka, Z.; Pérez, V.R.; López, A.B. Rationales for the selection of the best precursor for potassium doping of cobalt spinel based deN2O catalyst. Appl. Catal. B Environ. 2013, 136, 302–307. [Google Scholar] [CrossRef]
- Sun, M.; Wang, L.; Feng, B.; Zhang, Z.; Lu, G.; Guo, Y. The role of potassium in K/Co3O4 for soot combustion under loose contact. Catal. Today 2011, 175, 100–105. [Google Scholar] [CrossRef]
- Obalová, L.; Pacultová, K.; Balabánová, J.; Jirátová, K.; Bastl, Z.; Valášková, M.; Lacný, Z.; Kovanda, F. Effect of Mn/Al ratio in Co–Mn–Al mixed oxide catalysts prepared from hydrotalcite-like precursors on catalytic decomposition of N2O. Catal. Today 2007, 119, 233–238. [Google Scholar] [CrossRef]
- Ramírez, J.P.; Kapteijn, F.; Schöffel, K.; Moulijn, J.A. Formation and control of N2O in nitric acid production: Where do we stand today? Appl. Catal. B Environ. 2003, 44, 117–151. [Google Scholar] [CrossRef]
- Kapteijn, F.; Marbán, G.; Mirasol, J.R.; Moulijn, J.A. Kinetic Analysis of the Decomposition of Nitrous Oxide over ZSM 5 Catalysts. J. Catal. 1997, 167, 256–265. [Google Scholar] [CrossRef]
- Asano, K.; Ohnishi, C.; Iwamoto, S.; Shioya, Y.; Inoue, M. Potassium-doped Co3O4 catalyst for direct decomposition of N2O. Appl. Catal. B Environ. 2008, 78, 242–249. [Google Scholar] [CrossRef]
- Maniak, G.; Stelmachowski, P.; Zasada, F.; Piskorz, W.; Kotarba, A.; Sojka, Z. Guidelines for optimization of catalytic activity of 3d transition metal oxide catalysts in N2O decomposition by potassium promotion. Catal. Today 2011, 176, 369–372. [Google Scholar] [CrossRef]
- Maniak, G.; Stelmachowski, P.; Stanek, J.J.; Kotarba, A.; Sojka, Z. Catalytic properties in N2O decomposition of mixed cobalt–iron spinels. Catal. Commun. 2011, 15, 127–131. [Google Scholar] [CrossRef]
- Stelmachowski, P.; Maniak, G.; Kotarba, A.; Sojka, Z. Strong electronic promotion of Co3O4 towards N2O decomposition by surface alkali dopants. Catal. Commun. 2009, 10, 1062–1065. [Google Scholar] [CrossRef]
- Cheng, H.; Huang, Y.; Wang, A.; Li, L.; Wang, X.; Zhang, T. N2O decomposition over K-promoted Co-Al catalysts prepared from hydrotalcite-like precursors. Appl. Catal. B Environ. 2009, 89, 391–397. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, Q.; He, C.; Wang, Y.; Cheng, J.; Hao, Z. CoMOR zeolite catalyst prepared by buffered ion exchange for effective decomposition of nitrous oxide. J. Hazard. Mater. 2011, 192, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Pasha, N.; Lingaiah, N.; Babu, N.S.; Reddy, P.S.S.; Prasad, P.S. Studies on cesium doped cobalt oxide catalysts for direct N2O decomposition in the presence of oxygen and steam. Catal. Commun. 2008, 10, 132–136. [Google Scholar] [CrossRef]
- Da Cruz, R.; Mascarenhas, A.; Andrade, H.M.C. Co-ZSM-5 catalysts for N2O decomposition. Appl. Catal. B Environ. 1998, 18, 223–231. [Google Scholar] [CrossRef]
- Shen, Q.; Li, L.; Li, J.; Tian, H.; Hao, Z. A study on N2O catalytic decomposition over Co/MgO catalysts. J. Hazard. Mater. 2009, 163, 1332–1337. [Google Scholar] [CrossRef] [PubMed]
- Karásková, K.; Obalová, L.; Jirátová, K.; Kovanda, F. Effect of promoters in Co–Mn–Al mixed oxide catalyst on N2O decomposition. Chem. Eng. J. 2010, 160, 480–487. [Google Scholar] [CrossRef]
