Tuning the Selectivity of LaNiO3 Perovskites for CO2 Hydrogenation through Potassium Substitution

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
| CO2 + 4H2 → CH4 + 2H2O | Sabatier Reaction [6] | ΔH = −165.0 kJ mol−1 |
| CO2 + H2 → CO + H2O | Reverse Water Gas Shift [6] | ΔH = 41.15 kJ mol−1 |
2. Results and Discussion
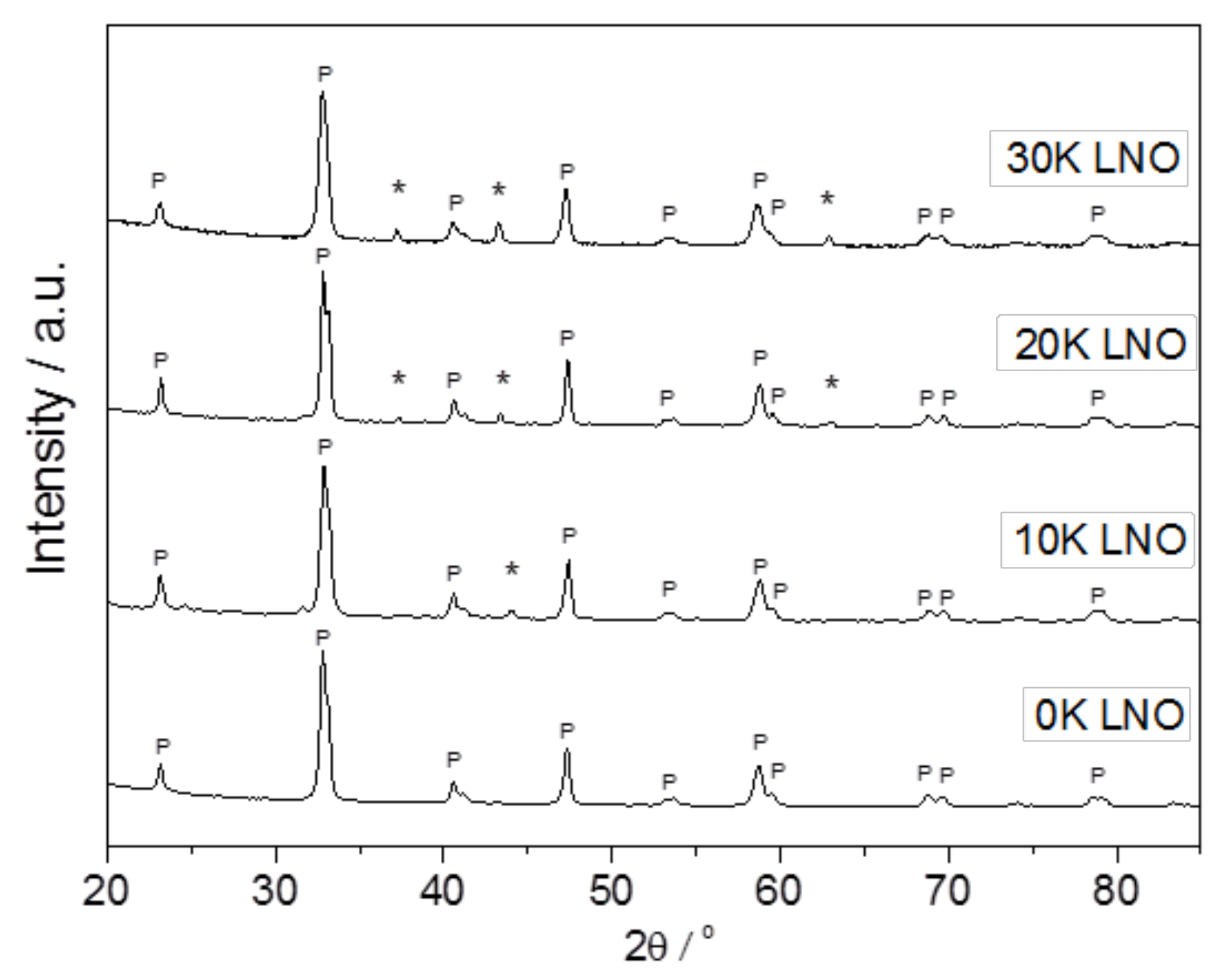
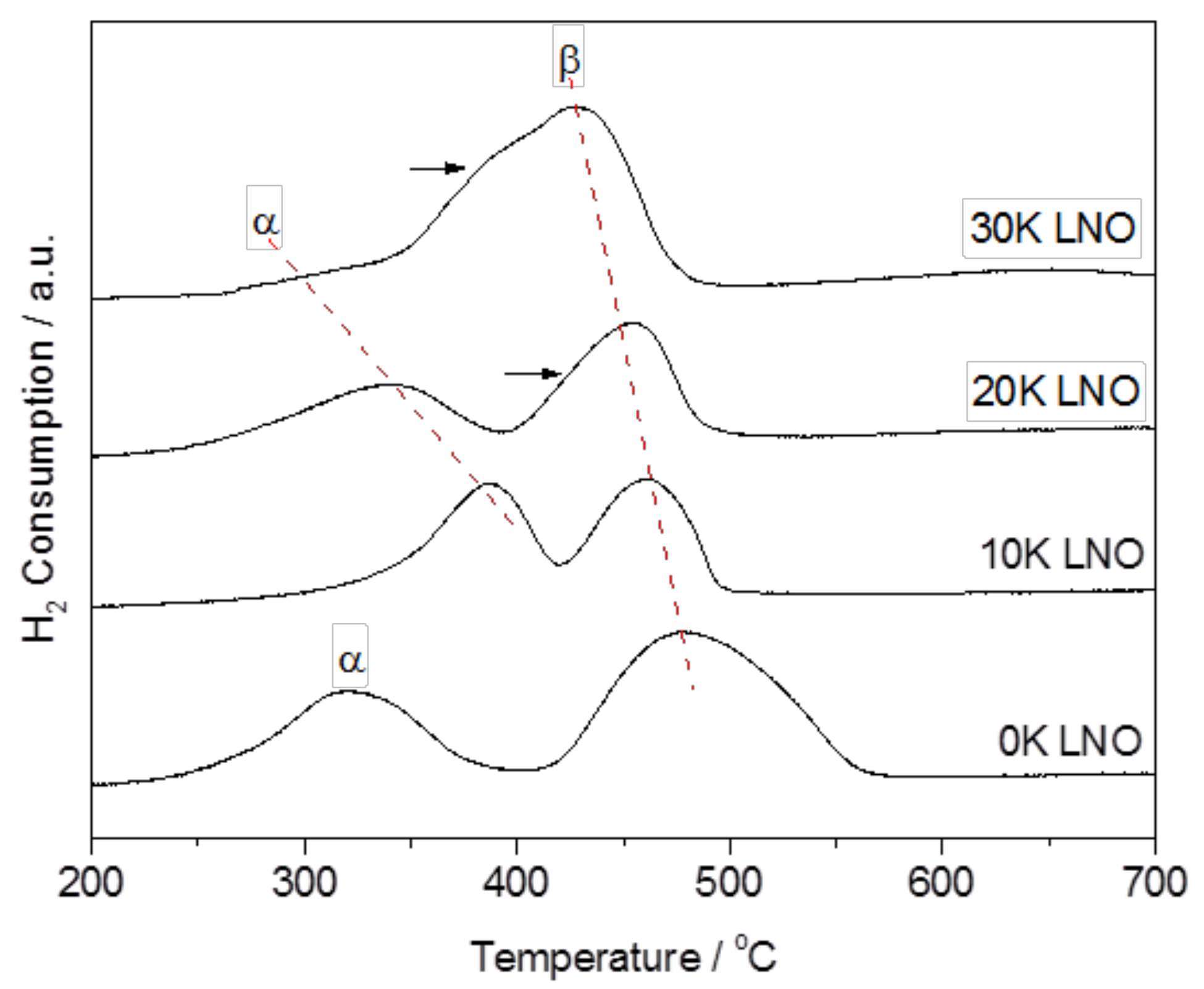
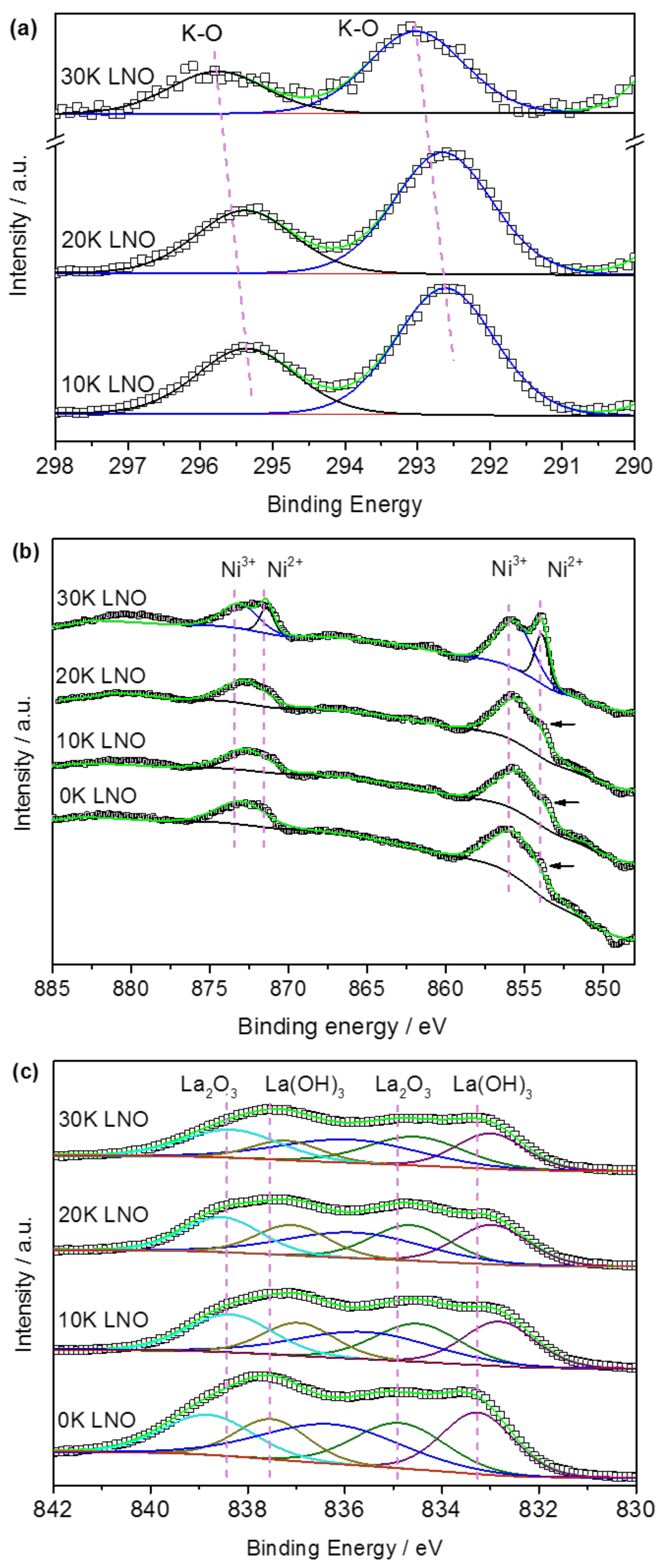
2.1. Material Characterization
2.2. Catalytic Performance
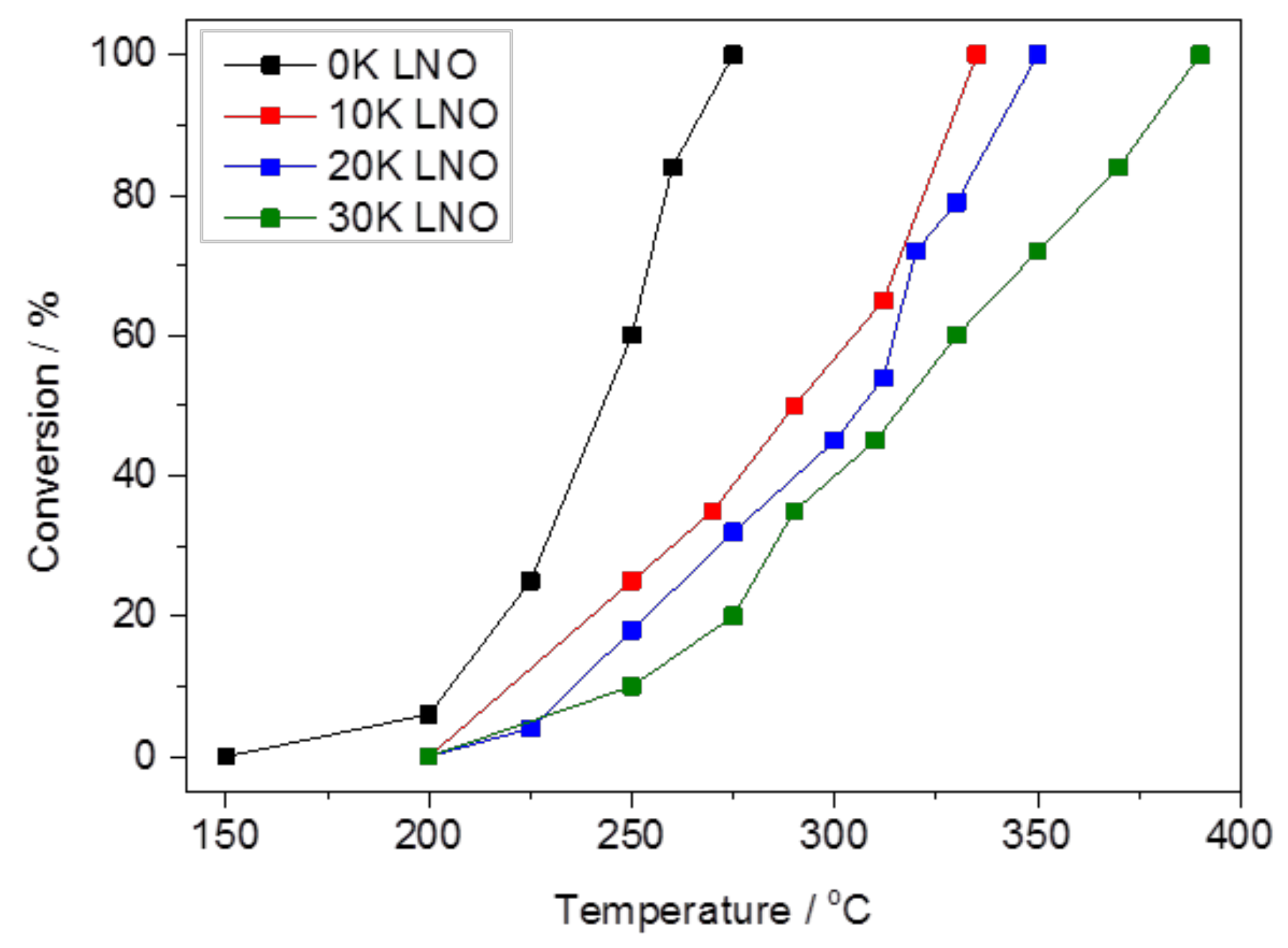
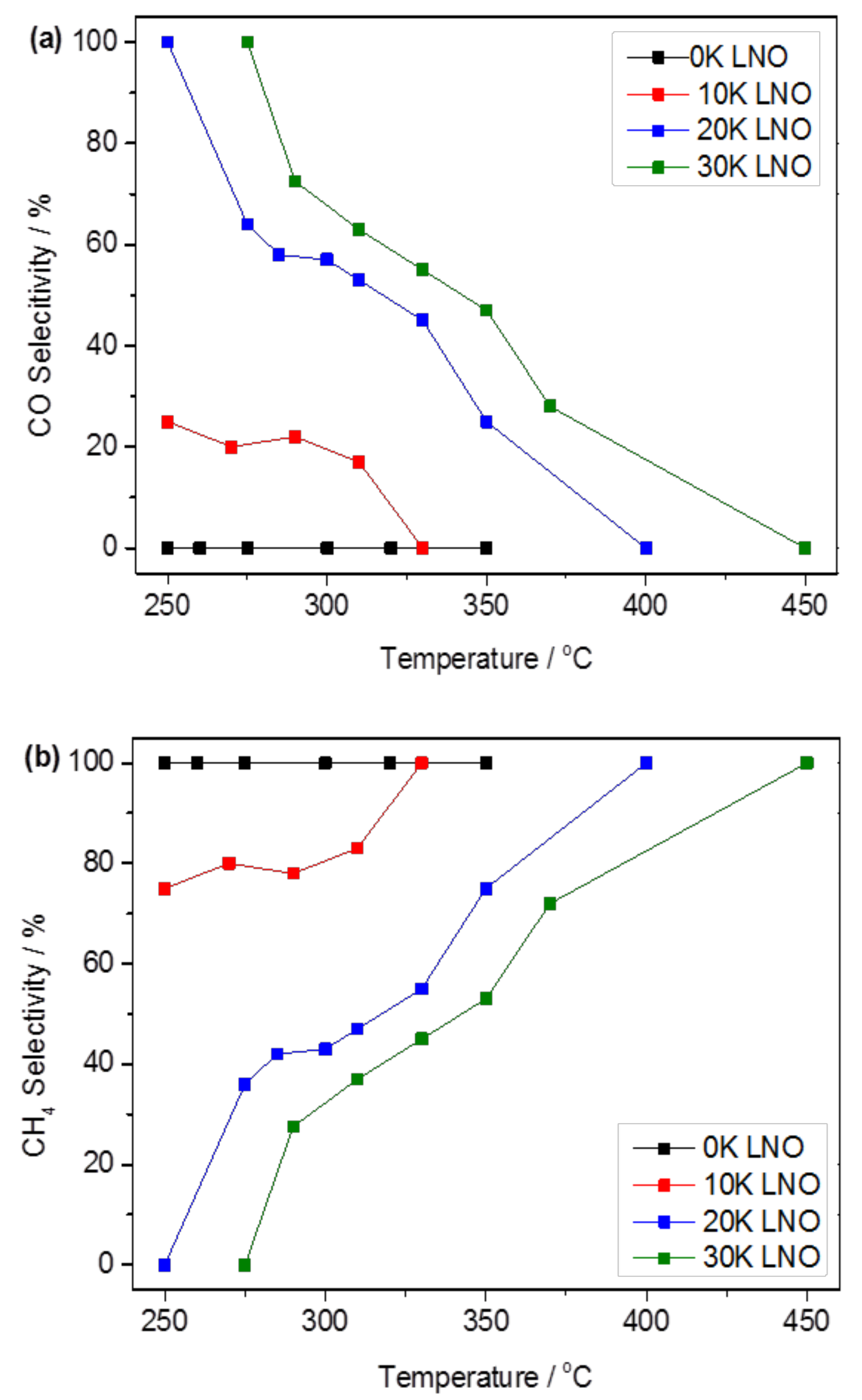
2.2.1. Carbon Dioxide Conversion and Reaction Selectivity
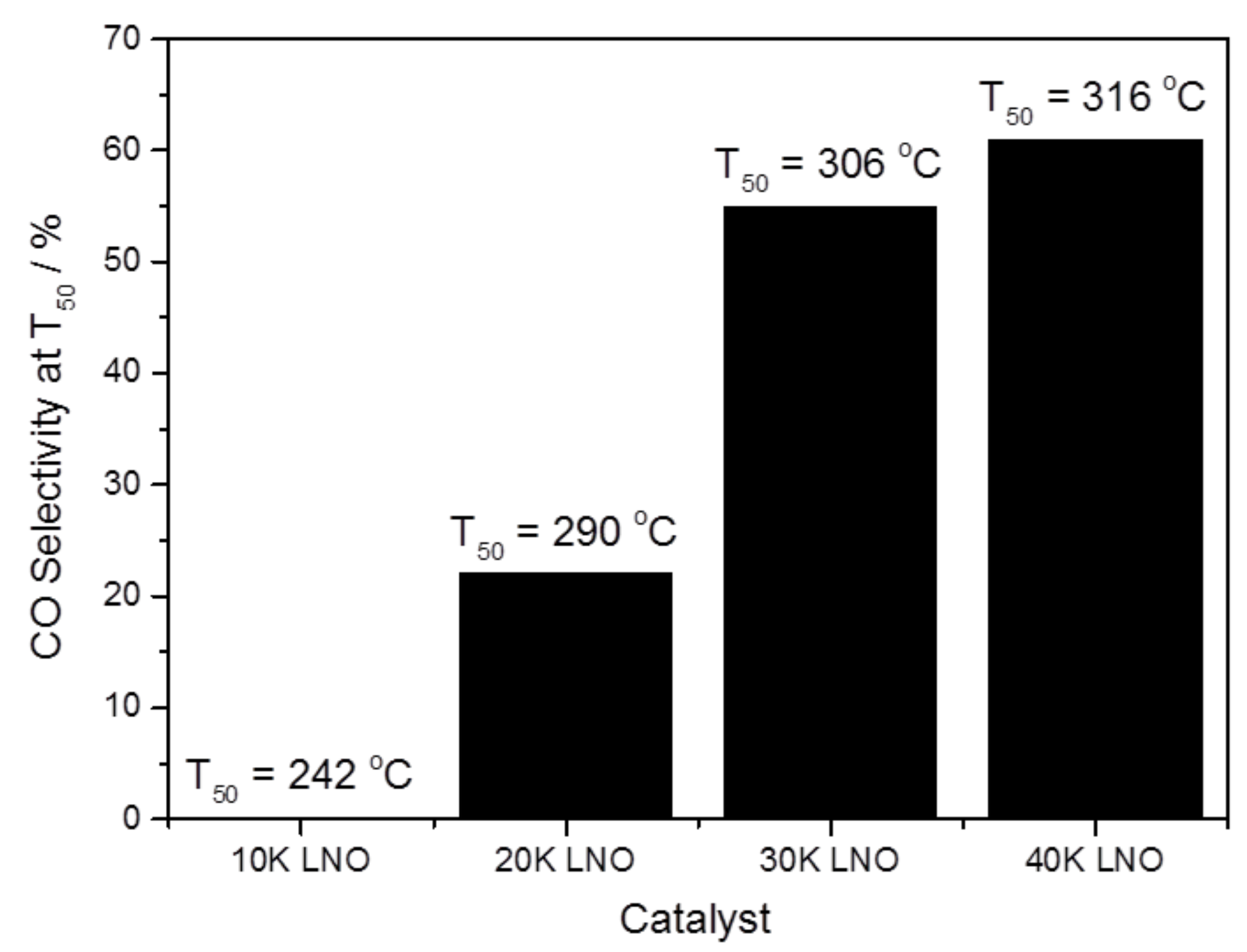
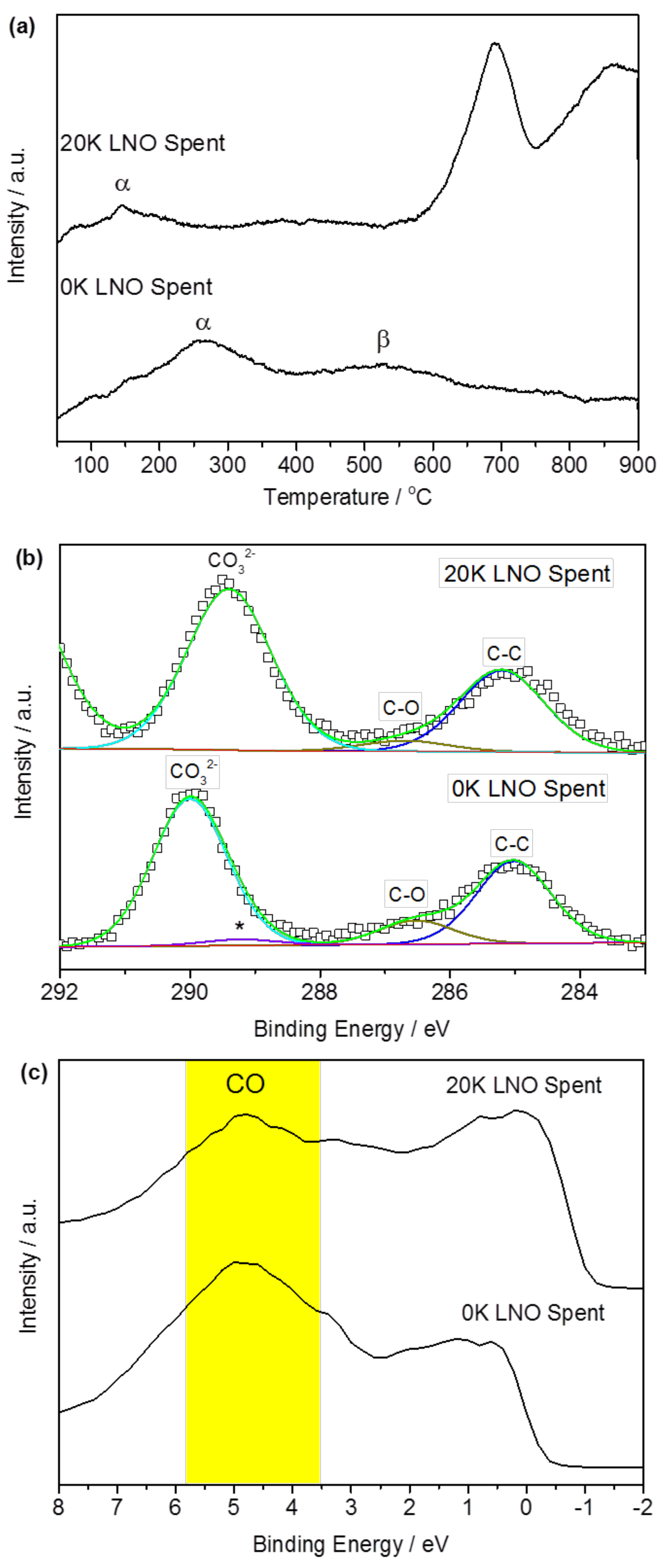
2.2.2. Role of Potassium Substitution in Changing Reaction Selectivity
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Catalytic Apparatus and Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Song, C. Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing. Catal. Today 2006, 115, 2–32. [Google Scholar] [CrossRef]
- Hunt, A.J.; Sin, E.H.K.; Marriott, R.; Clark, J.H. Generation, capture, and utilization of industrial carbon dioxide. ChemSusChem 2010, 3, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Xu, J.; Froment, G.F. Methane steam reforming, methanation and water-gas shift: I. Intrinsic kinetics. AIChE J. 1989, 35, 88–96. [Google Scholar] [CrossRef]
- Ohnishi, R.; Liu, S.; Dong, Q.; Wang, L.; Ichikawa, M. Catalytic dehydrocondensation of methane with CO and CO2 toward benzene and naphthalene on Mo/HZSM-5 and Fe/Co-Modified Mo/HZSM-5. J. Catal. 1999, 182, 92–103. [Google Scholar] [CrossRef]
