1. Introduction
Bladder Cancer (BC) is the tenth most common cancer in the United States, mostly affecting people older than 55 years. Bladder cancer (BC) exhibits a gender disparity, affecting men approximately four times more frequently than women [
1]. Furthermore, BC encompasses a broad spectrum of disease behavior, ranging from a slow-growing non-muscle-invasive form (NMIBC) to a highly aggressive muscle-invasive variant (MIBC). Although most BC patients are diagnosed with NMIBC, up to 25% of BC are identified as MIBC with substantial risk for mortality [
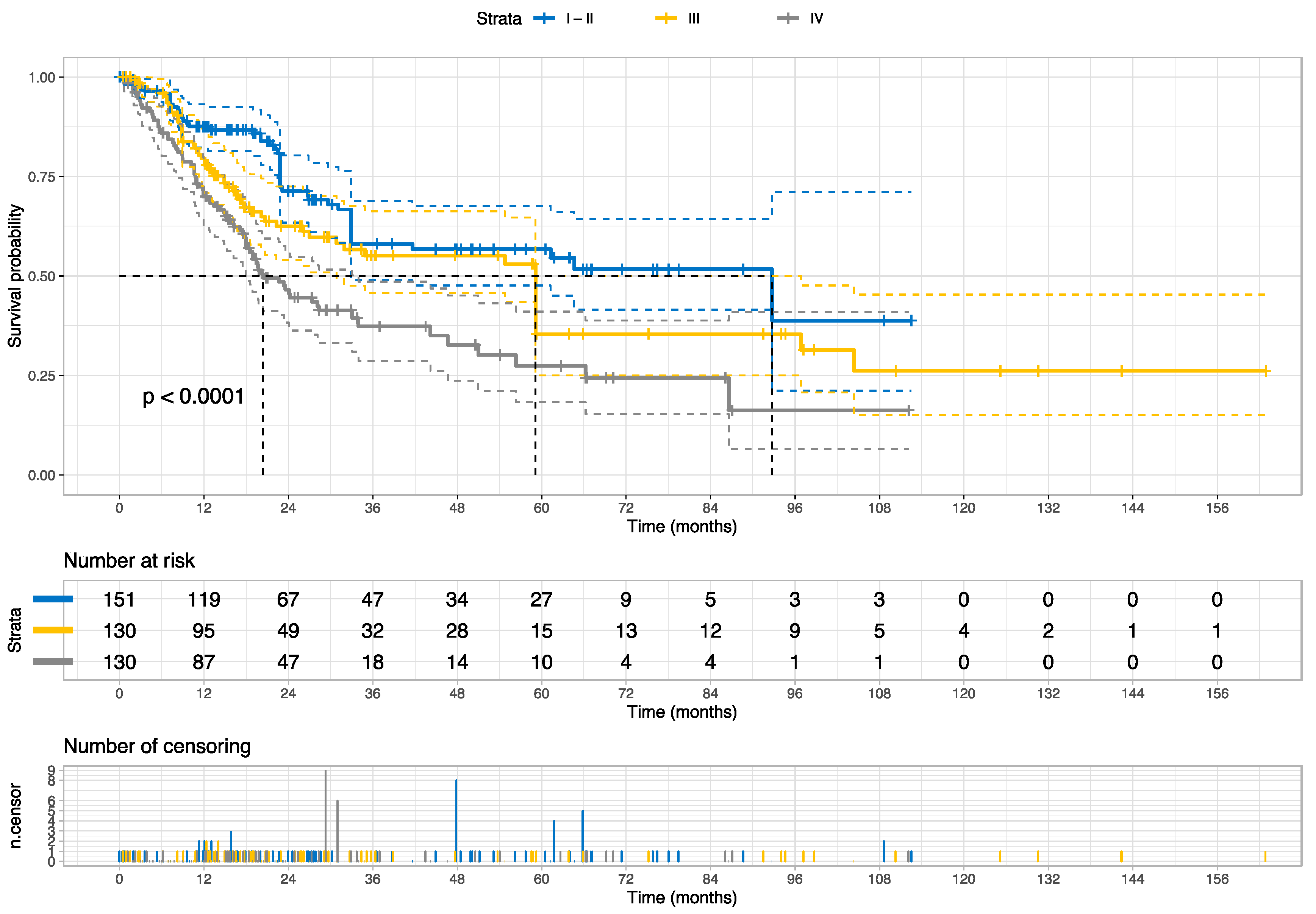
2]. BC cases with stage I or II show a 5-year relative survival rate of 96% or 70%, respectively, whereas 38 of 100 cases with stage III will survive 5 years; cases with stage IV have the poorest survival outcome with 6% of a 5-year relative survival rate. Moreover, BC reveals distinct multilevel molecular subtype profiles associated with prognosis and treatment responses [
3]. However, determining multilevel molecular subtype profiles (i.e., protein expression, gene mutation, mRNA, DNA methylation, and miRNA) requires a complex and expensive infrastructure likely unavailable in most cancer centers worldwide. Therefore, a cost-effective solution could ideally help to manage the patient selection according to their risk of having progressive cancers or to identify cases likely to benefit from certain treatment regimens.
Recent studies revealed the potential of deep learning (DL) to predict a new generation of digital biomarkers for detection, prognosis, molecular signature, and treatment response in different cancers, including bladder cancer [
4,
5,
6]. For instance, Woerl et al. reported the potential of DL to forecast the molecular subtypes of MIBC by analyzing hematoxylin and eosin (H&E) slides [
7]. As a proof-of-concept, Mundhada et al. have shown the DL capability to distinguish low-grade from high-grade histology [
8]. Zheng et al. purposed a DL framework to predict survival from histology images with BC [
9]. While deep learning holds immense potential, addressing certain tendencies that have arisen within its application is essential. Specifically, a majority of prior research treated confidence scores as equivalent to probability scores, disregarding the well-recognized problem of overconfidence in deep learning models [
10,
11]. Furthermore, these studies have not provided a feasible means of interpreting whether the feature distributions in the latent feature spaces reflect alterations in histological patterns that contribute to the prediction scores.
