
NEDD9 Overexpression Causes Hyperproliferation of Luminal Cells and Cooperates with HER2 Oncogene in Tumor Initiation: A Novel Prognostic Marker in Breast Cancer

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Conditional NEDD9 Knock-In (Floxed-STOP-NEDD9) Transgenic Strain and Locus Insertion
2.2. Generation of FVB/J-MMTV-Cre-NEDD9+/+ Mice for Mammary Gland-Specific Upregulation of NEDD9
2.3. PCR Screening Strategy for the Genotyping of the Homozygous NEDD9fx/fx Knock-In Mice
2.4. PCR Screening Strategy for the Genotyping of the Induced NEDD9 Knock-In Line (With Removed Transcription Stop Cassette)
2.5. PCR Screening Strategy for the Genotyping of the Homozygous NEDD9/Cre/Erbb2 Mice
2.6. Generation of Mouse Embryonic Fibroblasts with NEDD9 Knock-In
2.7. Quantification of Branching Density in Mammary Gland Whole Mounts
2.8. Cell Culture, Plasmids, and Other Reagents
2.9. Lentivirus Constructs, Cell Infection, and Transfection Reagents
2.10. Tumor Tissue Micro Array (TMA) and Patient Data
2.11. Immunohistochemistry and Scoring Procedures
2.12. Fluorescent Immunohistochemistry and Hematoxylin & Eosin (H&E) Staining
2.13. Histopathology Evaluation and Grading
2.14. Western Blotting
2.15. MTT-Based Proliferation and Viability Assay
2.16. Drug Treatment and Viability Assay
2.17. Acini Formation, Imaging, and Quantification Procedures
2.18. Kaplan–Meier Analysis and Drug Response Evaluation Using ROC Plotter
2.19. Statistical Analysis
3. Results
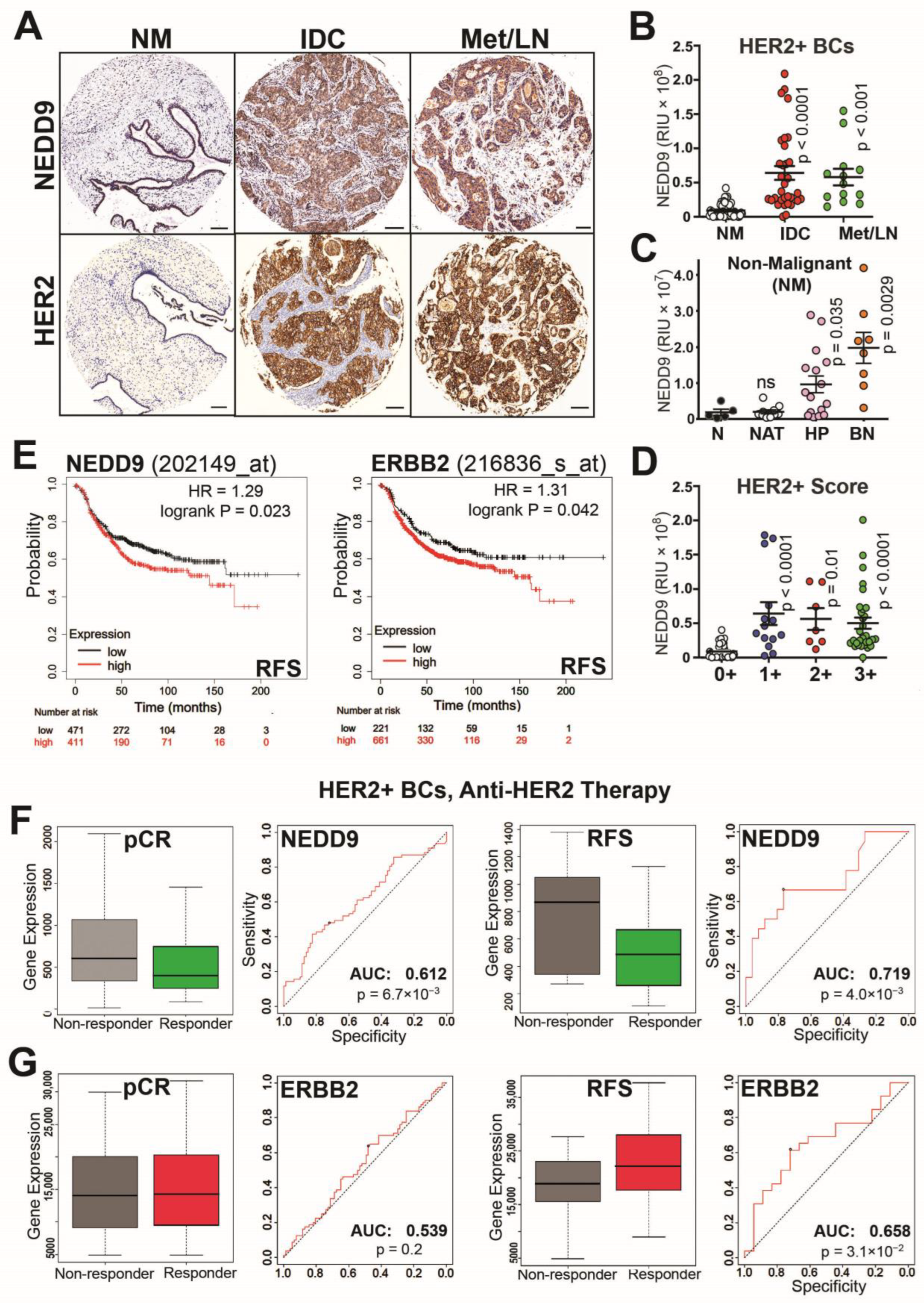
3.1. NEDD9 Expression Correlates with HER2+ Disease Progression and Treatment Outcomes
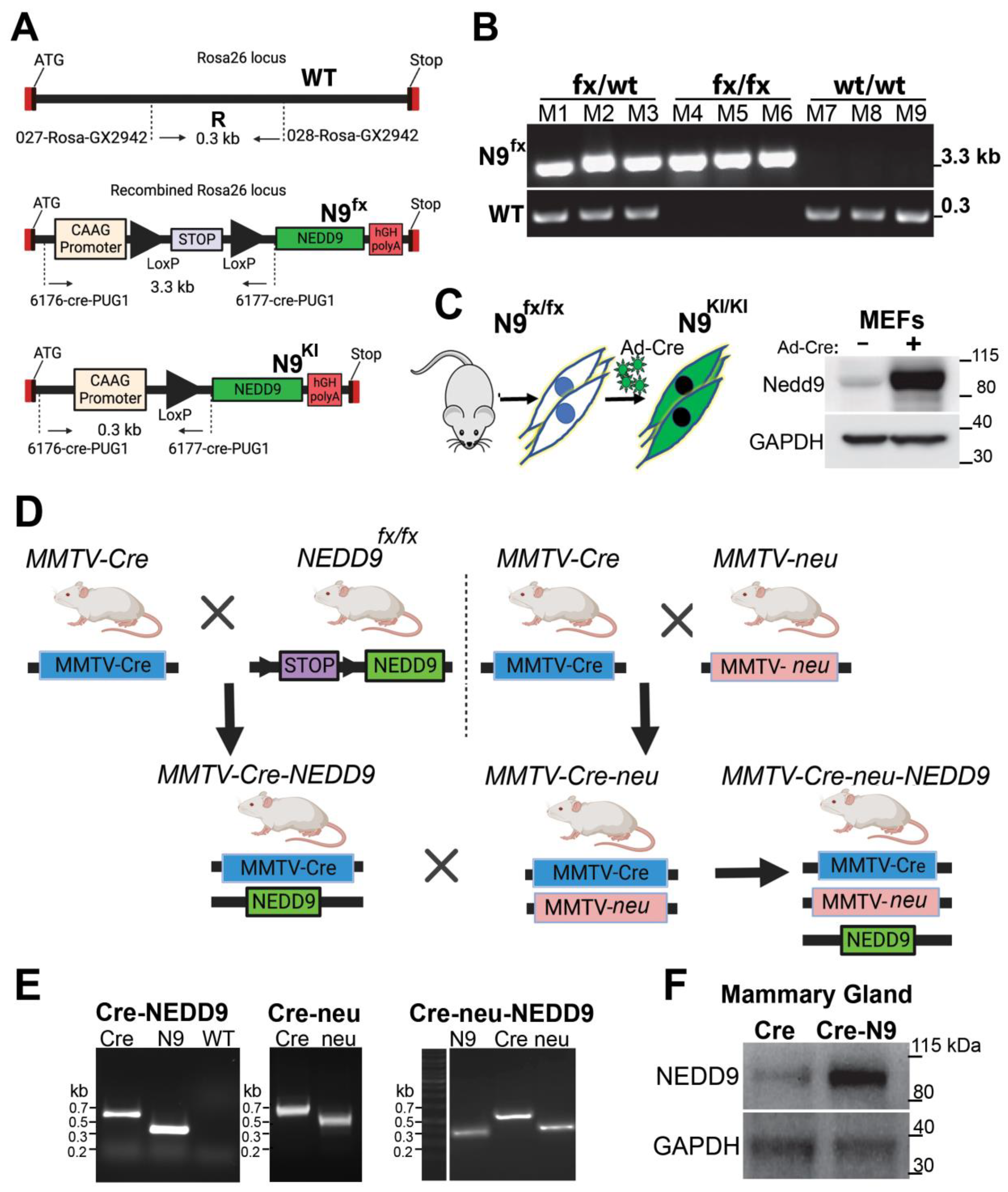
3.2. Generation of Conditional NEDD9 Knock-In (KI) Transgenic Mouse Model
3.3. Production and Analysis of Mammary Gland-Specific Expression of NEDD9
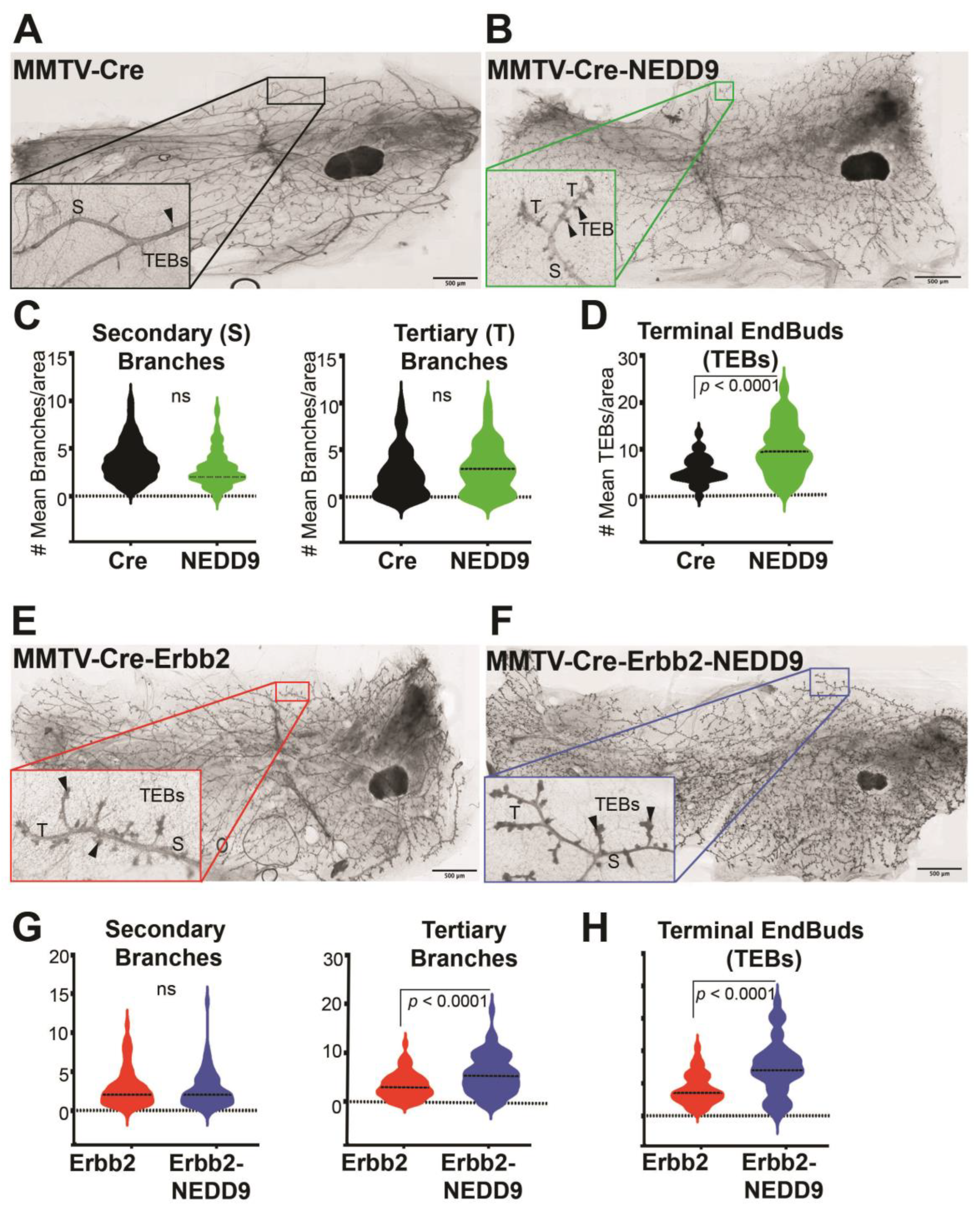
3.4. NEDD9 Overexpression Alters Mammary Gland Architecture by Increasing Mammary Gland Budding and Branching Morphogenesis and Cooperates with HER2
3.5. Cooperation between NEDD9 and HER2 Promotes Mammary Gland Branching Morphogenesis
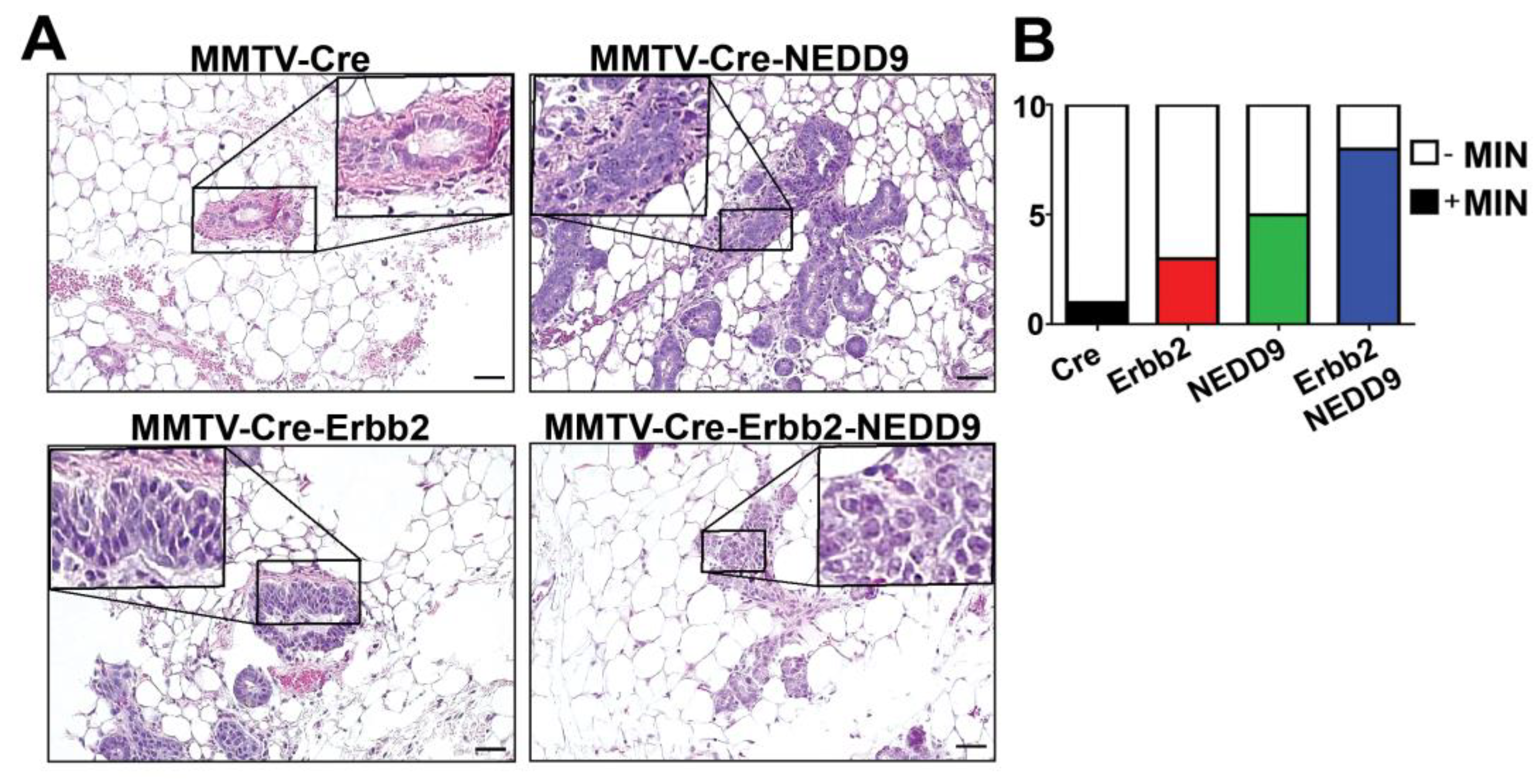
3.6. NEDD9 Overexpression Is Associated with Mammary Intra-Epithelia Neoplasia
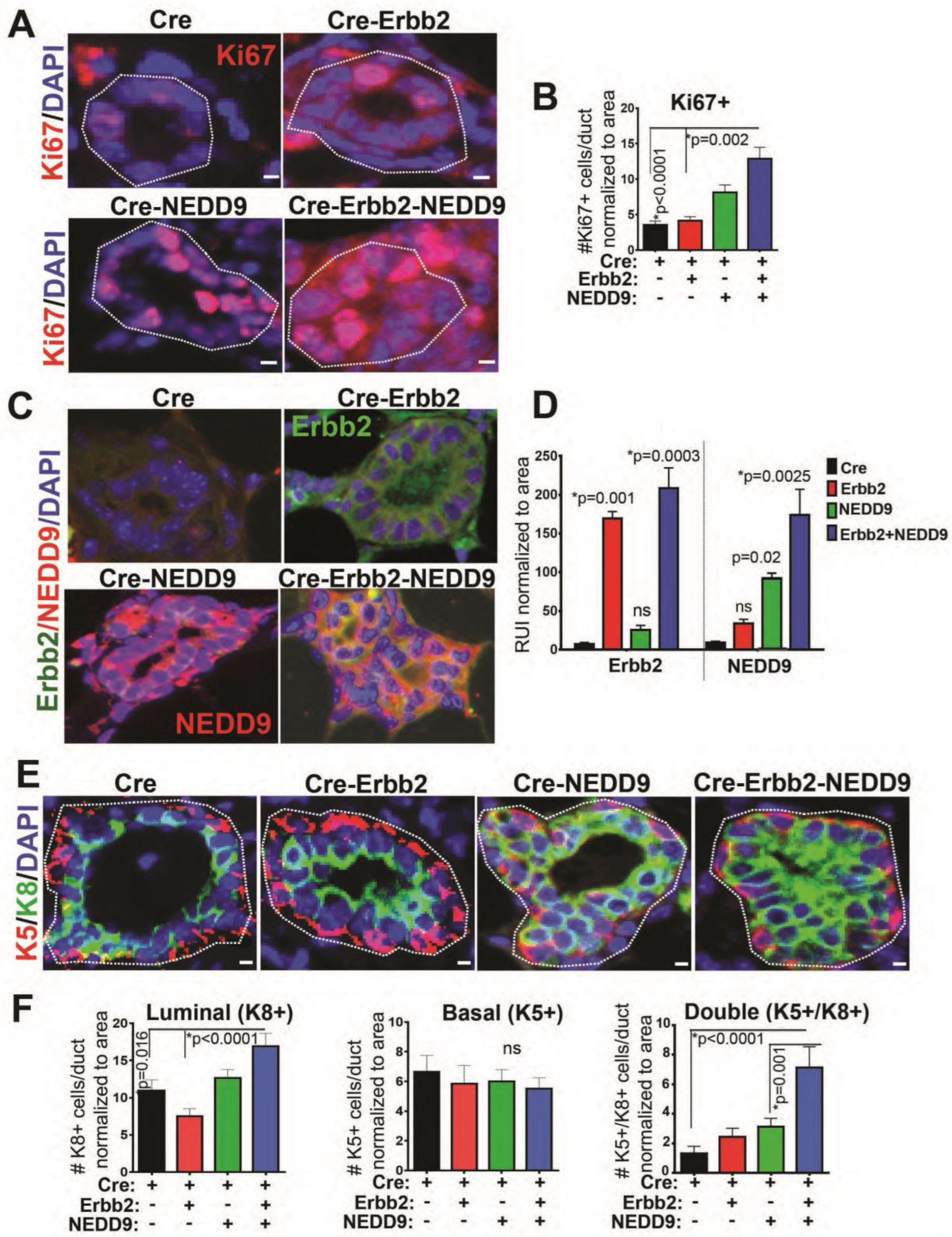
3.7. NEDD9 Overexpression Causes Hyperproliferation of Luminal Cells
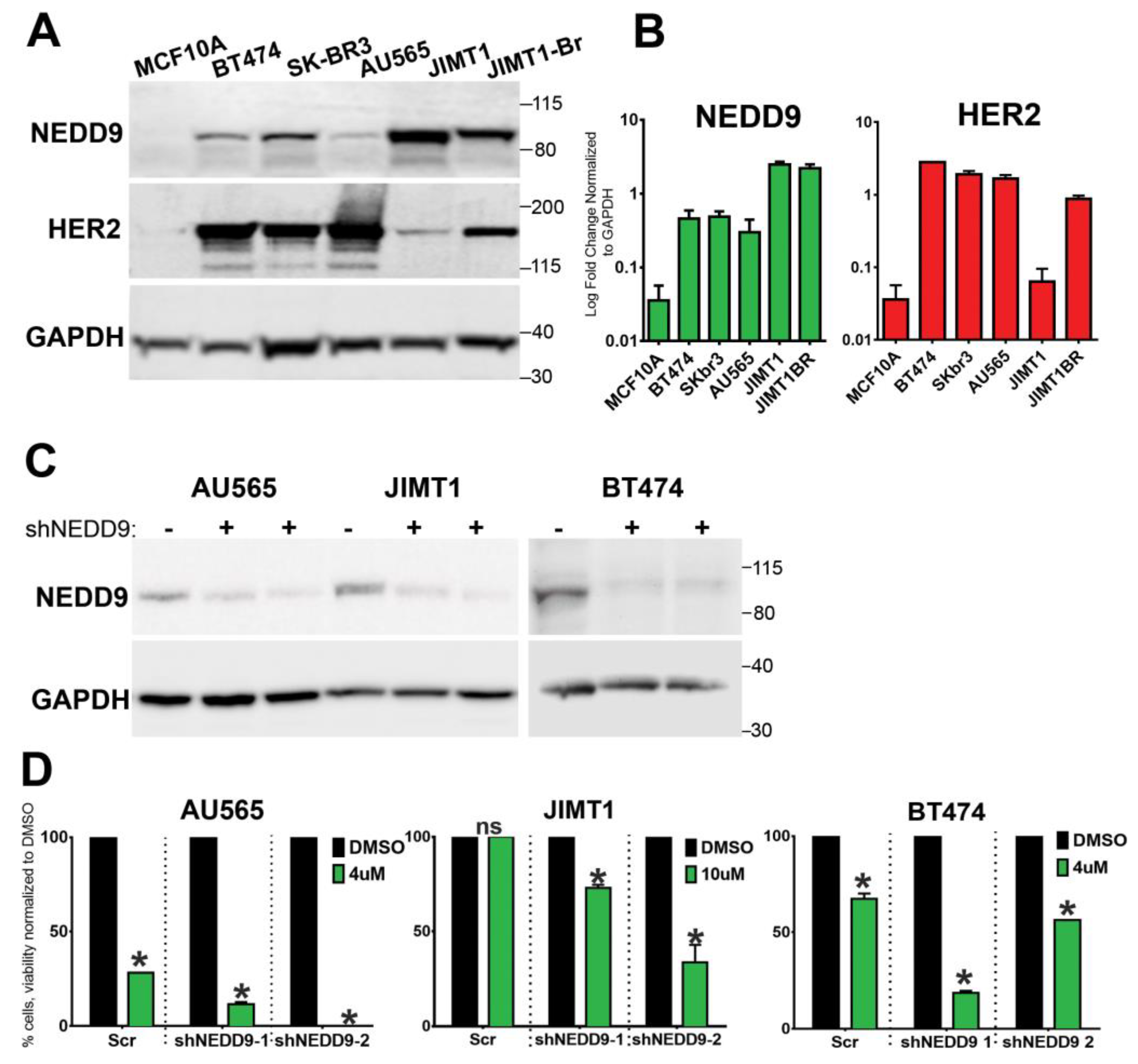
3.8. NEDD9 Is Overexpressed in Human HER2+ Breast Cancer Cell Lines
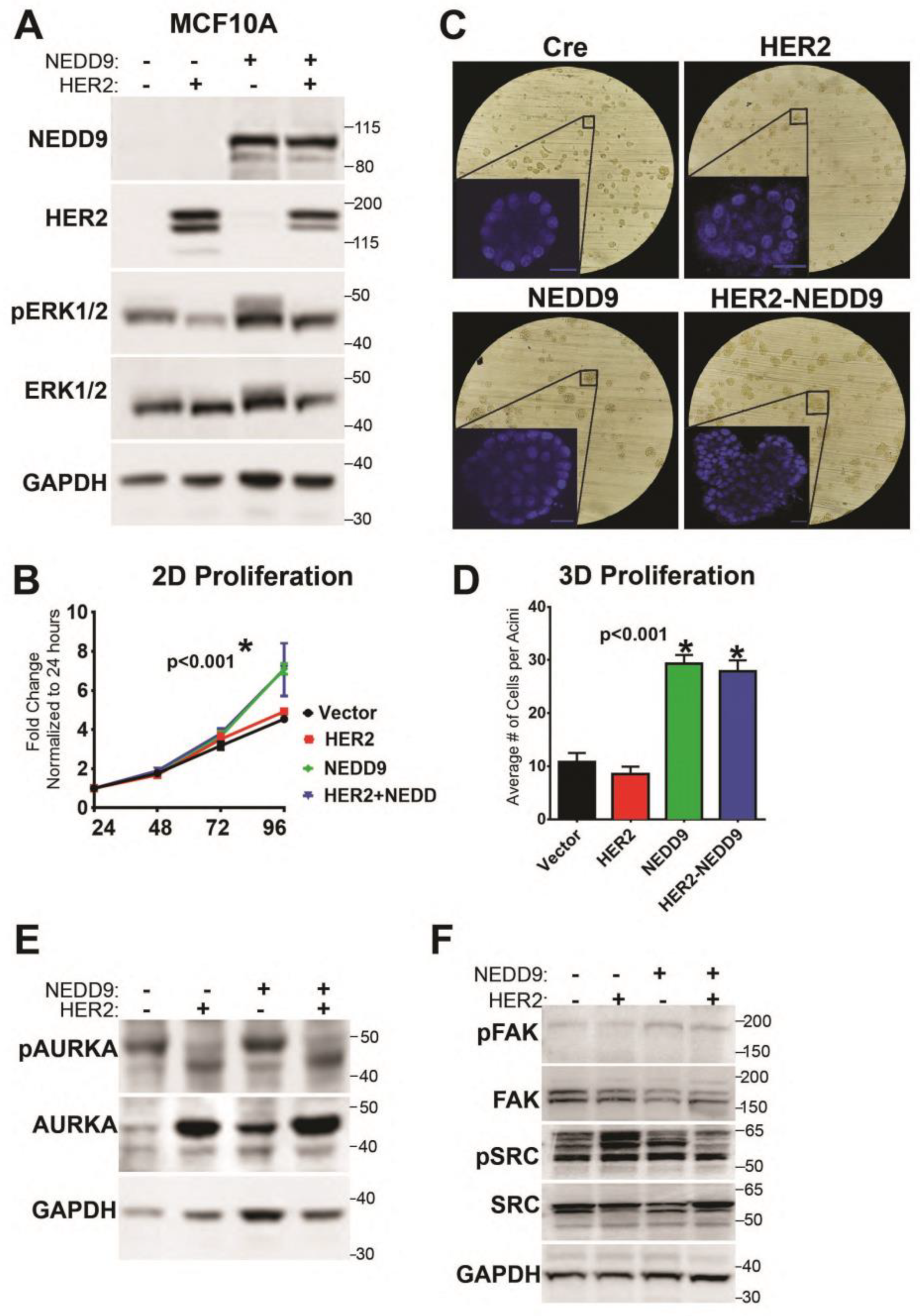
3.9. NEDD9 Overexpression in Normal Mammary Epithelial Cells Causes Increased Proliferation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures 2022; American Cancer Society: Atlanta, GA, USA, 2022. [Google Scholar]
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA A Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Gianni, L. HER2-positive breast cancer. Lancet 2017, 389, 2415–2429. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2012, 9, 16–32. [Google Scholar] [CrossRef]
- Muthuswamy, S.K. Trastuzumab resistance: All roads lead to SRC. Nat. Med. 2011, 17, 416. [Google Scholar] [CrossRef] [PubMed]
- Peiró, G.; Ortiz-Martínez, F.; Gallardo, A.; Pérez-Balaguer, A.; Sánchez-Payá, J.; Ponce, J.J.; Tibau, A.; López-Vilaro, L.; Escuin, D.; Adrover, E.; et al. Src, a potential target for overcoming trastuzumab resistance in HER2-positive breast carcinoma. Br. J. Cancer 2014, 111, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Belsches-Jablonski, A.P.; Biscardi, J.S.; Peavy, D.R.; Tice, D.A.; Romney, D.A.; Parsons, S.J. Src family kinases and HER2 interactions in human breast cancer cell growth and survival. Oncogene 2001, 20, 1465. [Google Scholar] [CrossRef]
- Shagisultanova, E.; Gaponova, A.V.; Gabbasov, R.; Nicolas, E.; Golemis, E.A. Preclinical and clinical studies of the NEDD9 scaffold protein in cancer and other diseases. Gene 2015, 567, 1–11. [Google Scholar] [CrossRef]
- Ahn, J.; Sanz-Moreno, V.; Marshall, C.J. The metastasis gene NEDD9 product acts through integrin β3 and Src to promote mesenchymal motility and inhibit amoeboid motility. J. Cell Sci. 2012, 125, 1814–1826. [Google Scholar] [CrossRef]
- Muller, W.J.; Arteaga, C.L.; Muthuswamy, S.K.; Siegel, P.M.; Webster, M.A.; Cardiff, R.D.; Meise, K.S.; Li, F.; Halter, S.A.; Coffey, R.J. Synergistic interaction of the Neu proto-oncogene product and transforming growth factor alpha in the mammary epithelium of transgenic mice. Mol. Cell. Biol. 1996, 16, 5726–5736. [Google Scholar] [CrossRef]
- Hsu, J.L.; Hung, M.-C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef]
- Sima, N.; Cheng, X.; Ye, F.; Ma, D.; Xie, X.; Lü, W. The Overexpression of Scaffolding Protein NEDD9 Promotes Migration and Invasion in Cervical Cancer via Tyrosine Phosphorylated FAK and SRC. PLoS ONE 2013, 8, e74594. [Google Scholar] [CrossRef]
- Izumchenko, E.; Singh, M.K.; Plotnikova, O.V.; Tikhmyanova, N.; Little, J.L.; Serebriiskii, I.G.; Seo, S.; Kurokawa, M.; Egleston, B.L.; Klein-Szanto, A.; et al. NEDD9 Promotes Oncogenic Signaling in Mammary Tumor Development. Cancer Res. 2009, 69, 7198–7206. [Google Scholar] [CrossRef]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Cabodi, S.; Tinnirello, A.; Di Stefano, P.; Bisarò, B.; Ambrosino, E.; Castellano, I.; Sapino, A.; Arisio, R.; Cavallo, F.; Forni, G.; et al. p130Cas as a New Regulator of Mammary Epithelial Cell Proliferation, Survival, and HER2-Neu Oncogene–Dependent Breast Tumorigenesis. Cancer Res. 2006, 66, 4672–4680. [Google Scholar] [CrossRef] [PubMed]
- Little, J.L.; Serzhanova, V.; Izumchenko, E.; Egleston, B.L.; Parise, E.; Klein-Szanto, A.J.; Loudon, G.; Shubina, M.; Seo, S.; Kurokawa, M.; et al. A requirement for Nedd9 in luminal progenitor cells prior to mammary tumorigenesis in MMTV-HER2/ErbB2 mice. Oncogene 2013, 33, 411. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bavarva, J.H.; Wang, Z.; Guo, J.; Qian, C.; Thibodeau, S.N.; Golemis, E.A.; Liu, W. HEF1, a novel target of Wnt signaling, promotes colonic cell migration and cancer progression. Oncogene 2011, 30, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Speranza, M.C.; Frattini, V.; Pisati, F.; Kapetis, D.; Porrati, P.; Eoli, M.; Pellegatta, S.; Finocchiaro, G. NEDD9, a novel target of miR-145, increases the invasiveness of glioblastoma. Oncotarget 2012, 3, 723–734. [Google Scholar] [CrossRef]
- Wang, H.; Mu, X.; Zhou, S.; Zhang, J.; Dai, J.; Tang, L.; Xiao, L.; Duan, Z.; Jia, L.; Chen, S. NEDD9 overexpression is associated with the progression of and an unfavorable prognosis in epithelial ovarian cancer. Hum. Pathol. 2014, 45, 401–408. [Google Scholar] [CrossRef]
- Li, G.; Robinson, G.W.; Lesche, R.; Martinez-Diaz, H.; Jiang, Z.; Rozengurt, N.; Wagner, K.-U.; Wu, D.-C.; Lane, T.F.; Liu, X.; et al. Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development 2002, 129, 4159–4170. [Google Scholar] [CrossRef]
- Zhong, J.; Baquiran, J.B.; Bonakdar, N.; Lees, J.; Ching, Y.W.; Pugacheva, E.; Fabry, B.; O’Neill, G.M. NEDD9 stabilizes focal adhesions, increases binding to the extra-cellular matrix and differentially effects 2D versus 3D cell migration. PloS ONE 2012, 7, e35058. [Google Scholar] [CrossRef] [PubMed]
- Jozefczuk, J.; Drews, K.; Adjaye, J. Preparation of mouse embryonic fibroblast cells suitable for culturing human embryonic and induced pluripotent stem cells. J. Vis. Exp. 2012, 64, 3854. [Google Scholar] [CrossRef]
- Ice, R.J.; McLaughlin, S.L.; Livengood, R.H.; Culp, M.V.; Eddy, E.R.; Ivanov, A.V.; Pugacheva, E.N. NEDD9 Depletion Destabilizes Aurora A Kinase and Heightens the Efficacy of Aurora A Inhibitors: Implications for Treatment of Metastatic Solid Tumors. Cancer Res. 2013, 73, 3168–3180. [Google Scholar] [CrossRef] [PubMed]
- Stanko, J.P.; Fenton, S.E. Quantifying Branching Density in Rat Mammary Gland Whole-mounts Using the Sholl Analysis Method. J. Vis. Exp. 2017, 125, 55789. [Google Scholar] [CrossRef]
- Lyle, L.T.; Lockman, P.R.; Adkins, C.E.; Mohammad, A.S.; Sechrest, E.; Hua, E.; Palmieri, D.; Liewehr, D.J.; Steinberg, S.M.; Kloc, W.; et al. Alterations in Pericyte Subpopulations Are Associated with Elevated Blood–Tumor Barrier Permeability in Experimental Brain Metastasis of Breast Cancer. Clin. Cancer Res. 2016, 22, 5287–5299. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Agazie, Y.M. Molecular mechanism for SHP2 in promoting HER2-induced signaling and transformation. J. Biol. Chem. 2009, 284, 12226–12234. [Google Scholar] [CrossRef] [PubMed]
- Addison, J.B.; Voronkova, M.A.; Fugett, J.H.; Lin, C.-C.; Linville, N.C.; Trinh, B.; Livengood, R.H.; Smolkin, M.B.; Schaller, M.D.; Ruppert, J.M.; et al. Functional Hierarchy and Cooperation of EMT Master Transcription Factors in Breast Cancer Metastasis. Mol. Cancer Res. 2021, 19, 784–798. [Google Scholar] [CrossRef]
- Tiscornia, G.; Singer, O.; Verma, I.M. Production and purification of lentiviral vectors. Nat. Protoc. 2006, 1, 241–245. [Google Scholar] [CrossRef]
- Whately, K.M.; Voronkova, M.A.; Maskey, A.; Gandhi, J.; Loskutov, J.; Choi, H.; Yanardag, S.; Chen, D.; Wen, S.; Margaryan, N.V.; et al. Nuclear Aurora-A kinase-induced hypoxia signaling drives early dissemination and metastasis in breast cancer: Implications for detection of metastatic tumors. Oncogene 2021, 40, 5651–5664. [Google Scholar] [CrossRef]
- Lánczky, A.; Győrffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Pugacheva, E.N.; Golemis, E.A. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat. Cell Biol. 2005, 7, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Brûlet, P.; Babinet, C.; Kemler, R.; Jacob, F. Monoclonal antibodies against trophectoderm-specific markers during mouse blastocyst formation. Proc. