The Role of STAT3 in Thyroid Cancer

Department of Biochemistry, Medical College of Wisconsin, 8701 Watertown Plank Road, Milwaukee, WI 53226, USA
*
Author to whom correspondence should be addressed.
Cancers 2014, 6(1), 526-544; https://doi.org/10.3390/cancers6010526
Submission received: 16 December 2013
/
Revised: 15 February 2014
/
Accepted: 27 February 2014
/
Published: 6 March 2014
(This article belongs to the Special Issue STAT3 Signalling in Cancer: Friend or Foe)
Abstract
:Thyroid cancer is the most common endocrine malignancy and its global incidence rates are rapidly increasing. Although the mortality of thyroid cancer is relatively low, its rate of recurrence or persistence is relatively high, contributing to incurability and morbidity of the disease. Thyroid cancer is mainly treated by surgery and radioiodine remnant ablation, which is effective only for non-metastasized primary tumors. Therefore, better understanding of the molecular targets available in this tumor is necessary. Similarly to many other tumor types, oncogenic molecular alterations in thyroid epithelium include aberrant signal transduction of the mitogen-activated protein kinase, phosphatidylinositol 3-kinase/AKT (also known as protein kinase B), NF-кB, and WNT/β-catenin pathways. However, the role of the Janus kinase (JAK)/signal transducer and activator of transcription (STAT3) pathway, a well-known mediator of tumorigenesis in different tumor types, is relatively less understood in thyroid cancer. Intriguingly, recent studies have demonstrated that, in thyroid cancer, the JAK/STAT3 pathway may function in the context of tumor suppression rather than promoting tumorigenesis. In this review, we provide an update of STAT3 function in thyroid cancer and discuss some of the evidences that support this hypothesis.
1. Introduction
Thyroid cancer is the most common neoplasm of the endocrine system, which originates from follicular thyrocytes or parafollicular C-cells of the thyroid gland (reviewed in [1]). Although relatively rare, thyroid cancer is the seventh most frequent human malignancy, and is increasing in incidence more rapidly than any other cancers. From 1995 to 2008, thyroid cancer age-adjusted incidence rates more than doubled in the U.S., whereas the incidence of most major cancers (lung, prostate, breast and colorectal) decreased during the same period [2]. Therefore, thyroid cancer is a growing health problem. Thyroid cancer is mainly treated by surgery and radioiodine remnant ablation, which is effective only for non-metastasized primary tumors. Metastatic and relapsed tumors are mostly incurable, requiring more advanced therapeutic modalities for patient survival.
Depending upon the cell origin and histological characteristics, thyroid carcinomas are generally classified to papillary thyroid carcinoma (PTC), follicular thyroid carcinoma (FTC), anaplastic thyroid carcinoma (ATC, poorly differentiated), and medullary thyroid cancer (MTC). As summarized in Table 1, most of these thyroid tumors arise from the follicular thyrocytes whereas MTC is the only C-cell-originated tumor, constituting the minor fraction of thyroid malignancies. While various alterations at molecular levels underlie the onset of thyroid carcinogenesis, activation of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)/AKT pathways, often in close connection and cooperation, constitute the major mechanisms for the development and progression of most thyroid tumors (Table 1). Nevertheless, activation of these pathways is attributed to different genetic or epigenetic alteration of oncogenes and tumor suppressors. In about 65% to 70% of the follicular thyrocyte-derived tumors, mutated B-Raf, Ras, phosphatase and tensin homolog (PTEN), or PIK3CA drives the activation of the MAPK and PI3K pathways [3,4,5]. Ras protein activator like 1 (RASAL1) is a major tumor suppressor in thyroid cancer and its alterations can affect the MAPK pathway activity [6]. Moreover, oncogenically mutated forms of REarranged during Transfection (RET) receptor tyrosine kinase are also often detected in certain thyroid tumors and are supposed to drive the pathways. For example, various mutations in the cell surface receptor domain or the cytoplasmic kinase domain constitutively activate RET in about 95% of hereditary MTC and about 50% of sporadic MTC cases (reviewed in [7,8]). In about 20% of PTC patients, the kinase domain of RET is often fused to various unrelated genes by chromosomal rearrangements (known as RET/PTC) and becomes constitutively active (reviewed in [9]). In addition, activation of other receptor tyrosine kinases including epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), and c-MET/hepatocyte growth factor receptor are also detected in a subset of MTC patients [10]. Despite the plethora of information on different signal transduction pathways in thyroid cancer, the information on the Janus kinase (JAK)/signal transducer and activator of transcription (STAT3) pathway is relatively limited. Intriguingly, emerging evidences suggest that the JAK/STAT3 pathway may have tumor suppressive effects on thyroid cancers, unlike its effects on many other cancer types.

{kind=link}
| Tumor type | Cells of origin | Prevalence (% of total thyroid cancers) | Frequently detected genetic/epigenetic alterations | Frequently detected aberrant pathway signaling |
|---|---|---|---|---|
| Papillary thyroid carcinoma (PTC) | Follicular thyrocytes | 80%–85% | BRAFV600E (45%) | MAPK pathway PI3K/AKT pathway IDH1-associated metabolic pathways |
| RET/PTC translocation (20%) | ||||
| IDH1 mutation (10%) | ||||
| EGFR mutation (5%) | ||||
| RASAL1 mutation or hypermethylation (3%) | ||||
| PTEN mutation (1%–2%) | ||||
| PIK3CA mutations (1%–2%) | ||||
| Follicular thyroid carcinoma (FTC) | Follicular thyrocytes | 10%–15% | PAX8/PPARγ rearrangement (40%–60%) | MAPK pathway PI3K/AKT pathway IDH1-associated metabolic pathways |
| HRAS, KRAS, or NRAS mutation (30%–45%) | ||||
| RASAL1 mutation or hypermethylation (32%) | ||||
| PTEN deletion (30%) | ||||
| PTEN mutation (10%–15%) | ||||
| PIK3CA mutation (5%–15%) | ||||
| IDH1 mutation (5%–15%) | ||||
| Poorly differentiated thyroid carcinoma (PDTC) | Follicular thyrocytes | 5%–10% | HRAS, KRAS, or NRAS mutation (20%–40%) | MAPK pathway PI3K/AKT pathway WNT/β-catenin pathway p53-regulated pathways |
| CTNNB1 mutation (25%) | ||||
| TP53 mutation (25%) | ||||
| Anaplastic thyroid carcinoma (ATC) | Follicular thyrocytes | 2%–3% | BRAFV600E mutation (25%–50%) | MAPK pathway PI3K/AKT pathway WNT/β-catenin pathway p53-regulated pathways IDH1- associated metabolic pathways |
| HRAS, KRAS, or NRAS (20%–30%) | ||||
| PTEN mutation (10%–20%) | ||||
| PIK3CA mutation (15%–25%) | ||||
| CTNNB1 mutation (60%–65%) | ||||
| TP53 mutation (70%–80%) | ||||
| IDH1 mutation (5%–15%) | ||||
| ALK mutation (10%) | ||||
| RASAL1 mutations or hypermethylation (33%) | ||||
| Medullary thyroid cancer (MTC) | Parafollicular C-cells | 2%–6% | RET mutation (99% of the familial, 30%–50% of the sporadic cases) | MAPK pathway |
| RAS mutation (10% of the sporadic cases) | PI3K/AKT pathway |
* This table was generated based on the reviews [1,10,11] and research articles [6,12,13,14,15,16]. For further details, refer to the cited reports. Abbreviations used are ALK, anaplastic lymphoma kinase; CTNNB1, cadherin-associated protein β1; EGFR; epithelial growth factor receptor; IDH, isocitrate dehydrogenase; MAPK, mitogen-activated protein kinase; PAX8, paired box 8; PI3K, phosphatidylinositol 3-kinase; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit α; PPARγ, peroxisome proliferator-activated receptor-gamma; PTEN, phosphatase and tensin homolog; RASAL1, Ras protein activator like 1; RET, REarranged in Transformation; RET/PTC, RET/papillary thyroid carcinoma.