Given the limitations of previous studies, our hypothesis posits that morphometrical patterns observed at the histological level are indicative of prognostic confidence scores, which are then associated with omics signatures specific to advanced bladder cancers. Our primary objective is to identify prognostic subgroups that reveal associations with molecular subtypes, utilizing histology images, including bladder cancers and weakly supervised learning. The major contribution of the current work is to provide a novel strategy that facilitates the development of interpretable prognostic scores derived from a collection of mixed histology patterns associated with molecular subtypes and potential treatment options for bladder cancers.
4. Discussion
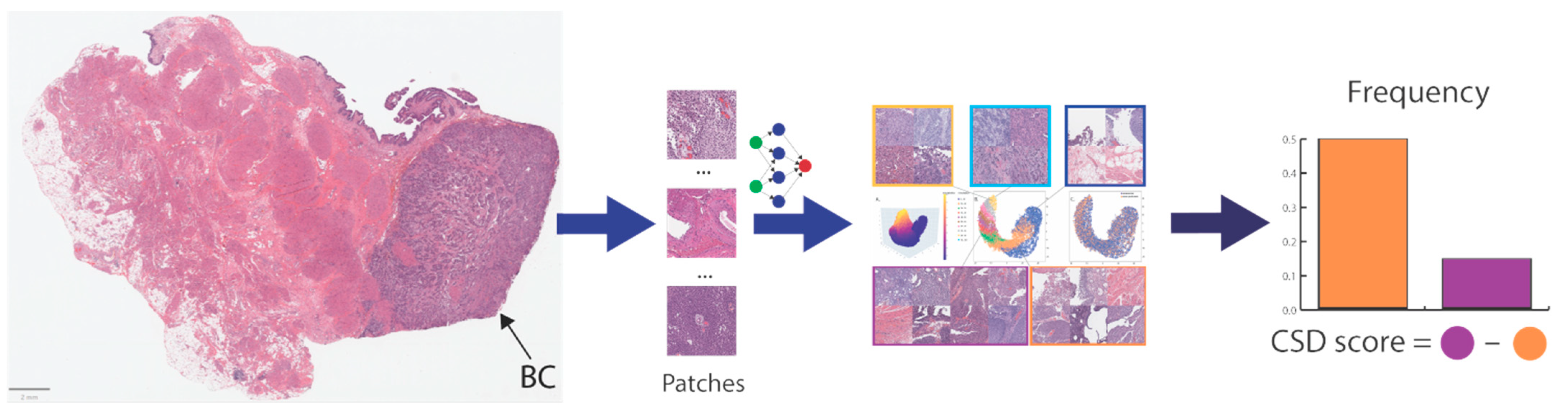
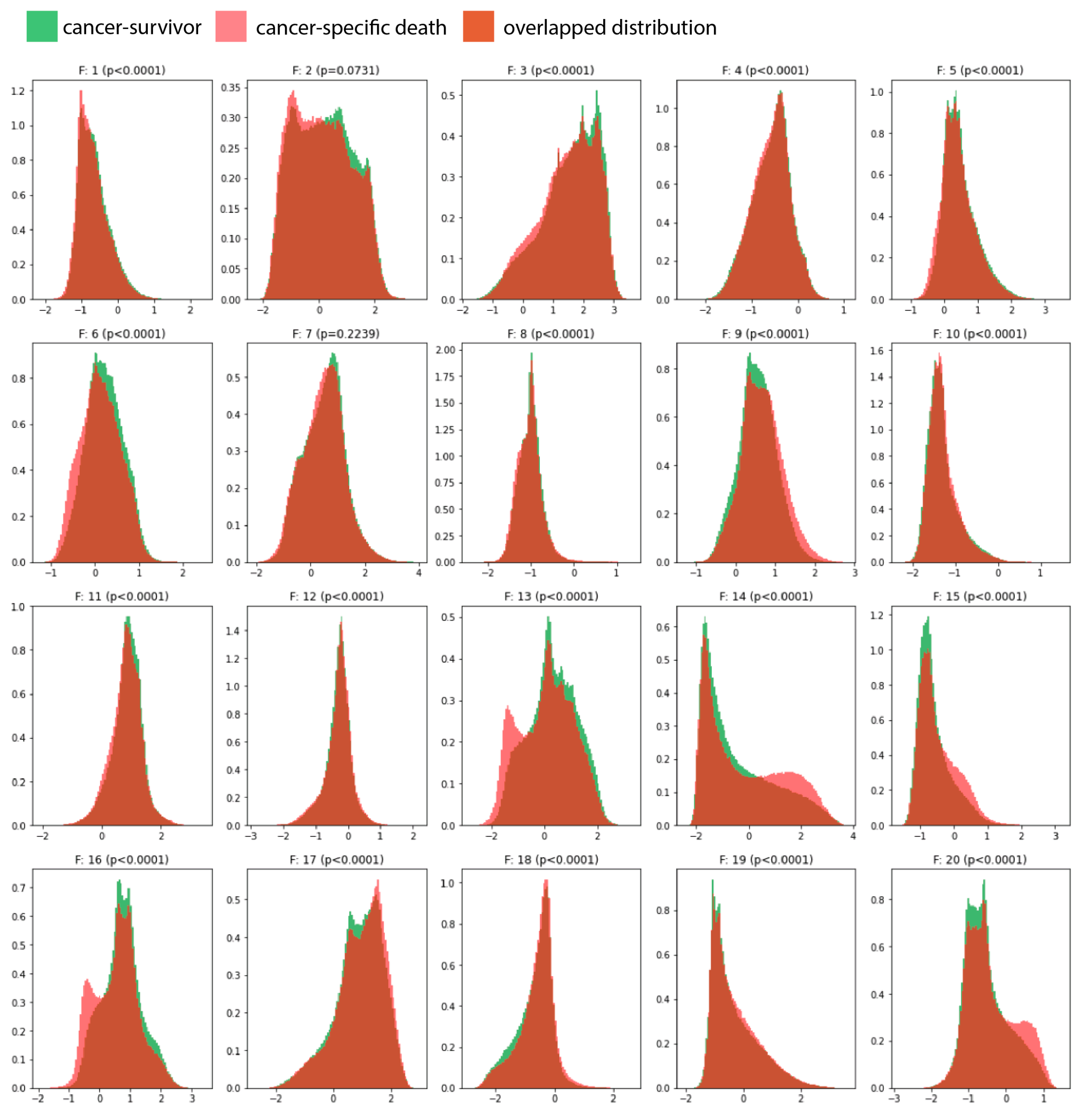
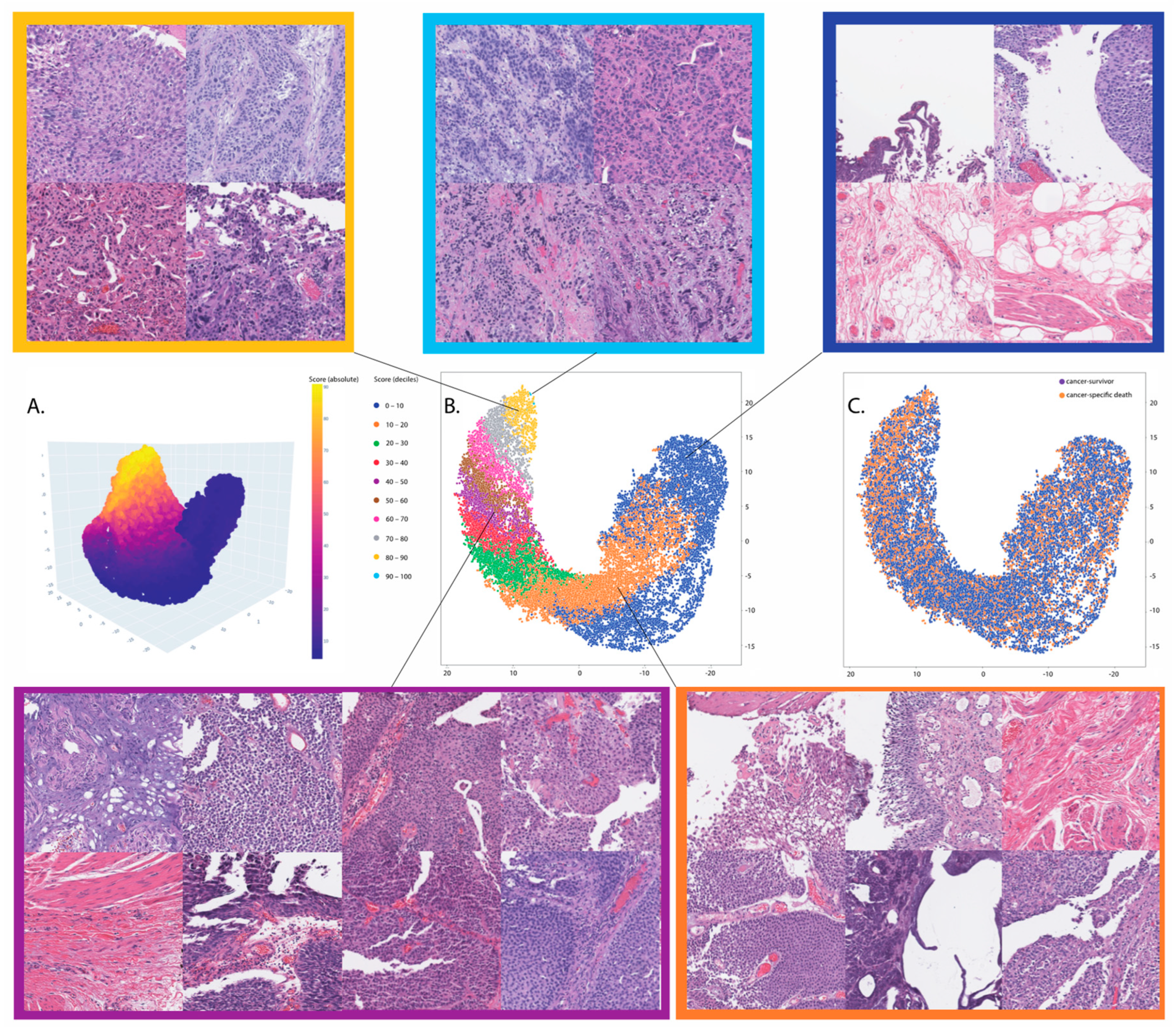
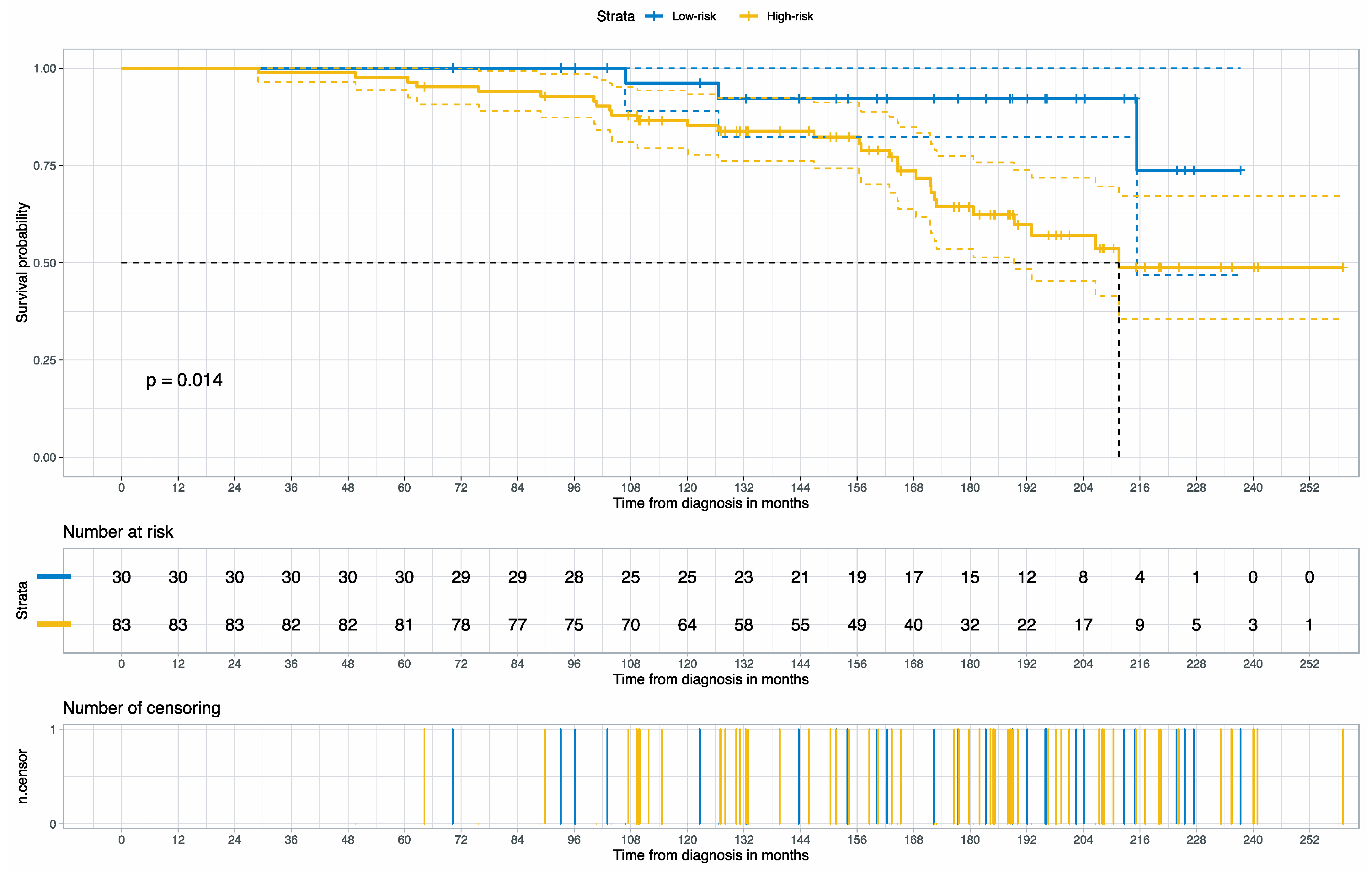
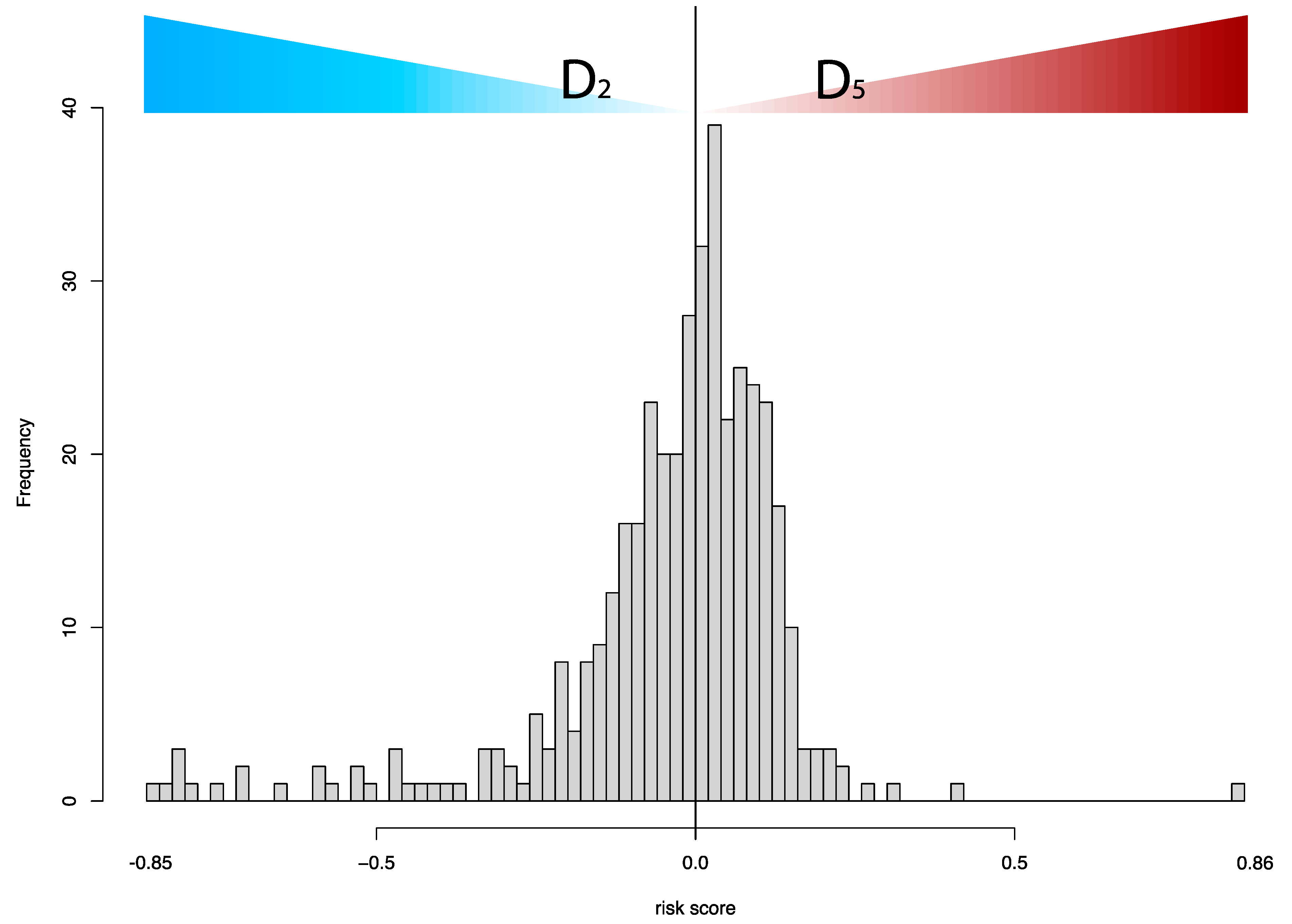
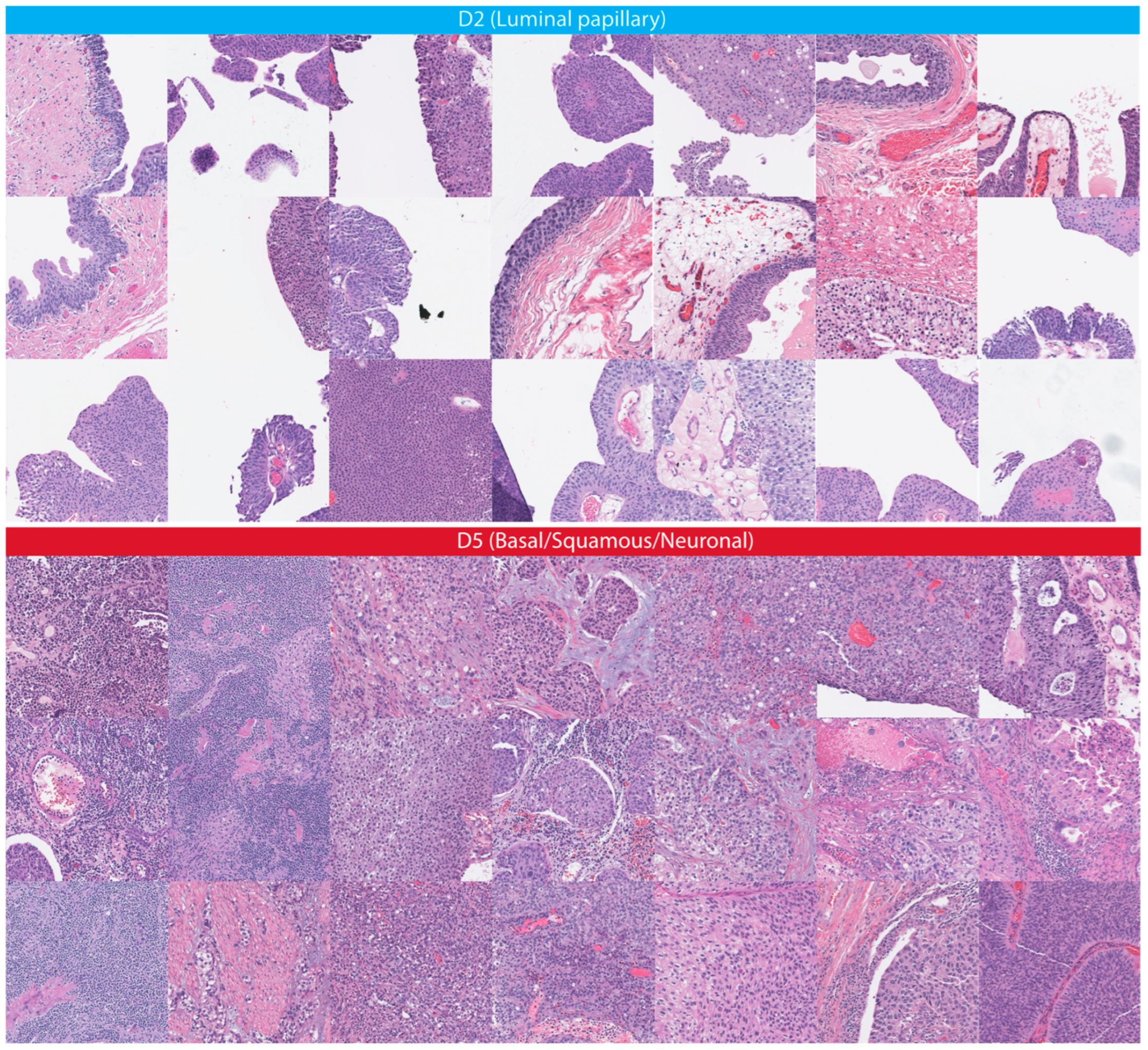
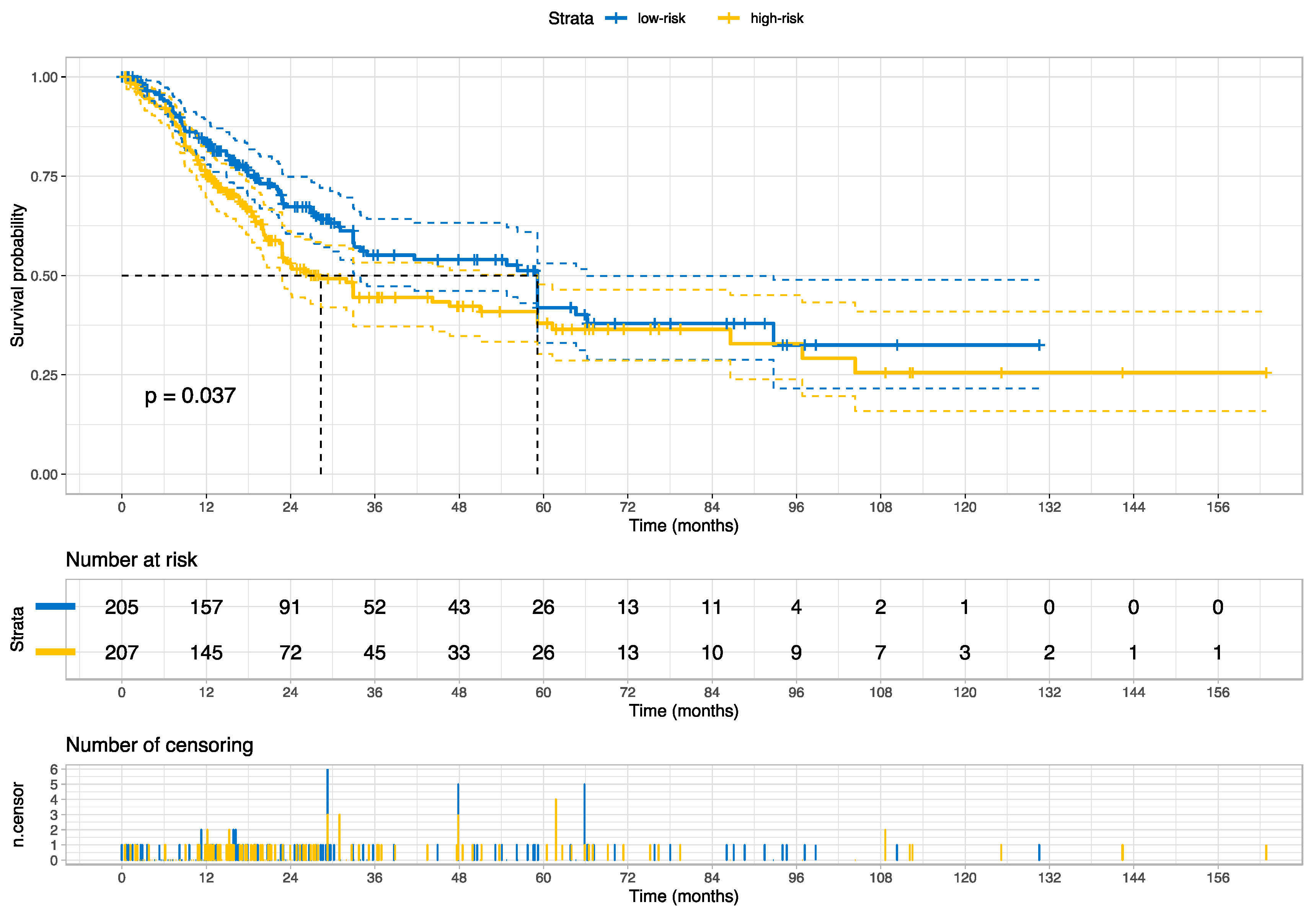
In this study, we developed and externally validated an AI-based algorithm that stratifies muscle-invasive bladder cancer by mortality risk directly from histology images. Moreover, our novel risk groups can reveal which histopathological pattern is dominant in tissues with bladder cancers. Our approach is feasible thanks to the intuitively well-sorted feature space generated by weakly supervised learning. This property has made it possible to discretize the feature space into ten small segments organized decile-wise, allowing us to evaluate the histopathological patterns for each prediction decile.
Earlier studies in bladder cancer applied deep learning to infer staging [
31], grade [
32,
33], recurrence risk [
34], FGFR3 mutation [
35], and specific molecular subtypes [
7] from histology images. Although some previous studies examined the prediction of molecular targets, the current study found that prognostic histopathological patterns for bladder cancer are rather associated with multi-omics profiles (i.e., transcriptomic, genomics, and epigenomics); these multi-omics profiles are already covering the specific molecular subtypes and the FGFR3 mutations investigated earlier, and we have shown that the accuracy of our risk groups for FGFR3 mutation is similar to the previous report, signifying the impact of multi-omics profiles as confounding factors on the results of earlier studies. In support of our findings, the BLCA-TCGA study (molecular characterization of bladder cancer) revealed that the molecular subtypes and signatures are linked with each other and distinct histopathologic patterns (e.g., papillary, basal/squamous) were connected with omics profiles that are prognostic and have different therapeutic targets [
3,
17]. A comparable study in Lung cancer reported that omics features are predictive of histology patterns as well [
36].
Although multiple studies identified the detection potential of single mutations or specific molecular subtypes from histology images [
37,
38,
39,
40,
41], the histopathological appearance is mainly driven by a collection of multifaceted molecular modulations and reflects the cancer malignancy and survival. Subsequently, establishing a direct association between a single molecular signature and histology images must be inadequate, given other confounders for bladder cancers.