- Klyushina, A.; Pacultová, K.; Karásková, K.; Jirátová, K.; Ritz, M.; Fridrichová, D.; Volodarskaja, A.; Obalová, L. Effect of preparation method on catalytic properties of Co-Mn-Al mixed oxides for N2O decomposition. J. Mol. Catal. A Chem. 2016, 425, 237–247. [Google Scholar] [CrossRef]
- Campa, M.C.; Indovina, V.; Pietrogiacomi, D. The dependence of catalytic activity for N2O decomposition on the exchange extent of cobalt or copper in Na-MOR, H-MOR and Na-MFI. Appl. Catal. B Environ. 2009, 91, 347–354. [Google Scholar] [CrossRef]
- Fellah, M.F.; Onal, I. N2O decomposition on Fe- and Co-ZSM-5: A density functional study. Catal. Today 2008, 137, 410–417. [Google Scholar] [CrossRef]
- Obalová, L.; Karásková, K.; Jirátová, K.; Kovanda, F. Effect of potassium in calcined Co–Mn–Al layered double hydroxide on the catalytic decomposition of N2O. Appl. Catal. B Environ. 2009, 90, 132–140. [Google Scholar] [CrossRef]
- Stelmachowski, P.; Ciura, K.; Grzybek, G. Morphology-dependent reactivity of cobalt oxide nanoparticles in N2O decomposition. Catal. Sci. Technol. 2016, 6, 5554–5560. [Google Scholar] [CrossRef]
- Gudyka, S.; Grzybek, G.; Gryboś, J.; Indyka, P.; Leszczyński, B.; Kotarba, A.; Sojka, Z. Enhancing the deN2O activity of the supported Co3O4|α-Al2O3 catalyst by glycerol-assisted shape engineering of the active phase at the nanoscale. Appl. Catal. B Environ. 2017, 201, 339–347. [Google Scholar] [CrossRef]
- Grzybek, G.; Wójcik, S.; Legutko, P.; Gryboś, J.; Indyka, P.; Leszczyński, B.; Kotarba, A.; Sojka, Z. Thermal stability and repartition of potassium promoter between the support and active phase in the K Co2.6Zn0.4O4|α Al2O3 catalyst for N2O decomposition: Crucial role of activation temperature on catalytic performance. Appl. Catal. B Environ. 2017, 205, 597–604. [Google Scholar] [CrossRef]
- Boroń, P.; Chmielarz, L.; Casale, S.; Calers, C.; Krafft, J.-M.; Dzwigaj, S. Effect of Co content on the catalytic activity of CoSiBEA zeolites in N2O decomposition and SCR of NO with ammonia. Catal. Today 2015, 258, 507–517. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, R.; Chen, B.; Li, Y.; Li, Y. Comparative study on the direct decomposition of nitrous oxide over M (Fe, Co, Cu)–BEA zeolites. J. Catal. 2012, 294, 99–112. [Google Scholar] [CrossRef]
- Smeets, P.J.; Meng, Q.; Corthals, S.; Leeman, H.; Schoonheydt, R.A. Co–ZSM-5 catalysts in the decomposition of N2O and the SCR of NO with CH4: Influence of preparation method and cobalt loading. Appl. Catal. B Environ. 2008, 84, 505–513. [Google Scholar] [CrossRef]
- Ghahri, A.; Golbabaei, F.; Vafajoo, L.; Mireskandari, S.M.; Yaseri, M.; Shahtaheri, S.J. Removal of Greenhouse Gas (N2O) by Catalytic Decomposition on Natural Clinoptilolite Zeolites Impregnated with Cobalt. Int. J. Environ. Res. 2017, 11, 327–337. [Google Scholar] [CrossRef]
- Obalová, L.; Karásková, K.; Wach, A.; Kustrowski, P.; Kutláková, K.M.; Michalik, S.; Jirátová, K. Alkali metals as promoters in Co–Mn–Al mixed oxide for N2O decomposition. Appl. Catal. A Gen. 2013, 462–463, 227–235. [Google Scholar] [CrossRef]
- Abu-Zied, B.M.; Soliman, S.A.; Abdellah, S.E. Pure and Ni-substituted Co3O4 spinel catalysts for direct N2O decomposition. Chin. J. Catal. 2014, 35, 1105–1112. [Google Scholar] [CrossRef]