2. The JAK/STAT Pathway
The JAK/STAT pathway mediates signal transduction induced by cytokine receptors. Upon cytokine binding to the receptors, the receptor-associated cytoplasmic tyrosine kinase JAK cross-phosphorylates each other on tyrosine for activation. Active JAKs phosphorylate receptors to generate the docking site for the latent cytoplasmic factors, STATs (reviewed in [17]). STATs are then phosphorylated by JAKs, dissociate from the receptor, dimerize, and translocate into the nucleus to induce expression of a variety of genes, so called “cytokine-responsive genes” [18,19]. A number of hematopoietin subfamily of cytokines can bind to the cytokine receptors and activate the JAK/STAT pathway. To mediate this cytokine receptor signaling, human genome encodes four known JAKs and at least six STATs (reviewed in [19,20]).
2.1. STAT3
STAT3 is a latent gene regulatory protein, serving as a key effector of cytokine receptor/JAK/STAT signaling. STAT3 was originally discovered, along with STAT1 and STAT2, in a protein complex responsible for the regulation of interferon-dependent transcription [21,22]. STAT3 was also discovered as a transcription factor that binds to Interleukin (IL)-6-responsive elements located in the promoter region of various acute-phase genes [23], and then it was shown to be phosphorylated on its Tyr705 in response to IL-6 [24]; the history of early discovery of STATs has been recently reviewed in detail elsewhere [25]. STAT3 is constitutively expressed in a wide range of tissues. Nonetheless, immunohistochemical analysis of phosphorylated STAT3 on Tyr705 revealed that its nuclear presence is not ubiquitous but is selectively detected in three major cell types in human tissues, suggesting its tissue-specific function [26]. These cell types are (i) lymphoid and accessary cells in immune, digestive, and respiratory systems; (ii) glandular, mucosal, and secretory epithelium in digestive system, endocrine and reproductive systems, and endothelium of circulatory system (heart, veins, and capillaries); (iii) proliferative and reabsorption-active cells in productive, urinary, and integumentary systems [26]. Unlike other STATs, STAT3 deletion in mice results in lethality between 6.5 and 7.5 days of early embryogenesis, indicating that STAT3 may have more fundamental role than other STATs [27,28,29]. Indeed, while regulating a number of genes involved in the inflammatory response, STAT3 can also regulate different genes involved in apoptosis, differentiation, and stem cell maintenance, which indicates its wider involvement in the maintenance of cellular homeostasis (reviewed in [30]).
2.2. Activation/Inactivation of STAT3
Phosphorylation of STAT3 on Tyr705 is the major mechanism of STAT3 activation, which is mediated by JAK upon stimulation of the heterodimeric gp130/cytokine-specific receptor complex by the IL-6 family of cytokines, including IL-6, leukemia inhibitory factor (LIF), ciliary neurotrophic factor, oncostatin M, IL-11, and cardiotrophin-1 (reviewed in [31]). STAT3 activation can also be mediated in a cytokine receptor-independent manner [17,20,32]. For example, activation of the receptor tyrosine kinases by epidermal growth factor or platelet-derived growth factor as well as expression of the oncogenic c-Src kinase or the small GTPase Ras can also induce STAT3 activation (additional signals that activate STAT3 are reviewed in [33]). Moreover, STAT3 phosphorylation on Tyr705 can also be mediated in a JAK-independent manner. For example, a recent work has demonstrated that the homodimer form of pyruvate kinase M2 can localize into the nucleus and phosphorylate STAT3 on Tyr705, which was necessary to upregulate MEK5 transcription and increase cell proliferation under a metabolic stress condition [34]. In addition to Tyr705 phosphorylation, Ser727 phosphorylation also has a role for STAT3 activity, particularly for maximizing the transcriptional activity of STAT3 [35,36]. Ser727 phosphorylation of STAT3 is mainly mediated by MAPK1/3 (also known as ERK1/2) in response to different growth factor signals, which often activate receptor tyrosine kinases. Therefore, STAT3 provides a converging point for the signal transduction mediated by cytokine receptors and receptor tyrosine kinases. Intriguingly, although phosphorylation of these two amino acid residues is expected to be synergistic for maximal STAT3 activation, it has been reported that Ser727 phosphorylation can prevent Tyr705 phosphorylation or even induces its dephosphorylation, suggesting the presence of “cross-regulation” to balance STAT3 activity between different pathways that activate STAT3 [35].
The activity of STAT3 should be precisely controlled for the maintenance of cellular homeostasis during development and in adults, which requires the participation of diverse negative regulators (reviewed in [32]). These negative regulators include (i) cytoplasmic tyrosine phosphatases, e.g., protein tyrosine phosphatase 1B, that dephosphorylate STAT proteins [37]; (ii) suppressors of cytokine signaling (SOCS) proteins that block the cytokine receptor [38]; (iii) proteins that inhibit activated STATs (PIAS) that interacts with tyrosine phosphorylated STATs and blocks their DNA binding [39]; (iv) naturally occurring truncated STAT proteins that can act as dominant-negative inhibitors by occupying DNA as non-functional transcription factor or by dimerizing with a wild-type STAT [40]. Loss of these negative regulators, particularly PIAS3 and SOCS, are known to contribute to abnormal STAT3 activation in leukemia, lymphoma, hepatocellular carcinoma and non-small cell lung carcinoma [41]. A study also demonstrated that SOCS-3 is frequently silenced by hypermethylation in human cancers [42].
2.3. Non-Canonical Activity of STAT3
Serving as a transcription factor upon activation via Tyr705 phosphorylation has been recognized as the canonical STAT3 function. However, STAT3 can also regulate transcription independently of Tyr705 phosphorylation. For example, similarly to the observation that unphosphorylated STAT1 could mediate LMP2 transcription in collaboration with IRF1 [43], unphosphorylated STAT3 can also regulate gene transcription in a complex with NF-κB [44,45,46,47]. In the STAT3/NF-κB complex, NF-κB provides DNA-binding and transactivation domains while STAT3 enables nuclear translocation of the complex [46]. Moreover, STAT3 is also subject to other posttranslational modifications in addition to phosphorylation and can mediate important cellular physiology independently of its transcriptional activating functions [25]. For example, non-tyrosine-phosphorylated and cytoplasmic-localized STAT3 could potentiate microtubule polymerization and cell movement by disrupting the interaction between microtubules and one of its partners, stathmin [48]. Recent studies have also demonstrated that STAT3 can localize into the mitochondria, where it controls the activity of the electron transport chain [49,50]. This STAT3 function in the mitochondria was shown to be important for Ras-induced transformation of mouse embryo fibroblasts [50]. These functions of STAT3 in respiration and Ras transformation required Ser727 phosphorylation but were independent of Tyr705 phosphorylation [50].
3. JAK/STAT Signaling in Different Cancer Types
STAT3 was originally identified as an oncogenic protein [51,52]. It has been shown that upregulated expression of STAT3 is associated with more malignant behavior of tumor cells and worse prognosis in a variety of human malignancies, e.g., cancers of the breast, prostate, ovary, lung, head and neck, esophagus, and brain [53,54,55,56,57,58,59,60,61]. In pancreatic cancer, STAT3 was shown to regulate angiogenesis and metastasis [62] and to promote cellular proliferation by accelerating G1/S-phase progression [63]. In gastric cancer, activation of STAT3 via the EGFR signaling pathway may contribute to tumor progression by increasing lymph node metastasis of the tumor [64]. Genetic/epigenetic alterations underlie aberrant STAT3 signaling in cancer and perturbation of positive or negative regulatory components in the JAK/STAT pathway that can cause a persistent STAT3 activation are often detected in different tumors (reviewed in [41]). Recent genome sequencing analyses have indeed revealed somatic mutations of the JAK/STAT pathway that occur in different tumor types [65,66,67]. Nonetheless, these mutations have not been reported yet in thyroid cancer.
STAT3 regulates many genes encoding cytokines, growth factors, and angiogenic factors, which in turn can activate STAT3 through their associated receptors. Therefore, persistent STAT3 activation in many cancers can establish a feed-forward loop between the tumor and the non-transformed stroma cells such as myeloid-derived suppressor cells, cancer-associated fibroblasts, and adipocytes (reviewed in [68,69]). In this manner, STAT3 can regulate a pro-carcinogenic inflammatory microenvironment and tumor immunity. For example, STAT3 activation in tumor cells can induce genes involved in the impairment of dendritic cell maturation, which results in the inhibition of T cell activation [18,70]. Conversely, blocking STAT3 activity in tumor cells induces production of the cell-extrinsic factors that enable dendritic cell maturation and function, leading to T cell activation and subsequent anti-tumor immune responses [18,70]. Recent studies have revealed STAT3 as a major player in the regulation of innate and adaptive tumor immunity and as a therapeutic target to enhance immune recognition and to break T cell tolerance of tumor cells [18,70]. Moreover, the observations that STAT3 silencing suppressed tumor cell growth more effectively in an in vivo microenvironment than in in vitro cultures strongly suggest that the role of STAT3 in facilitating tumorigenesis is mainly attributed to its effects on the tumor cell-extrinsic signaling [71,72].
Intriguingly, it has also been reported that high nuclear expression of Tyr705-phosphorylated STAT3 is correlated with improved survival, smaller tumors, or less aggressive histology in various tumors, including breast cancer [73,74], head and neck cancer [75], lung cancer [76], gastric cancer [77], soft tissue leiomyosarcoma [78], and advanced rectal cancer [79]. However, in pancreatic ductal adenocarcinoma, no association was seen between STAT3 phosphorylation and patient survival [80]. These studies suggest that STAT3 is not always oncogenic in cancer cells but it may behave in an opposing manner in certain tumor types. Indeed, the identity of STAT3 as an oncogene or a tumor suppressor has been questioned in different tumor types (reviewed in [30,81]).
4. STAT3 in Thyroid
4.1. STAT3 in Non-Malignant Thyroid Epithelium
STAT3 is highly expressed in normal thyroid gland. Intriguingly, STAT3 is more expressed in the right lobe than in the left lobe of thyroid, but the significance of this differential expression is currently unclear [82]. In normal thyroid glandular epithelium, Tyr705-phosphorylated STAT3 is distributed both in the cytoplasm and in the nucleus [26]. STAT3 appears to be important for thyroid function. As an effector of leptin signaling, STAT3 is involved in the regulation of neuroendocrine function of thyroid (reviewed in [83]). It has been shown that, upon stimulation of rat thyroid cells with thyroid-stimulating hormone, STAT3, but not STAT1, is rapidly phosphorylated on Tyr705 [84,85,86,87]. In this signaling, STAT3 is activated as a transcriptional activator and mediates gene expression required for the hormone-induced proliferative responses and immunomodulation [84,85,86,87].
Expression of STAT3 and its activity is altered in different thyroid diseases, showing different patterns from the expression of other STATs. For example, when surgical specimens from the patients diagnosed as having Hashimoto's disease or focal lymphocytic thyroiditis were analyzed, activated STAT1 and STAT5 were mainly detected in infiltrating cells of hematopoietic origin whereas STAT3 expression was restricted to epithelial cells [88]. This cell-type-specific expression pattern of STAT3 suggests its distinct role in growth and proliferation of regenerating thyroid follicles. Moreover, STAT3 was colocalized with the anti-apoptotic protein Bcl-2 and expression of Tyr705-phosphorylated STAT3 was associated with low levels of stromal fibrosis, suggesting that STAT3 serves as a protective factor in remodeling of the inflamed thyroid gland [88]. Of note, persisting cytokine-mediated activation of STAT3 was detected in non-lesional adjacent thyroid tissues after thyroidectomy due to different types of diseases. This STAT3 activation was associated with cytoplasmic localization of p53, which has been suggested as a risk factor for the development of neoplastic diseases [89].
4.2. STAT3 in Thyroid Cancer
Although aberrant STAT3 activity mediates oncogenic signaling in many different cancers, STAT3 is also known for its tumor suppressive effects (reviewed in [30]). Indeed, studies including ours have demonstrated that STAT3 can mediate tumor suppressive signal transduction in different tumor types [90,91,92,93,94]. STAT3 therefore has opposing effects on tumorigenesis, and these paradoxical STAT3 effects are also observed in certain thyroid cancer types.
4.2.1. Expression of STAT3 in Thyroid Cancer
Increased expression and activation of STAT3 is detected in the tissue specimens of thyroid cancers, including lymphatic metastasis of PTC [95]. Consistent with this, a histological analysis revealed that STAT3 is distinctively expressed in PTC but not in follicular tumors, suggesting that STAT3 activation may be involved in the establishment of the papillary phenotype [96]. Moreover, a recent study detected increased nuclear-localization of Tyr705 phosphorylated STAT3 in about 57% (63 of 110) human primary PTC cases, preferentially in association with the tumor stroma [90]. On the contrary, a study has demonstrated that STAT3 activity, as determined by its affinity to DNA, is significantly lower in PTC cells than those of surrounding normal thyroid tissues [97]. In this study, tumor size larger than 2 cm was the only clinicopathologic parameter associated with lower STAT3 activity. Moreover, this study revealed an inverse-correlation between tumor size and STAT3 activity, specifically in B-RafV600E-positive cases [97]. Consistent with this finding, increased STAT3 activity was inversely correlated with tumor size and the presence of distant metastases in another study [90]. However in contrast to the former study, this study detected increased STAT3 phosphorylation in multiple human primary PTC cases. These studies suggest that STAT3 expression is altered in selective types of thyroid cancer in correlation with low degree of tumor malignancy.
4.2.2. Mechanism of STAT3 Activation in Thyroid Cancer
It was originally discovered that, in the NIH3T3 model of in vitro tumorigenesis, the oncogene RET/PTC could induce Tyr705 phosphorylation of STAT3 [98]. In this system, STAT3 activation facilitated RET/PTC-mediated cellular transformation by regulating expression of vascular endothelial growth factor, cyclin D1, and intercellular adhesion molecule 1. This observation suggested a possibility that RET/PTC may induce STAT3 activation. Intriguingly, in the human anaplastic thyroid cancer line ARO, STAT3 could directly interact with ectopically expressed RET/PTC1 and phosphorylation of its Tyr705 did not require either JAK or c-Src kinase, indicating a possibility of direct STAT3 activation by the oncogenic kinase [98]. A recent report demonstrated that constitutive STAT3 activation can be mediated via the IL-6/gp130/JAK autocrine signaling pathway in different thyroid cancer lines that express RET/PTC, B-RafV600E, or mutated Ras [90]. Apart from Tyr705 phosphorylation, it has also been shown that Ser727 phosphorylation can be induced by cyclin-dependent kinase 5 in response to the activation of human epidermal growth factor receptor 2 (HER2/erbB2) in the MTC cell line, TT [99]. Intriguingly, while STAT3 Tyr705 phosphorylation was not affected under this condition, overexpression of mutant STAT3 (Ser727 to Ala) dominant-negatively inhibited cyclin-dependent kinase 5-mediated TT cell proliferation, suggesting a specific role of STAT3 for HER2/cyclin-dependent kinase 5 signaling. In addition to these mechanisms, STAT3 in thyroid cancer appears to be regulated via an unexpected mechanism. In KAT-18 cells, a primary culture of anaplastic thyroid cancer, a mutant p53 that harbors a gain of mutation, i.e., TP53G199V missense mutation, affected STAT3 expression status and mediated anti-apoptotic function by transcriptionally upregulating STAT3 [100]. It may also be possible that STAT3 activation in PTC is partly attributed to the mutations in receptor tyrosine kinase since receptor tyrosine kinase can activate STAT3 pathway [33] and activation mutations of epidermal growth factor receptor gene occur in PTC [101].