- Wang, L.; Ohnishi, R.; Ichikawa, M. Selective dehydroaromatization of methane toward benzene on Re/HZSM-5 catalysts and effects of CO/CO2 Addition. J. Catal. 2000, 190, 276–283. [Google Scholar] [CrossRef]
- Liu, Z.; Nutt, M.A.; Iglesia, E. The effects of CO2, CO and H2 Co-reactants on methane reactions catalyzed by Mo/H-ZSM-5. Catal. Lett. 2002, 81, 271–279. [Google Scholar] [CrossRef]
- Arandiyan, H.; Wang, Y.; Scott, J.; Mesgari, S.; Dai, H.; Amal, R. In situ exsolution of bimetallic Rh–Ni nanoalloys: A highly efficient catalyst for CO2 methanation. ACS Appl. Mater. Interfaces 2018, 10, 16352–16357. [Google Scholar] [CrossRef]
- Arandiyan, H.; Wang, Y.; Sun, H.; Rezaei, M.; Dai, H. Ordered meso- and macroporous perovskite oxide catalysts for emerging applications. Chem. Commun. 2018, 54, 6484–6502. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Qi, G.; Dahlberg, K.; Li, W. Strontium-doped perovskites rival platinum catalysts for treating NOx in simulated diesel exhaust. Science 2010, 327, 1624–1627. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Arandiyan, H.; Scott, J.; Akia, M.; Dai, H.; Deng, J.; Aguey-Zinsou, K.-F.; Amal, R. High performance Au–Pd supported on 3D hybrid strontium-substituted lanthanum manganite perovskite catalyst for methane combustion. ACS Catal. 2016, 6, 6935–6947. [Google Scholar] [CrossRef]
- Fukuhara, C.; Hayakawa, K.; Suzuki, Y.; Kawasaki, W.; Watanabe, R. A novel nickel-based structured catalyst for CO2 methanation: A honeycomb-type Ni/CeO2 catalyst to transform greenhouse gas into useful resources. Appl. Catal. A 2017, 532, 12–18. [Google Scholar] [CrossRef]
- Ashok, J.; Ang, M.L.; Kawi, S. Enhanced activity of CO2 methanation over Ni/CeO2-ZrO2 catalysts: Influence of preparation methods. Catal. Today 2017, 281, 304–311. [Google Scholar] [CrossRef]