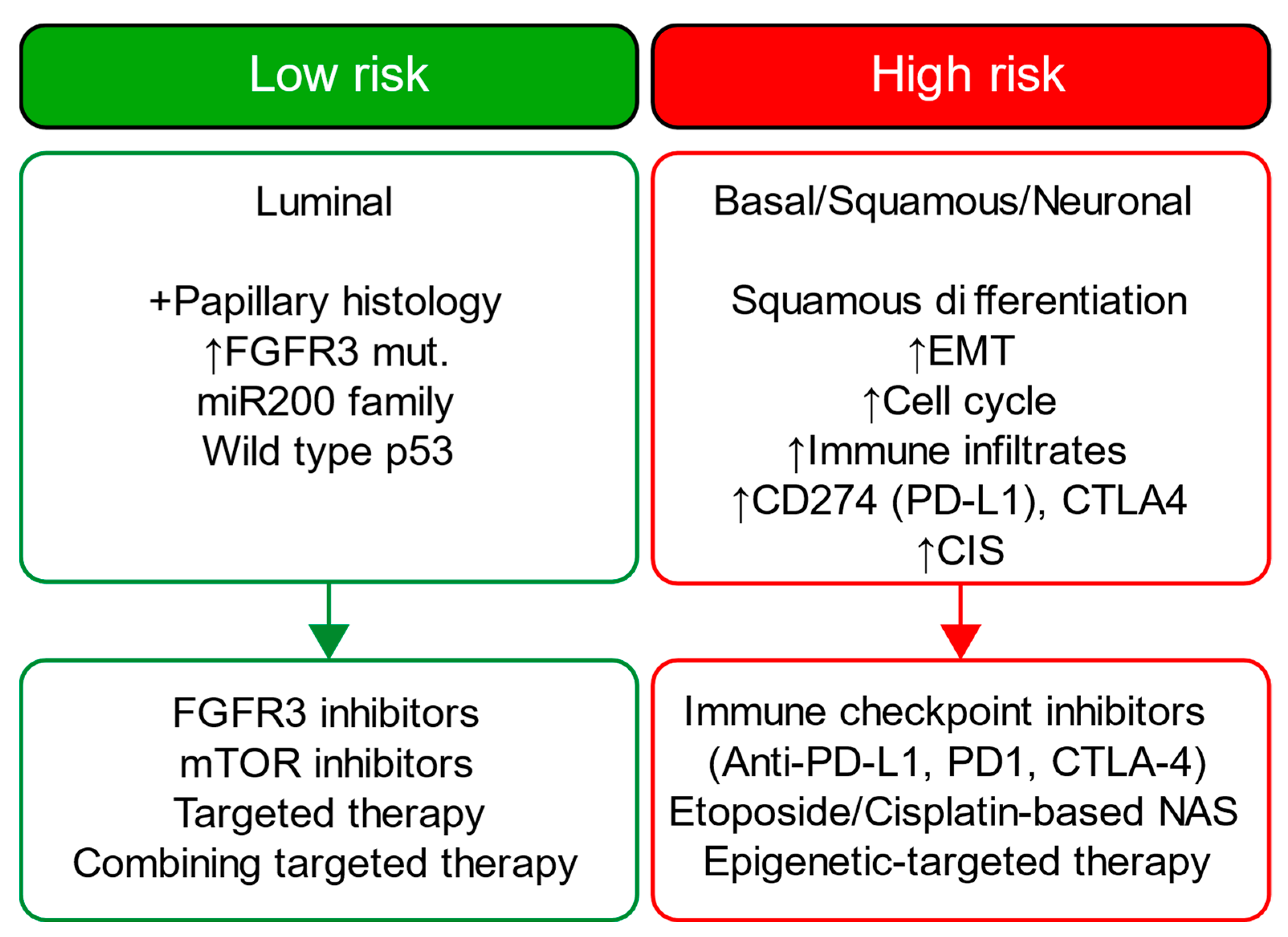
Our novel risk groups are linked with therapeutic targets like FGFR3 (erdafitinib) [
42], ERBB3 (afatinib) [
43], PI(3)K (LY294002, other mTOR inhibitors) [
44,
45], and TSC1 (nab-sirolimus, study no.: NCT05103358) [
3] as well as with female gender-biased gene mutations like KDM6A mutation (a histone lysine demethylase) [
46]. Accordingly, our novel risk group holds a potential clinical utility in pre-screening for mono- and combinational target therapies (
Figure 9). This potential will be more evident once prospective randomized studies to validate the clinical utility of our approach for patient selection in the real-world clinical setting are available.
A detailed examination of the multi-omics profiles associated with our risk groups reveals unique molecular regulatory profiles at the microRNA, lncRNA, and DNA methylation levels. We found that the low-risk group is linked with molecular subtypes with good survival for coding and non-coding RNAs or DNA methylation. These multi-omics subtypes are associated with papillary tumors, high FGFR3 mutations and miR-200 levels, and low Epithelial–Mesenchymal Transition (EMT) scores, CD274 (PD-L1) and PDCD1 (PD-1) level [
17]. In contrast, the high-risk group is linked with molecular subtypes with poor survival for coding and non-coding RNA, which are further associated with lymphocyte infiltration, the high expression of CIS (carcinoma in situ) signature genes, CD274 (PD-L1) and PDCD1 (PD-1) levels, high TP53 mutations and EMT scores [
17]. The high-risk group is additionally linked to cluster 2 for DNA hypomethylation, which has more DNA hypermethylation signals (more gene inactivation) than cluster 4, which is linked with the low-risk group [
17]. Our data further facilitates deriving a hypothesis that the low-risk group, with favorable multi-omics profiles, is likely more responsive to different targeted therapies than the high-risk group, and the high-risk group may benefit from immune checkpoint inhibitors (i.e., anti-PD-1 or PD-L1); our data also suggest that epigenetic therapy could be a potential therapeutic option for our high-risk group.
Figure 9 summarizes each risk group’s molecular characteristics and potential treatment options.
Comparable studies utilized activation maps or tiles with top scores to interpret the model inference. However, the trustworthiness of activation maps could be more questionable as deep neural network classifiers have an opportunistic nature, and the existing saliency methods inherit a high risk for misinterpretation, limited reproducibility, and sparse visualization [
47,
48]. Moreover, it should be considered that tiles with top scores ignore the variance in histology patterns between two categories after thresholding predictions, as evident by our data on the correlation between histology patterns and prediction deciles.