- Del Río, L.; Marbán, G. Stainless steel wire mesh supported potassium doped cobalt oxide catalysts for the catalytic decomposition of nitrous oxide. Appl. Catal. B Environ. 2012, 126, 39–46. [Google Scholar] [CrossRef]
- Karásková, K.; Obalová, L.; Kovanda, F. N2O catalytic decomposition and temperature programmed desorption tests on alkali metals promoted Co–Mn–Al mixed oxide. Catal. Today 2011, 176, 208–211. [Google Scholar] [CrossRef]
- Žaneta, C.; Obalová, L.; Kovanda, F.; Legut, D.; Titov, A.; Ritz, M.; Fridrichová, D.; Michalik, S.; Kuśtrowski, P.; Jirátová, K. Effect of precursor synthesis on catalytic activity of Co3O4 in N2O decomposition. Catal. Today 2015, 257, 18–25. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, C.; He, H.; Teraoka, Y. Catalytic decomposition of N2O over CeO2 promoted Co3O4 spinel catalyst. Appl. Catal. B Environ. 2007, 75, 167–174. [Google Scholar] [CrossRef]
- Piskorz, W.; Zasada, F.; Stelmachowski, P.; Kotarba, A.; Sojka, Z. Decomposition of N2O over the surface of cobalt spinel: A DFT account of reactivity experiments. Catal. Today 2008, 137, 418–422. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, C.; He, H.; Teraoka, Y. Promotion effect of residual K on the decomposition of N2O over cobalt–cerium mixed oxide catalyst. Catal. Today 2007, 126, 449–455. [Google Scholar] [CrossRef]
- Klegova, A.; Inayat, A.; Indyka, P.; Gryboś, J.; Sojka, Z.; Pacultová, K.; Schwieger, W.; Volodarskaja, A.; Kuśtrowski, P.; Rokicińska, A.; et al. Cobalt mixed oxides deposited on the SiC open-cell foams for nitrous oxide decomposition. Appl. Catal. B Environ. 2019, 255, 255. [Google Scholar] [CrossRef]
- Iwanek, E.; Krawczyk, K.; Petryk, J.; Sobczak, J.W.; Kaszkur, Z. Direct nitrous oxide decomposition with CoOx CeO2 catalysts. Appl. Catal. B Environ. 2011, 106, 416–422. [Google Scholar] [CrossRef]
- Thangavelu, K.; Parameswari, K.; Kuppusamy, K.; Haldorai, Y. A simple and facile method to synthesize Co3O4 nanoparticles from metal benzoate dihydrazinate complex as a precursor. Mater. Lett. 2011, 65, 1482–1484. [Google Scholar] [CrossRef]
- Russo, N.; Fino, D.; Saracco, G.; Specchia, V. N2O catalytic decomposition over various spinel-type oxides. Catal. Today 2007, 119, 228–232. [Google Scholar] [CrossRef]
- Grzybek, G.; Stelmachowski, P.; Indyka, P.; Inger, M.; Wilk, M.; Kotarba, A.; Sojka, Z. Cobalt–zinc spinel dispersed over cordierite monoliths for catalytic N2O abatement from nitric acid plants. Catal. Today 2015, 257, 93–97. [Google Scholar] [CrossRef]
- Zhang, R.; Hedjazi, K.; Chen, B.; Li, Y.; Lei, Z.; Liu, N. M(Fe, Co)-BEA washcoated honeycomb cordierite for N 2 O direct decomposition. Catal. Today 2016, 273, 273–285. [Google Scholar] [CrossRef]
- Jirátová, K.; Balabánová, J.; Kovanda, F.; Klegová, A.; Obalová, L.; Fajgar, R. Cobalt Oxides Supported Over Ceria–Zirconia Coated Cordierite Monoliths as Catalysts for Deep Oxidation of Ethanol and N2O Decomposition. Catal. Lett. 2017, 147, 1379–1391. [Google Scholar] [CrossRef]
- Klyushina, A.; Pacultová, K.; Krejčová, S.; Słowik, G.; Jirátová, K.; Kovanda, F.; Ryczkowski, J.; Obalová, L. Advantages of stainless steel sieves as support for catalytic N2O decomposition over K doped Co3O4. Catal. Today 2015, 257 Pt 1, 2–10. [Google Scholar] [CrossRef]
- Klegová, A.; Pacultová, K.; Fridrichová, D.; Volodarskaja, A.; Kovanda, F.; Jirátová, K. Cobalt Oxide Catalysts on Commercial Supports for N2O Decomposition. Chem. Eng. Technol. 2017, 40, 981–990. [Google Scholar] [CrossRef]
- Pacultová, K.; Klegova, A.; Kiška, T.; Fridrichová, D.; Martaus, A.; Rokicińska, A.; Kuśtrowski, P.; Obalová, L. Effect of support on the catalytic activity of Co3O4-Cs deposited on open-cell ceramic foams for N2O decomposition. Mater. Res. Bull. 2020, 129, 110892. [Google Scholar] [CrossRef]
- Rutkowska, M.; Piwowarska, Z.; Micek, E.; Chmielarz, L. Hierarchical Fe-, Cu- and Co-Beta zeolites obtained by mesotemplate-free method. Part I: Synthesis and catalytic activity in N2O decomposition. Microporous Mesoporous Mater. 2015, 209, 54–65. [Google Scholar] [CrossRef]
- Sadek, R.; Chalupka, K.A.; Mierczynski, P.; Rynkowski, J.; Gurgul, J.; Dzwigaj, S. Cobalt Based Catalysts Supported on Two Kinds of Beta Zeolite for Application in Fischer-Tropsch Synthesis. Catalysts 2019, 9, 497. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.-H.; Ryu, J.-H.; Kim, J.-H.; Jang, I.H.; Kim, A.R.; Han, G.Y.; Bae, J.W.; Ha, K.-S. Role of ZSM5 Distribution on Co/SiO2 Fischer–Tropsch Catalyst for the Production of C5–C22 Hydrocarbons. Energy Fuels 2012, 26, 6061–6069. [Google Scholar] [CrossRef]
- Kurian, M.; Thankachan, S.; Nair, D.S.; Aswathy, E.K.; Babu, A.; Thomas, A.; KT, B.K. Structural, magnetic, and acidic properties of cobalt ferrite nanoparticles synthesised by wet chemical methods. J. Adv. Ceram. 2015, 4, 199–205. [Google Scholar] [CrossRef] [Green Version]







| Sample | Chemical Composition (wt.%) | Si/Al Ratio (mol/mol) | Co/AM * Ratio | SSA ** (m2/g) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Si | Al | K | Na | Ca | Fe | Co | Mn | ||||
| AA-S | 33.4 | 5.5 | 7.2 | 2.9 | 2.3 | 0.9 | - | - | 5.8 | - | 15.0 |
| AA-N | 37.8 | 6.3 | 1.6 | 0.3 | 1.7 | 1.1 | - | - | 5.8 | - | 23.4 |
| AA-D | 42.0 | 3.4 | 1.1 | 0.2 | 0.5 | 0.7 | - | - | 11.8 | - | 111.7 |
| AA-S-Co | 30.5 | 5.1 | 6.6 | 2.6 | 2.1 | 0.9 | 6.4 | - | 5.7 | 0.4 | 13.5 |
| AA-N-Co | 35.0 | 6.0 | 1.4 | 0.2 | 1.6 | 1.0 | 5.7 | - | 5.6 | 2.1 | 27.7 |
| AA-D-Co | 38.7 | 3.5 | 1.1 | 0.2 | 0.4 | 0.7 | 5.4 | - | 10.6 | 2.5 | 49.9 |
| AA-IE-Co | 32.0 | 5.2 | 4.1 | 0.5 | 1.7 | 0.9 | 6.3 | - | 5.9 | 0.9 | 115.8 |
| AA-IE-CoMn | 31.9 | 5.2 | 4.2 | 0.5 | 1.7 | 0.9 | 5.4 | 0.8 | 5.9 | 0.7 | 118.6 |
| Sample | cSUM (μmol/g) | Tmax1 (°C) | cmax1 (μmol/g) | Pmax1 * (%) | Tmax2 (°C) | cmax2 (μmol/g) | Pmax2 * (%) | Tmax3 (°C) | cmax3 (μmol/g) | Pmax3 * (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| AA-S-Co | 501 | 175 | 501 | 100 | - | - | - | - | - | - |
| AA-N-Co | 1542 | 173 | 1313 | 85 | 390 | 229 | 15 | - | - | - |
| AA-D-Co | 593 | 158 | 446 | 75 | 255 | 70 | 12 | 382 | 77 | 13 |
| AA-IE-Co | 1446 | 178 | 1379 | 95 | 247 | 27 | 2 | 362 | 40 | 3 |
| AA-IE-MnCo | 1211 | 187 | 1211 | 100 | - | - | - | - | - | - |