4.2.3. Effects of STAT3 on Thyroid Cancer: Are They Tumor Suppressive?
Although STAT3 was previously reported for its effects to promote tumor cell survival and proliferation in the context of RET/PTC-tumorigenesis of NIH3T3 or TP53G199V signaling in an anaplastic thyroid cancer cell line [98,100], multiple studies have also identified STAT3 as a tumor suppressor in different thyroid cancer types. For example, a recent study demonstrated that, although shRNA-mediated STAT3 knockdown did not affect in vitro growth of representative thyroid cancer cell lines that express high STAT3 activity, it led to the generation of larger tumors than control when STAT3-depleted tumors cells were xenografted in mice. Consistent with this result, STAT3 deficiency facilitated tumorigenesis in a murine model of B-RafV600E-induced PTC [90]. To mediate tumor suppression, STAT3 increased transcription of the tumor suppresser insulin-like growth factor binding protein 7 and negatively regulated aerobic glycolysis, decreasing energy metabolism in cancer cells [90]. Therefore, STAT3 appears to function as a tumor suppressor in the background of PTC.
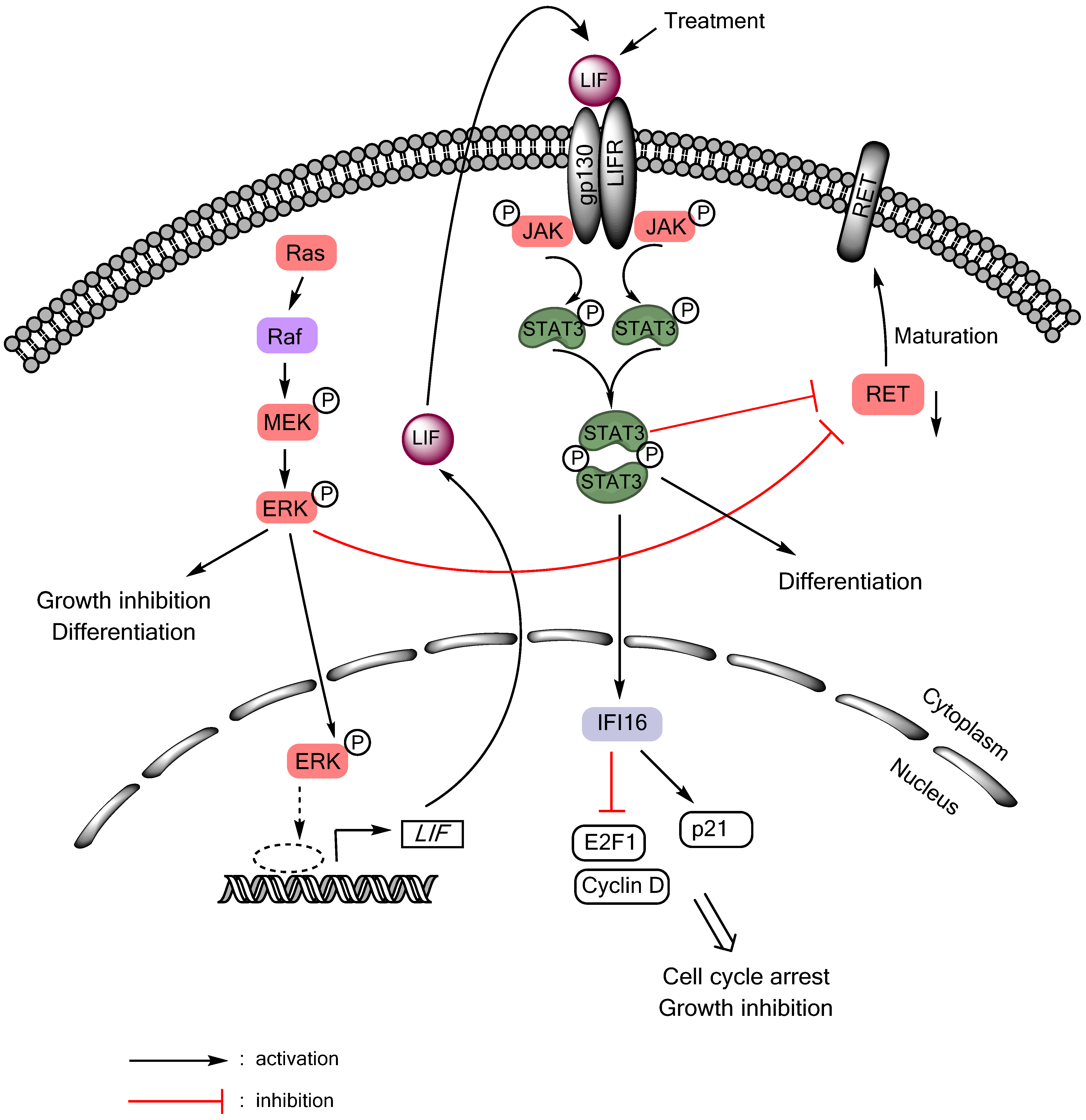
Autocrine/paracrine tumor suppressive signaling of the gp130/JAK/STAT3 pathway can also be activated in MTC cells in response to Ras or Raf activation (Figure 1). Although aberrant Ras or Raf activity is a central feature of many epithelial cancers, sustained activation of Ras/Raf elicits senescence-like growth arrest responses, referred to as “oncogene-induced senescence”, in primary cultured normal cells and premalignant lesions [102,103]. These phenomena are now interpreted as innate tumor-suppressive responses, which are triggered as a fail-safe anti-tumorigenic mechanism by aberrant cell proliferation signals. Intriguingly, similar responses can be induced in certain types of malignant cancer that are not transformed by Ras/Raf, including MTC [91,104,105,106,107], leading to a hypothesis that these tumor types may retain functional tumor suppressive mechanism against Ras/Raf oncogenesis. Upon Ras/Raf activation, MTC cells undergo growth arrest and differentiation within 48 hours, as manifested by cell cycle arrest in G0/G1 phases, increased calcitonin gene expression, and silenced expression of the oncogenic RET [104,106,108]. Prolonged cell culture under this condition eventually leads to expression of the senescence marker, senescence-associated β-galactosidase [109]. In different cell types, the Ras/Raf pathway mediates growth arrest by controlling key cell cycle regulatory and tumor suppressive proteins, including Rb, E2F1, cyclin-dependent kinase inhibitors, and TP53. Our previous studies indicate that, in MTC cells, this tumor suppressive signaling is connected to LIF-controlled extracellular mechanisms (Figure 1). Using biochemical analysis of the culture medium conditioned by MTC cell expressing exogenously introduced oncogenic c-Raf kinase domain, we identified LIF as being essential and sufficient to mediate the growth inhibitory signaling activated by Ras/Raf [91]. LIF mediated this growth inhibitory effect through the gp130/JAK/STAT3 pathway since anti-gp130 blocking antibody or dominant negative STAT3 blocked the effect of LIF. Subsequent analysis of STAT3-mediated gene expression programs identified IFI16 as an effector of STAT3, which regulates the S-phase transcription factor E2F1 and the cyclin-dependent kinase inhibitor p21CIP1 to induce cell cycle arrest in MTC cells [110]. This LIF/gp130/JAK/STAT3-mediated growth inhibitory signaling is conserved in different MTC cell lines regardless of the mutational status of RET [109], and could also be activated by interleukin-1β via the Raf/MEK/ERK pathway [92]. Importantly, recombinant LIF could activate STAT3 and downregulate RET and induce tumor suppression in human MTC cells xenografted in mice [111], suggesting its potential as a therapeutic reagent to treat MTC. While these studies support the tumor suppressive role of STAT3 in the context of MTC, a recent study showed that AZD1480, a small molecule inhibitor of JAK1/2, could also inhibit the growth of MTC cell lines [112]. Therefore, it may be possible that either too high or too low STAT3 activity triggers growth inhibitory responses in MTC cells. Of note, AZD1480 also showed efficacy in the STAT3-deficeint cell line, TPC-1, suggesting that the drug effect could be independent of STAT3 [112]. Together, these studies highlight the complexities of the JAK/STAT signaling in thyroid cancers.