We applied the neural architecture search to achieve a data-driven architecture design with a better trade-off between accuracy, interpretability, and model complexity. In our study, only 20 feature representations (i.e., the 2D feature maps of the last convolutional layer) are sufficient to derive accurate predictions from histology images and correspond, for example, to 4% of feature representations of ResNet18 [
49] (i.e., 512 features), an off-the-shelf model commonly used in medical imaging research. Reducing the feature representation is associated with a better computation cost for downstream analysis and improved human interpretation of these features. Moreover, our approach helps visualize and analyze three-dimensional representative features that preserve topological information at reasonable computation costs (e.g., analysis of 8,000,000 data points required ~30 min using parallel computing). In contrast, comparable studies that utilized off-the-shelf models are limited by extremely reduced feature granularity (1D) with loss of topological information for downstream analysis, given the high computation cost to analyze a large number of 3D representative features that these models have. Accordingly, comparable studies excluded the most information from the feature representation to conduct downstream analysis. In contrast, our approach preserves the high granularity of the feature representation for downstream analysis and consequently improves the interpretability of our AI model.
Despite the strengths of our study, it is essential to acknowledge certain limitations. Firstly, using slide images introduces potential variability in image quality due to factors such as diverse scanning technologies, staining variations, and image artifacts. These variations can introduce inconsistencies that may impact the accuracy and reliability of image analysis and interpretation. Nevertheless, we took measures to mitigate this concern by using PlexusNET to address the domain shift [
16], conducting a comprehensive manual review involving domain experts and validating our findings on multicentric datasets. Additionally, we employed feature visualization techniques to identify the potential impact of artifacts and reviewed for the staining variations on the selected histological patterns. Secondly, it is crucial to recognize that TCGA slide images offer a glimpse of a specific tumor region or patient sample, which may not fully capture the complex intra- and inter-tumor heterogeneity. Tumors can exhibit spatial and molecular heterogeneity, resulting in significant variations between different regions within the same tumor or among tumors of the same type. Analyzing only a subset of slide images may provide an incomplete representation of tumor characteristics. Nonetheless, it is noteworthy that the TCGA and PLCO study followed good research practices, aiming to select the most representative samples from each patient according to the existing technical feasibility. Moreover, the quality of survival data of TCGA was validated for overall survival analyses [
18]. The good research practices and the data quality help mitigate this limitation to some extent. It is important to emphasize that TCGA slide images, obtained through the TCGA project, do not directly correspond to the specific sampling areas used for molecular examination. These images are prepared using Hematoxylin and Eosin (HE) staining, a common technique for histological analysis. In contrast, molecular examinations and profiling involve separate samples or portions of the tumor that undergo different processing steps. TCGA employs distinct protocols for various analyses, including genomic, transcriptomic, and proteomic profiling, which are not directly applied to the same tissue sections used for generating slide images. These protocols often utilize specialized techniques, such as DNA sequencing or protein expression analysis, requiring separate tissue preparation and processing. Hence, it is crucial to note that TCGA slide images, while they provide valuable histological information, do not directly correlate with the specific regions of the tumor that underwent molecular examination. Rather, they serve as representative snapshots of the tumor’s morphology and architecture, offering valuable context for researchers analyzing the genomic and molecular data obtained from the TCGA project. We preferred slide images with formalin-fixed paraffin-embedded (FFPE) tissues as this approach offers standardized staining and more reliable histology images. In contrast, the process of preparing and staining frozen tissue slides are demanding and often result in associated artifacts; freezing can cause structural changes and cellular damage, while its staining consistency can be challenging due to variations in tissue quality and protocols [
50,
51,
52]. Finally, histology images from frozen sections are also snapshots, contrary to a common misconception that assumes these images are direct complements to the entire TCGA samples.
The current study introduces a novel AI-based risk grouping system for survival derived from bladder cancer H&E slides. We show the linkage between our risk groups and multi-omics profiles for muscle-invasive bladder cancers. We highlight the concerns with predicting single molecular signatures (e.g., FGFR3) from histology images. While our approach has been rigorously tested and validated in the context of bladder cancer, its applicability extends beyond this specific disease.
Challenges and Future Directions
The present work underscores the significance of associating feature space distributions with prediction scores for the purpose of developing an interpretable scoring system for the mortality prediction. One of the prevailing challenges within the medical domain pertains to the divergence between the development dataset and unseen cohorts, which poses a persistent issue for existing algorithms. In response to this challenge, we have introduced a normalization strategy tailored for out-of-distribution cohorts, which seeks to mitigate skewness, following the principles of the central limit theorem. Our proposed normalization technique necessitates the utilization of a representative cohort to ensure the reliability of outcomes. Furthermore, we have put forth a continuous normalization approach with instantaneous threshold adjustments; this, however, requires either a latency period or initial representative data for accurate normalization. Another challenge that need to be addressed is the application boundary of our approach. The application boundary is generally determined by the image quality as well as the cohort characterization of the development set. One of the foremost challenges lies in harmonizing and integrating multi-omics data, including transcriptomics, genomics, and epigenomics. Future research should focus on developing robust methodologies and computational tools to streamline such a process, including all available data types. Integrating multi-omics analysis into the clinical workflow is a significant challenge.
Future efforts will focus on validating our approach for clinical utility to optimize the treatment management for bladder cancer. Digital biomarkers, such as histomics, have the potential to serve as companion variables for disease staging and patient selection. Future research should also explore integration with Electronic Health Records (EHRs) and decision support systems, ensuring clinicians can access and utilize the integrated data efficiently. Integrating multi-omics data can further our understanding of disease mechanisms, potentially leading to breakthroughs in treatment and prevention. Yet, it is not clear whether omics strategies provide superior clinical benefits compared to a single data modality. Finally, possessing a scoring system that captures the omics features of the underlying disease from a single image modality (in our case, FFPE histology images) may help justify customizing the molecular profiling in the clinical setting.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 Low-Risk or
Low-Risk or  High-Risk Group
High-Risk Group