- Abdolrahmani, M.; Parvari, M.; Habibpoor, M. Effect of copper substitution and preparation methods on the LaMnO3±δ structure and catalysis of methane combustion and CO oxidation. Chin. J. Catal. 2010, 31, 394–403. [Google Scholar] [CrossRef]
- Singh, S.; Zubenko, D.; Rosen, B.A. Influence of LaNiO3 shape on its solid-phase crystallization into coke-free reforming catalysts. ACS Catal. 2016, 6, 4199–4205. [Google Scholar] [CrossRef]
- Fierro, J.L.G.; Tascón, J.M.D.; Tejuca, L.G. Surface properties of LaNiO3: Kinetic studies of reduction and of oxygen adsorption. J. Catal. 1985, 93, 83–91. [Google Scholar] [CrossRef]
- Weatherbee, G.D.; Bartholomew, C.H. Hydrogenation of CO2 on group VIII metals: II. Kinetics and mechanism of CO2 hydrogenation on nickel. J. Catal. 1982, 77, 460–472. [Google Scholar] [CrossRef]
- Lin, W.; Stocker, K.M.; Schatz, G.C. Mechanisms of hydrogen-assisted CO2 reduction on nickel. J. Am. Chem. Soc. 2017, 139, 4663–4666. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, S.; Zhang, L.; Chen, Z.; Wang, M.; Wang, S. A facile method to promote LaMnO3 perovskite catalyst for combustion of methane. Catal. Commun. 2017, 97, 88–92. [Google Scholar] [CrossRef]
- Chen, D.; Chen, C.; Baiyee, Z.M.; Shao, Z.; Ciucci, F. Nonstoichiometric oxides as low-cost and highly-efficient oxygen reduction/evolution catalysts for low-temperature electrochemical devices. Chem. Rev. 2015, 115, 9869–9921. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Duan, H.; Su, X.; Chen, X.; Huang, Y.; Chen, X.; Delgado, J.J.; Zhang, T. Promoting role of potassium in the reverse water gas shift reaction on Pt/mullite catalyst. Catal. Today 2017, 281 Pt 2, 319–326. [Google Scholar] [CrossRef]
- Yang, X.L.; Su, X.; Chen, X.D.; Duan, H.M.; Liang, B.L.; Liu, Q.G.; Liu, X.Y.; Ren, Y.J.; Huang, Y.Q.; Zhang, T. Promotion effects of potassium on the activity and selectivity of Pt/zeolite catalysts for reverse water gas shift reaction. Appl. Catal. B 2017, 216, 95–105. [Google Scholar] [CrossRef]
- Amoyal, M.; Vidruk-Nehemya, R.; Landau, M.V.; Herskowitz, M. Effect of potassium on the active phases of Fe catalysts for carbon dioxide conversion to liquid fuels through hydrogenation. J. Catal. 2017, 348, 29–39. [Google Scholar] [CrossRef]
- Bansode, A.; Tidona, B.; von Rohr, P.R.; Urakawa, A. Impact of K and Ba promoters on CO2 hydrogenation over Cu/Al2O3 catalysts at high pressure. Catal. Sci. Technol. 2013, 3, 767–778. [Google Scholar] [CrossRef]
- Xu, J.; Liu, J.; Zhao, Z.; Xu, C.; Zheng, J.; Duan, A.; Jiang, G. Easy synthesis of three-dimensionally ordered macroporous La1−xKxCoO3 catalysts and their high activities for the catalytic combustion of soot. J. Catal. 2011, 282, 1–12. [Google Scholar] [CrossRef]
- Feng, N.; Chen, C.; Meng, J.; Wu, Y.; Liu, G.; Wang, L.; Wan, H.; Guan, G. Facile synthesis of three-dimensionally ordered macroporous silicon-doped La0.8K0.2CoO3 perovskite catalysts for soot combustion. Catal. Sci. Technol. 2016, 6, 7718–7728. [Google Scholar] [CrossRef]
- de Lima, S.M.; da Silva, A.M.; da Costa, L.O.O.; Assaf, J.M.; Mattos, L.V.; Sarkari, R.; Venugopal, A.; Noronha, F.B. Hydrogen production through oxidative steam reforming of ethanol over Ni-based catalysts derived from La1−xCexNiO3 perovskite-type oxides. Appl. Catal. B 2012, 121, 1–9. [Google Scholar] [CrossRef]