5. Conclusions
Cellular context can influence the response to a signal and, indeed, STAT3 can either promote or suppress cell growth depending upon cell types. It is currently unclear how STAT3 can mediate these opposing effects. Because oncogenic stress can elicit growth inhibitory responses as an innate tumor defense mechanism, STAT3 signaling may also be networked with a similar tumor defense mechanism, which is activated in the face of aberrant STAT3 activation. It appears that this context is relevant in a subset of thyroid cancer, including PTC and MTC. Intriguingly, the effect of the Ras/Raf pathway is clearly in contrast in these two thyroid tumor types in that the pathway is constitutively active and promotes PTC cell proliferation whereas its basal activity is relatively low while its constitutive activation can induce growth inhibition in MTC cells. Nevertheless, the effect of Ras/Raf activation on STAT3 was consistent in these two tumor types in that, in both MTC and PTC, Ras/Raf activates STAT3 via the IL-6 family cytokine-mediated gp130/JAK pathway and STAT3 operates in the context of tumor suppression. It is possible that STAT3 signaling is developmental biologically programmed to be tumor suppressive in thyroid epithelium. Of note, while STAT3 played a role as a transcription factor in both thyroid cancer types, the role of STAT3 in the mitochondria was also important to mediate tumor suppressive signaling in PTC [90]. It is currently unclear whether this non-canonical STAT3 function is also important to mediate tumor suppression of MTC cells.
Figure 1.
LIF/JAK/STAT3-mediated growth inhibitory signaling in medullary thyroid cancer. Activation of the Ras/Raf/MEK/ERK pathway induces RET downregulation, growth inhibition, and differentiation in MTC cells by inducing expression and secretion of LIF, which activates STAT3 in an autocrine/paracrine mode.
Figure 1.
LIF/JAK/STAT3-mediated growth inhibitory signaling in medullary thyroid cancer. Activation of the Ras/Raf/MEK/ERK pathway induces RET downregulation, growth inhibition, and differentiation in MTC cells by inducing expression and secretion of LIF, which activates STAT3 in an autocrine/paracrine mode.

It is an intriguing question why the activity of Ras/Raf induces the IL-6 family cytokine-activated gp130/JAK/STAT3 pathway. Noteworthy is that an emerging issue in the study of the Ras/Raf-induced growth inhibitory signaling is the involvement of diverse extracellular soluble factors. It has been reported that cells undergoing Ras/Raf-induced senescence secrete soluble factors that can help reinforce senescence-like growth arrest responses [113,114]. Interestingly, these factors (which include insulin-like growth factor binding protein-7, plasminogen-activator inhibitor-1, and CXCR2-binding chemokines such as interleukin-8 or growth-regulated oncogene-α) were identified in a cell-specific manner. Perhaps, certain thyroid cell types may retain similar mechanisms which may be activated in response to aberrant Ras/Raf activation. Nevertheless, noteworthy is that STAT3 activity is upregulated in PTC, suggesting as-yet-unidentified benefit of STAT3 upregulation for PTC development.
Acknowledgments
This work was supported by American Cancer Society (RSGM-10-189-01-TBE) and the National Cancer Institute (R01CA138441) to Jong-In Park. The authors wish to apolo-gize to those whose work is not cited owing to space limitations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Sprague, B.L.; Warren Andersen, S.; Trentham-Dietz, A. Thyroid cancer incidence and socioeconomic indicators of health care access. Cancer Causes Control 2008, 19, 585–593. [Google Scholar] [CrossRef]
- Cohen, Y.; Xing, M.; Mambo, E.; Guo, Z.; Wu, G.; Trink, B.; Beller, U.; Westra, W.H.; Ladenson, P.W.; Sidransky, D. Braf mutation in papillary thyroid carcinoma. J. Natl. Cancer Inst. 2003, 95, 625–627. [Google Scholar] [CrossRef]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of braf mutations in thyroid cancer: Genetic evidence for constitutive activation of the ret/ptc-ras-braf signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar]
- Hou, P.; Liu, D.; Shan, Y.; Hu, S.; Studeman, K.; Condouris, S.; Wang, Y.; Trink, A.; El-Naggar, A.K.; Tallini, G.; et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/akt pathway in thyroid cancer. Clin. Cancer Res. 2007, 13, 1161–1170. [Google Scholar] [CrossRef]
- Liu, D.; Yang, C.; Bojdani, E.; Murugan, A.K.; Xing, M. Identification of rasal1 as a major tumor suppressor gene in thyroid cancer. J. Natl. Cancer Inst. 2013, 105, 1617–1627. [Google Scholar] [CrossRef]
- Kouvaraki, M.A.; Shapiro, S.E.; Perrier, N.D.; Cote, G.J.; Gagel, R.F.; Hoff, A.O.; Sherman, S.I.; Lee, J.E.; Evans, D.B. Ret proto-oncogene: A review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid 2005, 15, 531–544. [Google Scholar] [CrossRef]
- Ichihara, M.; Murakumo, Y.; Takahashi, M. Ret and neuroendocrine tumors. Cancer Lett. 2004, 204, 197–211. [Google Scholar] [CrossRef]
- Nikiforov, Y.E. Thyroid carcinoma: Molecular pathways and therapeutic targets. Mod. Pathol. 2008, 21, S37–S43. [Google Scholar] [CrossRef]
- Giunti, S.; Antonelli, A.; Amorosi, A.; Santarpia, L. Cellular signaling pathway alterations and potential targeted therapies for medullary thyroid carcinoma. Int. J. Endocrinol. 2013. [Google Scholar] [CrossRef]
- Pinchot, S.N.; Kunnimalaiyaan, M.; Sippel, R.S.; Chen, H. Medullary thyroid carcinoma: Targeted therapies and future directions. J. Oncol. 2009. [Google Scholar] [CrossRef]
- Agrawal, N.; Jiao, Y.; Sausen, M.; Leary, R.; Bettegowda, C.; Roberts, N.J.; Bhan, S.; Ho, A.S.; Khan, Z.; Bishop, J.; et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in ret and ras. J. Clin. Endocrinol. Metab. 2012, 98, E364–E369. [Google Scholar]
- Ciampi, R.; Mian, C.; Fugazzola, L.; Cosci, B.; Romei, C.; Barollo, S.; Cirello, V.; Bottici, V.; Marconcini, G.; Rosa, P.M.; et al. Evidence of a low prevalence of ras mutations in a large medullary thyroid cancer series. Thyroid 2013, 23, 50–57. [Google Scholar] [CrossRef]
- Liu, Z.; Hou, P.; Ji, M.; Guan, H.; Studeman, K.; Jensen, K.; Vasko, V.; El-Naggar, A.K.; Xing, M. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J. Clin. Endocrinol. Metab. 2008, 93, 3106–3116. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Wald, A.I.; Roy, S.; Durso, M.B.; Nikiforov, Y.E. Targeted next-generation sequencing panel (thyroseq) for detection of mutations in thyroid cancer. J. Clin. Endocrinol. Metab. 2013, 98, E1852–E1860. [Google Scholar] [CrossRef]
- Boichard, A.; Croux, L.; Al Ghuzlan, A.; Broutin, S.; Dupuy, C.; Leboulleux, S.; Schlumberger, M.; Bidart, J.M.; Lacroix, L. Somatic ras mutations occur in a large proportion of sporadic ret-negative medullary thyroid carcinomas and extend to a previously unidentified exon. J. Clin. Endocrinol. Metab. 2012, 97, E2031–E2035. [Google Scholar] [CrossRef]