| Sample | cSUM (μmol/g) | Tmax1 (°C) | cmax1 (μmol/g) | Pmax1 * (%) | Tmax2 (°C) | cmax2 (μmol/g) | Pmax2 * (%) | Tmax3 (°C) | cmax3 (μmol/g) | Pmax3 * (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| AA-S-Co | 213 | 105 | 42 | 20 | 186 | 161 | 76 | 321 | 10 | 14 |
| AA-N-Co | 125 | 110 | 82 | 66 | 363 | 14 | 11 | 437 | 30 | 23 |
| AA-D-Co | 156 | 112 | 36 | 23 | 204 | 44 | 28 | 289 | 76 | 49 |
| AA-IE-Co | 306 | 111 | 215 | 70 | 338 | 91 | 30 | - | - | - |
| AA-IE-MnCo | 408 | 113 | 227 | 56 | 273 | 181 | 44 | - | - | - |
| Sample | cSUM (μmol/g) | Tmax1 (°C) | cmax1 (μmol/g) | Pmax1 * (%) | Tmax2 (°C) | cmax2 (μmol/g) | Pmax2 * (%) | Tmax3 (°C) | cmax3 (μmol/g) | Pmax3 * (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| AA-S-Co | 360 | 289 | 77 | 21 | 330 | 218 | 61 | 478 | 65 | 19 |
| AA-N-Co | 875 | 304 | 704 | 80 | 483 | 171 | 20 | - | - | - |
| AA-D-Co | 260 | 284 | 76 | 29 | 306 | 184 | 71 | - | - | |
| AA-IE-Co | 176 | 222 | 60 | 34 | 418 | 116 | 66 | - | - | - |
| AA-IE-MnCo | 500 | 239 | 337 | 67 | 303 | 163 | 33 | - | - | - |
| Sample | k390 (m3 s−1 kg−1) | k390 * (m3 s−1 m−2) | EA (J mol−1) | EA */EA |
|---|---|---|---|---|
| AA-S-Co | 6.59 × 10−4 | 4.88 × 10−2 | 60,750 | 2.29 |
| AA-N-Co | 1.27 × 10−3 | 4.58 × 10−2 | 87,322 | 1.52 |
| AA-D-Co | 1.89 × 10−4 | 3.80 × 10−3 | 74,589 | 1.77 |
| AA-S-Co * | 1.82 × 10−3 | 1.35 × 10−1 | 139,135 | - |
| AA-N-Co * | 3.65 × 10−4 | 1.32 × 10−2 | 132,484 | - |
| AA-D-Co * | 1.84 × 10−4 | 3.69 × 10−3 | 131,677 | - |
| AA-IE-Co | 5.97 × 10−4 | 5.15 × 10−3 | 69,927 | - |
| AA-IE-MnCo | 8.75 × 10−4 | 7.38 × 10−3 | 71,050 | - |
| Sample | Chemical Composition (wt.%) | Si/Al Ratio (mol/mol) | SSA * (m2/g) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Si | Al | K | Fe | Ca | Na | Mg | Ti | |||
| ECO 50 | 35.2 | 6.7 | 3.5 | 1.3 | 3.1 | 0.2 | 0.5 | 0.1 | 5.2 | 26.4 |
| γ-Al2O3 | 0.1 | 51.8 | - | - | - | 0.4 | 0.3 | - | - | 309.0 |
| Parameter | Value |
|---|---|
| Silicate modulus of the alkaline activator | Ms = 1.51 |
| Water coefficient: | w = 0.7 |
| Alkali content | Me2O * = 8.2 wt.% |
| Alkali molar ratio | Na2O:K2O = 0.56 |
| Catalyst | Support | Preparation Method |
|---|---|---|
| Al2O3-Co | Commercial (γ-Al2O3) | IWI |
| AA-S-Co | AZF (AA-S) | IWI |
| AA-N-Co | Ion exchanged AZF (AA-N) | IWI |
| AA-D-Co | Acid leached AZF (AA-D) | IWI |
| AA-IE-Co | AZF (AA-S) | DIE |
| AA-IE-CoMn | AZF (AA-S) | DIE |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tišler, Z.; Klegová, A.; Svobodová, E.; Šafář, J.; Strejcová, K.; Kohout, J.; Šlang, S.; Pacultová, K.; Rodríguez-Padrón, D.; Bulánek, R. Cobalt Based Catalysts on Alkali-Activated Zeolite Foams for N2O Decomposition. Catalysts 2020, 10, 1398. https://doi.org/10.3390/catal10121398
Tišler Z, Klegová A, Svobodová E, Šafář J, Strejcová K, Kohout J, Šlang S, Pacultová K, Rodríguez-Padrón D, Bulánek R. Cobalt Based Catalysts on Alkali-Activated Zeolite Foams for N2O Decomposition. Catalysts. 2020; 10(12):1398. https://doi.org/10.3390/catal10121398
Chicago/Turabian StyleTišler, Zdeněk, Anna Klegová, Eliška Svobodová, Jan Šafář, Kateřina Strejcová, Jan Kohout, Stanislav Šlang, Kateřina Pacultová, Daily Rodríguez-Padrón, and Roman Bulánek. 2020. "Cobalt Based Catalysts on Alkali-Activated Zeolite Foams for N2O Decomposition" Catalysts 10, no. 12: 1398. https://doi.org/10.3390/catal10121398