- Silva, P.P.; Ferreira, R.A.R.; Noronha, F.B.; Hori, C.E. Hydrogen production from steam and oxidative steam reforming of liquefied petroleum gas over cerium and strontium doped LaNiO3 catalysts. Catal. Today 2017, 289, 211–221. [Google Scholar] [CrossRef]
- Seaton, N.A. Determination of the connectivity of porous solids from nitrogen sorption measurements. Chem. Eng. Sci. 1991, 46, 1895–1909. [Google Scholar] [CrossRef]
- Zhen, W.; Li, B.; Lu, G.; Ma, J. Enhancing catalytic activity and stability for CO2 methanation on Ni-Ru/Al2O3 via modulating impregnation sequence and controlling surface active species. RSC Adv. 2014, 4, 16472–16479. [Google Scholar] [CrossRef]
- Maneerung, T.; Hidajat, K.; Kawi, S. K-doped LaNiO3 perovskite for high-temperature water-gas shift of reformate gas: Role of potassium on suppressing methanation. Int. J. Hydrogen Energy 2017, 42, 9840–9857. [Google Scholar] [CrossRef]
- Miyakoshi, A.; Ueno, A.; Ichikawa, M. XPS and TPD characterization of manganese-substituted iron–potassium oxide catalysts which are selective for dehydrogenation of ethylbenzene into styrene. Appl. Catal. A 2001, 219, 249–258. [Google Scholar] [CrossRef]
- Sawyer, R.; Nesbitt, H.W.; Secco, R.A. High resolution X-ray Photoelectron Spectroscopy (XPS) study of K2O–SiO2 glasses: Evidence for three types of O and at least two types of Si. J. Non-Cryst. Solids 2012, 358, 290–302. [Google Scholar] [CrossRef]
- Misra, D.; Kundu, T.K. Transport properties and metal–insulator transition in oxygen deficient LaNiO3: A density functional theory study. Mater. Res. Express 2016, 3, 095701. [Google Scholar] [CrossRef]
- Mickevičius, S.; Grebinskij, S.; Bondarenka, V.; Vengalis, B.; Šliužienė, K.; Orlowski, B.A.; Osinniy, V.; Drube, W. Investigation of epitaxial LaNiO3−x thin films by high-energy XPS. J. Alloys Compd. 2006, 423, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Kesavan, J.K.; Luisetto, I.; Tuti, S.; Meneghini, C.; Iucci, G.; Battocchio, C.; Mobilio, S.; Casciardi, S.; Sisto, R. Nickel supported on YSZ: The effect of Ni particle size on the catalytic activity for CO2 methanation. J. CO2 Util. 2018, 23, 200–211. [Google Scholar] [CrossRef]
- Su, X.; Yang, X.; Zhao, B.; Huang, Y. Designing of highly selective and high-temperature endurable RWGS heterogeneous catalysts: Recent advances and the future directions. J. Energy Chem. 2017, 26, 854–867. [Google Scholar] [CrossRef]
- Sun, F.-m.; Yan, C.-f.; Wang, Z.-d.; Guo, C.-q.; Huang, S.-l. Ni/Ce–Zr–O catalyst for high CO2 conversion during reverse water gas shift reaction (RWGS). Int. J. Hydrogen Energy 2015, 40, 15985–15993. [Google Scholar] [CrossRef]
- Tada, S.; Shimizu, T.; Kameyama, H.; Haneda, T.; Kikuchi, R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int. J. Hydrogen Energy 2012, 37, 5527–5531. [Google Scholar] [CrossRef]
- Ullah, S.; Lovell, E.C.; Wong, R.J.; Tan, T.H.; Scott, J.A.; Amal, R. Light enhanced CO2 reduction to CH4 using non-precious transition metal catalysts. ACS Sustain. Chem. Eng. 2020, 8, 5056–5066. [Google Scholar] [CrossRef]
- García-García, I.; Lovell, E.C.; Wong, R.J.; Barrio, V.L.; Scott, J.; Cambra, J.F.; Amal, R. Silver-Based Plasmonic Catalysts for Carbon Dioxide Reduction. ACS Sustain. Chem. Eng. 2020, 8, 1879–1887. [Google Scholar] [CrossRef]
- Jantarang, S.; Lovell, E.C.; Tan, T.H.; Scott, J.; Amal, R. Role of support in photothermal carbon dioxide hydrogenation catalysed by Ni/CexTiyO2. Prog. Nat. Sci. Mat. Int. 2018, 28, 168–177. [Google Scholar] [CrossRef]
- Tejuca, L.G.; Fierro, J.L.G. XPS and TPD probe techniques for the study of LaNiO3 perovskite oxide. Thermochim. Acta 1989, 147, 361–375. [Google Scholar] [CrossRef]
- Day, J.P.; Pearson, R.G.; Basolo, F. Kinetics and mechanism of the thermal decomposition of nickel tetracarbonyl. J. Am. Chem. Soc. 1968, 90, 6933–6938. [Google Scholar] [CrossRef]
- Lehman, R.L.; Gentry, J.S.; Glumac, N.G. Thermal stability of potassium carbonate near its melting point. Thermochim. Acta 1998, 316, 1–9. [Google Scholar] [CrossRef]
- Zagli, A.E.; Falconer, J.L.; Keenan, C.A. Methanation on supported nickel catalysts using temperature programmed heating. J. Catal. 1979, 56, 453–467. [Google Scholar] [CrossRef]
- Peebles, D.E.; Goodman, D.W.; White, J.M. Methanation of carbon dioxide on nickel(100) and the effects of surface modifiers. J. Phys. Chem. 1983, 87, 4378–4387. [Google Scholar] [CrossRef]
- Dwivedi, N.; Yeo, R.J.; Satyanarayana, N.; Kundu, S.; Tripathy, S.; Bhatia, C.S. Understanding the role of nitrogen in plasma-assisted surface modification of magnetic recording media with and without ultrathin carbon overcoats. Sci. Rep. 2015, 5, 7772. [Google Scholar] [CrossRef] [Green Version]
- Glatzel, P.; Singh, J.; Kvashnina, K.O.; van Bokhoven, J.A. In Situ Characterization of the 5d Density of States of Pt Nanoparticles upon Adsorption of CO. J. Am. Chem. Soc. 2010, 132, 2555–2557. [Google Scholar] [CrossRef]
- Chen, C.-S.; Cheng, W.-H.; Lin, S.-S. Mechanism of CO formation in reverse water–gas shift reaction over Cu/Al2O3 catalyst. Catal. Lett. 2000, 68, 45–48. [Google Scholar] [CrossRef]
- Wong Roong, J.; Tsounis, C.; Scott, J.; Low Gary, K.C.; Amal, R. Promoting catalytic oxygen activation by localized surface plasmon resonance: Effect of visible light pre-treatment and bimetallic interactions. ChemCatChem 2017, 10, 287–295. [Google Scholar] [CrossRef]
- Wong, R.J.; Scott, J.; Kappen, P.; Low, G.K.C.; Hart, J.N.; Amal, R. Enhancing bimetallic synergy with light: The effect of UV light pre-treatment on catalytic oxygen activation by bimetallic Au–Pt nanoparticles on a TiO2 support. Catal. Sci. Technol. 2017, 7, 4792–4805. [Google Scholar] [CrossRef]
- Irusta, S.; Pina, M.P.; Menéndez, M.; Santamarı́a, J. Catalytic combustion of volatile organic compounds over La-based perovskites. J. Catal. 1998, 179, 400–412. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | BET Surface Area (m2 g−1) | K Surface Amount a,b (at. %) | Overall K Amount b,c (at. %) |
|---|---|---|---|
| 0K LNO | 11 | 0 | 0 |
| 10K LNO | 6.5 | 30.4 | 0.56 |
| 20K LNO | 5.2 | 23.8 | 1.3 |
| 30K LNO | 7.1 | 3.56 | 2.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsounis, C.; Wang, Y.; Arandiyan, H.; Wong, R.J.; Toe, C.Y.; Amal, R.; Scott, J. Tuning the Selectivity of LaNiO3 Perovskites for CO2 Hydrogenation through Potassium Substitution. Catalysts 2020, 10, 409. https://doi.org/10.3390/catal10040409
Tsounis C, Wang Y, Arandiyan H, Wong RJ, Toe CY, Amal R, Scott J. Tuning the Selectivity of LaNiO3 Perovskites for CO2 Hydrogenation through Potassium Substitution. Catalysts. 2020; 10(4):409. https://doi.org/10.3390/catal10040409
Chicago/Turabian StyleTsounis, Constantine, Yuan Wang, Hamidreza Arandiyan, Roong Jien Wong, Cui Ying Toe, Rose Amal, and Jason Scott. 2020. "Tuning the Selectivity of LaNiO3 Perovskites for CO2 Hydrogenation through Potassium Substitution" Catalysts 10, no. 4: 409. https://doi.org/10.3390/catal10040409