- Mohr, A.; Chatain, N.; Domoszlai, T.; Rinis, N.; Sommerauer, M.; Vogt, M.; Muller-Newen, G. Dynamics and non-canonical aspects of jak/stat signalling. Eur. J. Cell Biol. 2012, 91, 524–532. [Google Scholar] [CrossRef]
- Gamero, A.M.; Young, H.A.; Wiltrout, R.H. Inactivation of stat3 in tumor cells: Releasing a brake on immune responses against cancer? Cancer Cell 2004, 5, 111–112. [Google Scholar] [CrossRef]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the jak/stat pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar]
- Pellegrini, S.; Dusanter-Fourt, I. The structure, regulation and function of the janus kinases (jaks) and the signal transducers and activators of transcription (stats). Eur. J. Biochem. 1997, 248, 615–633. [Google Scholar]
- Levy, D.E.; Kessler, D.S.; Pine, R.; Darnell, J.E., Jr. Cytoplasmic activation of isgf3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro. Genes Dev. 1989, 3, 1362–1371. [Google Scholar] [CrossRef]
- Levy, D.E.; Kessler, D.S.; Pine, R.; Reich, N.; Darnell, J.E., Jr. Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control. Genes Dev. 1988, 2, 383–393. [Google Scholar] [CrossRef]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of aprf, a novel ifn-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A stat family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar]
- Stark, G.R.; Darnell, J.E., Jr. The jak-stat pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef]
- Chen, C.L.; Hsieh, F.C.; Lin, J. Systemic evaluation of total stat3 and stat3 tyrosine phosphorylation in normal human tissues. Exp. Mol. Pathol. 2006, 80, 295–305. [Google Scholar] [CrossRef]
- Raz, R.; Lee, C.K.; Cannizzaro, L.A.; d’Eustachio, P.; Levy, D.E. Essential role of stat3 for embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 1999, 96, 2846–2851. [Google Scholar] [CrossRef]
- Takeda, T.; Kurachi, H.; Yamamoto, T.; Homma, H.; Morishige, K.; Miyake, A.; Murata, Y. Participation of jak, stat and unknown proteins in human placental lactogen-induced signaling: A unique signaling pathway different from prolactin and growth hormone. J. Endocrinol. 1997, 153, R1–R3. [Google Scholar] [CrossRef]
- Akira, S. Functional roles of stat family proteins: Lessons from knockout mice. Stem Cells 1999, 17, 138–146. [Google Scholar] [CrossRef]
- Resemann, H.K.; Watson, C.J.; Lloyd-Lewis, B. The stat3 paradox: A killer and an oncogene. Mol. Cell. Endocrinol. 2014, 382, 603–611. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. Jak-stat signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: How intimate is the relationship? An. N. Y. Acad. Sci. 2009, 1171, 59–76. [Google Scholar]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase m2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef]
- Chung, J.; Uchida, E.; Grammer, T.C.; Blenis, J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell. Biol. 1997, 17, 6508–6516. [Google Scholar]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by stat1 and stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Zabolotny, J.M.; Bence-Hanulec, K.K.; Stricker-Krongrad, A.; Haj, F.; Wang, Y.; Minokoshi, Y.; Kim, Y.B.; Elmquist, J.K.; Tartaglia, L.A.; Kahn, B.B.; et al. Ptp1b regulates leptin signal transduction in vivo. Dev. Cell 2002, 2, 489–495. [Google Scholar] [CrossRef]
- Krebs, D.L.; Hilton, D.J. Socs proteins: Negative regulators of cytokine signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of stat3 signal transduction by pias3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Huso, D.L.; Nathans, D.; Desiderio, S. Specific ablation of stat3beta distorts the pattern of stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell 2002, 108, 331–344. [Google Scholar] [CrossRef]
- Groner, B.; Lucks, P.; Borghouts, C. The function of stat3 in tumor cells and their microenvironment. Semin. Cell Dev. Biol. 2008, 19, 341–350. [Google Scholar] [CrossRef]
- He, B.; You, L.; Uematsu, K.; Zang, K.; Xu, Z.; Lee, A.Y.; Costello, J.F.; McCormick, F.; Jablons, D.M. Socs-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 14133–14138. [Google Scholar] [CrossRef]
- Chatterjee-Kishore, M.; Wright, K.L.; Ting, J.P.; Stark, G.R. How stat1 mediates constitutive gene expression: A complex of unphosphorylated stat1 and irf1 supports transcription of the lmp2 gene. EMBO J. 2000, 19, 4111–4122. [Google Scholar] [CrossRef]
- Liu, Z.; Hazan-Halevy, I.; Harris, D.M.; Li, P.; Ferrajoli, A.; Faderl, S.; Keating, M.J.; Estrov, Z. Stat-3 activates nf-kappab in chronic lymphocytic leukemia cells. Mol. Cancer Res. 2011, 9, 507–515. [Google Scholar] [CrossRef]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated stat3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated stat3 accumulates in response to il-6 and activates transcription by binding to nfkappab. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef]
- Yang, J.; Stark, G.R. Roles of unphosphorylated stats in signaling. Cell Res. 2008, 18, 443–451. [Google Scholar] [CrossRef]
- Gao, S.P.; Bromberg, J.F. Touched and moved by stat3. Sci. STKE 2006, 2006, pe30. [Google Scholar]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial stat3 supports ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. Stats in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Ai, T.; Wang, Z.; Zhang, M.; Zhang, L.; Wang, N.; Li, W.; Song, L. Expression and prognostic relevance of stat3 and cyclin d1 in non-small cell lung cancer. Int. J. Biol. Markers 2012, 27, e132–e138. [Google Scholar] [CrossRef]
- Birner, P.; Toumangelova-Uzeir, K.; Natchev, S.; Guentchev, M. Stat3 tyrosine phosphorylation influences survival in glioblastoma. J. Neurooncol. 2010, 100, 339–343. [Google Scholar]
- Liu, X.; He, Z.; Li, C.H.; Huang, G.; Ding, C.; Liu, H. Correlation analysis of jak-stat pathway components on prognosis of patients with prostate cancer. Pathol. Oncol. Res. 2012, 18, 17–23. [Google Scholar] [CrossRef]
- Masuda, M.; Ruan, H.Y.; Ito, A.; Nakashima, T.; Toh, S.; Wakasaki, T.; Yasumatsu, R.; Kutratomi, Y.; Komune, S.; Weinstein, I.B. Signal transducers and activators of transcription 3 up-regulates vascular endothelial growth factor production and tumor angiogenesis in head and neck squamous cell carcinoma. Oral Oncol. 2007, 43, 785–790. [Google Scholar] [CrossRef]
- Min, H.; Wei-hong, Z. Constitutive activation of signal transducer and activator of transcription 3 in epithelial ovarian carcinoma. J. Obstet. Gynaecol. Res. 2009, 35, 918–925. [Google Scholar] [CrossRef]
- Schoppmann, S.F.; Jesch, B.; Friedrich, J.; Jomrich, G.; Maroske, F.; Birner, P. Phosphorylation of signal transducer and activator of transcription 3 (stat3) correlates with her-2 status, carbonic anhydrase 9 expression and prognosis in esophageal cancer. Clin. Exp. Metastasis 2012, 29, 615–624. [Google Scholar] [CrossRef]
- Sheen-Chen, S.M.; Huang, C.C.; Tang, R.P.; Chou, F.F.; Eng, H.L. Prognostic value of signal transducers and activators of transcription 3 in breast cancer. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2286–2290. [Google Scholar] [CrossRef]
- Yakata, Y.; Nakayama, T.; Yoshizaki, A.; Kusaba, T.; Inoue, K.; Sekine, I. Expression of p-stat3 in human gastric carcinoma: Significant correlation in tumour invasion and prognosis. Int. J. Oncol. 2007, 30, 437–442. [Google Scholar]
- Chen, Y.; Wang, J.; Wang, X.; Liu, X.; Li, H.; Lv, Q.; Zhu, J.; Wei, B.; Tang, Y. Stat3, a poor survival predicator, is associated with lymph node metastasis from breast cancer. J. Breast Cancer 2013, 16, 40–49. [Google Scholar] [CrossRef]
- Wei, D.; Le, X.; Zheng, L.; Wang, L.; Frey, J.A.; Gao, A.C.; Peng, Z.; Huang, S.; Xiong, H.Q.; Abbruzzese, J.L.; et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 2003, 22, 319–329. [Google Scholar] [CrossRef]
- Scholz, A.; Heinze, S.; Detjen, K.M.; Peters, M.; Welzel, M.; Hauff, P.; Schirner, M.; Wiedenmann, B.; Rosewicz, S. Activated signal transducer and activator of transcription 3 (stat3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology 2003, 125, 891–905. [Google Scholar] [CrossRef]
- Deng, J.; Liang, H.; Zhang, R.; Sun, D.; Pan, Y.; Liu, Y.; Zhang, L.; Hao, X. Stat3 is associated with lymph node metastasis in gastric cancer. Tumour Biol. 2013, 34, 2791–2800. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal jak2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Nhieu, J.T.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating stat3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Rebouissou, S.; Amessou, M.; Couchy, G.; Poussin, K.; Imbeaud, S.; Pilati, C.; Izard, T.; Balabaud, C.; Bioulac-Sage, P.; Zucman-Rossi, J. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 2009, 457, 200–204. [Google Scholar] [CrossRef] [Green Version]
- Bournazou, E.; Bromberg, J. Targeting the tumor microenvironment: Jak-stat3 signaling. JAKSTAT 2013. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. Stats in cancer inflammation and immunity: A leading role for stat3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The jak2 inhibitor azd1480 potently blocks stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar] [CrossRef]
- Xin, H.; Herrmann, A.; Reckamp, K.; Zhang, W.; Pal, S.; Hedvat, M.; Zhang, C.; Liang, W.; Scuto, A.; Weng, S.; et al. Antiangiogenic and antimetastatic activity of jak inhibitor azd1480. Cancer Res. 2011, 71, 6601–6610. [Google Scholar] [CrossRef]
- Sonnenblick, A.; Uziely, B.; Nechushtan, H.; Kadouri, L.; Galun, E.; Axelrod, J.H.; Katz, D.; Daum, H.; Hamburger, T.; Maly, B.; et al. Tumor stat3 tyrosine phosphorylation status, as a predictor of benefit from adjuvant chemotherapy for breast cancer. Breast Cancer Res. Treat. 2013, 138, 407–413. [Google Scholar] [CrossRef]
- Dolled-Filhart, M.; Camp, R.L.; Kowalski, D.P.; Smith, B.L.; Rimm, D.L. Tissue microarray analysis of signal transducers and activators of transcription 3 (stat3) and phospho-stat3 (tyr705) in node-negative breast cancer shows nuclear localization is associated with a better prognosis. Clin. Cancer Res. 2003, 9, 594–600. [Google Scholar]
- Pectasides, E.; Egloff, A.M.; Sasaki, C.; Kountourakis, P.; Burtness, B.; Fountzilas, G.; Dafni, U.; Zaramboukas, T.; Rampias, T.; Rimm, D.; et al. Nuclear localization of signal transducer and activator of transcription 3 in head and neck squamous cell carcinoma is associated with a better prognosis. Clin. Cancer Res. 2010, 16, 2427–2434. [Google Scholar] [CrossRef]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the egfr kinase domain mediate stat3 activation via il-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef]
- Woo, S.; Lee, B.L.; Yoon, J.; Cho, S.J.; Baik, T.K.; Chang, M.S.; Lee, H.E.; Park, J.W.; Kim, Y.H.; Kim, W.H. Constitutive activation of signal transducers and activators of transcription 3 correlates with better prognosis, cell proliferation and hypoxia-inducible factor-1alpha in human gastric cancer. Pathobiology 2011, 78, 295–301. [Google Scholar] [CrossRef]
- Setsu, N.; Kohashi, K.; Endo, M.; Yamamoto, H.; Tamiya, S.; Takahashi, Y.; Yamada, Y.; Ishii, T.; Matsuda, S.; Yokoyama, R.; et al. Phosphorylation of signal transducer and activator of transcription 3 in soft tissue leiomyosarcoma is associated with a better prognosis. Int. J. Cancer 2013, 132, 109–115. [Google Scholar] [CrossRef]
- Monnien, F.; Zaki, H.; Borg, C.; Mougin, C.; Bosset, J.F.; Mercier, M.; Arbez-Gindre, F.; Kantelip, B. Prognostic value of phosphorylated stat3 in advanced rectal cancer: A study from 104 french patients included in the eortc 22921 trial. J. Clin. Pathol. 2010, 63, 873–878. [Google Scholar] [CrossRef]
- Koperek, O.; Aumayr, K.; Schindl, M.; Werba, G.; Soleiman, A.; Schoppmann, S.; Sahora, K.; Birner, P. Phosphorylation of stat3 correlates with her2 status, but not with survival in pancreatic ductal adenocarcinoma. APMIS 2013. [Google Scholar] [CrossRef]
- De la Iglesia, N.; Puram, S.V.; Bonni, A. Stat3 regulation of glioblastoma pathogenesis. Curr. Mol. Med. 2009, 9, 580–590. [Google Scholar] [CrossRef]
- Albi, E.; Curcio, F.; Spelat, R.; Lazzarini, R.; Loreti, E.; Ferri, I.; Ambesi-Impiombato, F.S. The thyroid lobes: The different twins. Arch. Biochem. Biophys. 2012, 518, 16–22. [Google Scholar] [CrossRef]
- Bates, S.H.; Myers, M.G. The role of leptin→stat3 signaling in neuroendocrine function: An integrative perspective. J. Mol. Med. 2004, 82, 12–20. [Google Scholar] [CrossRef]
- Chung, J.; Park, E.S.; Kim, D.; Suh, J.M.; Chung, H.K.; Kim, J.; Kim, H.; Park, S.J.; Kwon, O.Y.; Ro, H.K.; et al. Thyrotropin modulates interferon-gamma-mediated intercellular adhesion molecule-1 gene expression by inhibiting janus kinase-1 and signal transducer and activator of transcription-1 activation in thyroid cells. Endocrinology 2000, 141, 2090–2097. [Google Scholar]
- Park, E.S.; Kim, H.; Suh, J.M.; Park, S.J.; Kwon, O.Y.; Kim, Y.K.; Ro, H.K.; Cho, B.Y.; Chung, J.; Shong, M. Thyrotropin induces socs-1 (suppressor of cytokine signaling-1) and socs-3 in frtl-5 thyroid cells. Mol. Endocrinol. 2000, 14, 440–448. [Google Scholar] [CrossRef]
- Park, E.S.; Kim, H.; Suh, J.M.; Park, S.J.; You, S.H.; Chung, H.K.; Lee, K.W.; Kwon, O.Y.; Cho, B.Y.; Kim, Y.K.; et al. Involvement of jak/stat (janus kinase/signal transducer and activator of transcription) in the thyrotropin signaling pathway. Mol. Endocrinol. 2000, 14, 662–670. [Google Scholar] [CrossRef]
- Kim, H.; Suh, J.M.; Hwang, E.S.; Kim, D.W.; Chung, H.K.; Song, J.H.; Hwang, J.H.; Park, K.C.; Ro, H.K.; Jo, E.K.; et al. Thyrotropin-mediated repression of class ii trans-activator expression in thyroid cells: Involvement of stat3 and suppressor of cytokine signaling. J. Immunol. 2003, 171, 616–627. [Google Scholar]
- Staab, J.; Barth, P.J.; Meyer, T. Cell-type-specific expression of stat transcription factors in tissue samples from patients with lymphocytic thyroiditis. Endocr. Pathol. 2012, 23, 141–150. [Google Scholar] [CrossRef]
- Ardito, G.; Revelli, L.; Boninsegna, A.; Sgambato, A.; Moschella, F.; Marzola, M.C.; Giustozzi, E.; Avenia, N.; Castelli, M.; Rubello, D. Immunohistochemical evaluation of inflammatory and proliferative markers in adjacent normal thyroid tissue in patients undergoing total thyroidectomy: Results of a preliminary study. J. Exp. Clin. Cancer Res. 2010, 29, 77. [Google Scholar] [CrossRef]
- Couto, J.P.; Daly, L.; Almeida, A.; Knauf, J.A.; Fagin, J.A.; Sobrinho-Simoes, M.; Lima, J.; Maximo, V.; Soares, P.; Lyden, D.; et al. Stat3 negatively regulates thyroid tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E2361–E2370. [Google Scholar] [CrossRef]
- Park, J.I.; Strock, C.J.; Ball, D.W.; Nelkin, B.D. The ras/raf/mek/extracellular signal-regulated kinase pathway induces autocrine-paracrine growth inhibition via the leukemia inhibitory factor/jak/stat pathway. Mol. Cell. Biol. 2003, 23, 543–554. [Google Scholar] [CrossRef]
- Park, J.I.; Strock, C.J.; Ball, D.W.; Nelkin, B.D. Interleukin-1beta can mediate growth arrest and differentiation via the leukemia inhibitory factor/jak/stat pathway in medullary thyroid carcinoma cells. Cytokine 2005, 29, 125–134. [Google Scholar] [CrossRef]
- Vindrieux, D.; Augert, A.; Girard, C.A.; Gitenay, D.; Lallet-Daher, H.; Wiel, C.; Le Calve, B.; Gras, B.; Ferrand, M.; Verbeke, S.; et al. Pla2r1 mediates tumor suppression by activating jak2. Cancer Res. 2013, 73, 6334–6345. [Google Scholar] [CrossRef]
- Lu, C.; Kerbel, R.S. Interleukin-6 undergoes transition from paracrine growth inhibitor to autocrine stimulator during human melanoma progression. J. Cell Biol. 1993, 120, 1281–1288. [Google Scholar] [CrossRef]
- Zhang, J.; Gill, A.; Atmore, B.; Johns, A.; Delbridge, L.; Lai, R.; McMullen, T. Upregulation of the signal transducers and activators of transcription 3 (stat3) pathway in lymphatic metastases of papillary thyroid cancer. Int. J. Clin. Exp. Pathol. 2011, 4, 356–362. [Google Scholar]
- Trovato, M.; Grosso, M.; Vitarelli, E.; Ruggeri, R.M.; Alesci, S.; Trimarchi, F.; Barresi, G.; Benvenga, S. Distinctive expression of stat3 in papillary thyroid carcinomas and a subset of follicular adenomas. Histol. Histopathol. 2003, 18, 393–399. [Google Scholar]
- Kim, W.G.; Choi, H.J.; Kim, W.B.; Kim, E.Y.; Yim, J.H.; Kim, T.Y.; Gong, G.; Kim, S.Y.; Chung, N.; Shong, Y.K. Basal stat3 activities are negatively correlated with tumor size in papillary thyroid carcinomas. J. Endocrinol. Investig. 2012, 35, 413–418. [Google Scholar]
- Hwang, J.H.; Kim, D.W.; Suh, J.M.; Kim, H.; Song, J.H.; Hwang, E.S.; Park, K.C.; Chung, H.K.; Kim, J.M.; Lee, T.H.; et al. Activation of signal transducer and activator of transcription 3 by oncogenic ret/ptc (rearranged in transformation/papillary thyroid carcinoma) tyrosine kinase: Roles in specific gene regulation and cellular transformation. Mol. Endocrinol. 2003, 17, 1155–1166. [Google Scholar] [CrossRef]
- Lin, H.; Chen, M.C.; Chiu, C.Y.; Song, Y.M.; Lin, S.Y. Cdk5 regulates stat3 activation and cell proliferation in medullary thyroid carcinoma cells. J. Biol. Chem. 2007, 282, 2776–2784. [Google Scholar] [CrossRef]
- Kim, T.H.; Lee, S.Y.; Rho, J.H.; Jeong, N.Y.; Soung, Y.H.; Jo, W.S.; Kang, D.Y.; Kim, S.H.; Yoo, Y.H. Mutant p53 (g199v) gains antiapoptotic function through signal transducer and activator of transcription 3 in anaplastic thyroid cancer cells. Mol. Cancer Res. 2009, 7, 1645–1654. [Google Scholar] [CrossRef]
- Masago, K.; Asato, R.; Fujita, S.; Hirano, S.; Tamura, Y.; Kanda, T.; Mio, T.; Katakami, N.; Mishima, M.; Ito, J. Epidermal growth factor receptor gene mutations in papillary thyroid carcinoma. Int. J. Cancer 2009, 124, 2744–2749. [Google Scholar] [CrossRef]
- Mooi, W.J.; Peeper, D.S. Oncogene-induced cell senescence—Halting on the road to cancer. N. Engl. J. Med. 2006, 355, 1037–1046. [Google Scholar] [CrossRef]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef]
- Nakagawa, T.; Mabry, M.; de Bustros, A.; Ihle, J.N.; Nelkin, B.D.; Baylin, S.B. Introduction of v-ha-ras oncogene induces differentiation of cultured human medullary thyroid carcinoma cells. Proc. Natl. Acad. Sci. USA 1987, 84, 5923–5927. [Google Scholar] [CrossRef]
- Wood, K.W.; Qi, H.; D’Arcangelo, G.; Armstrong, R.C.; Roberts, T.M.; Halegoua, S. The cytoplasmic raf oncogene induces a neuronal phenotype in pc12 cells: A potential role for cellular raf kinases in neuronal growth factor signal transduction. Proc. Natl. Acad. Sci. USA 1993, 90, 5016–5020. [Google Scholar] [CrossRef]
- Carson, E.B.; McMahon, M.; Baylin, S.B.; Nelkin, B.D. Ret gene silencing is associated with raf-1-induced medullary thyroid carcinoma cell differentiation. Cancer Res. 1995, 55, 2048–2052. [Google Scholar]
- Park, J.I.; Powers, J.F.; Tischler, A.S.; Strock, C.J.; Ball, D.W.; Nelkin, B.D. Gdnf-induced leukemia inhibitory factor can mediate differentiation via the mek/erk pathway in pheochromocytoma cells derived from nf1-heterozygous knockout mice. Exp. Cell Res. 2005, 303, 79–88. [Google Scholar]
- Carson-Walter, E.B.; Smith, D.P.; Ponder, B.A.; Baylin, S.B.; Nelkin, B.D. Post-transcriptional silencing of ret occurs, but is not required, during raf-1 mediated differentiation of medullary thyroid carcinoma cells. Oncogene 1998, 17, 367–376. [Google Scholar]
- Arthan, D.; Hong, S.K.; Park, J.I. Leukemia inhibitory factor can mediate ras/raf/mek/erk-induced growth inhibitory signaling in medullary thyroid cancer cells. Cancer Lett. 2010, 297, 31–41. [Google Scholar] [CrossRef]
- Kim, E.J.; Park, J.I.; Nelkin, B.D. Ifi16 is an essential mediator of growth inhibition, but not differentiation, induced by the leukemia inhibitory factor/jak/stat pathway in medullary thyroid carcinoma cells. J. Biol. Chem. 2005, 280, 4913–4920. [Google Scholar]
- Starenki, D.; Singh, N.K.; Jensen, D.R.; Peterson, F.C.; Park, J.I. Recombinant leukemia inhibitory factor suppresses human medullary thyroid carcinoma cell line xenografts in mice. Cancer Lett. 2013, 339, 144–151. [Google Scholar] [CrossRef]
- Couto, J.P.; Almeida, A.; Daly, L.; Sobrinho-Simoes, M.; Bromberg, J.F.; Soares, P. Azd1480 blocks growth and tumorigenesis of ret- activated thyroid cancer cell lines. PLoS One 2012, 7, e46869. [Google Scholar]
- Cichowski, K.; Hahn, W.C. Unexpected pieces to the senescence puzzle. Cell 2008, 133, 958–961. [Google Scholar] [CrossRef]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: Sms-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Sosonkina, N.; Starenki, D.; Park, J.-I. The Role of STAT3 in Thyroid Cancer. Cancers 2014, 6, 526-544. https://doi.org/10.3390/cancers6010526
AMA Style
Sosonkina N, Starenki D, Park J-I. The Role of STAT3 in Thyroid Cancer. Cancers. 2014; 6(1):526-544. https://doi.org/10.3390/cancers6010526
Chicago/Turabian StyleSosonkina, Nadiya, Dmytro Starenki, and Jong-In Park. 2014. "The Role of STAT3 in Thyroid Cancer" Cancers 6, no. 1: 526-544. https://doi.org/10.3390/cancers6010526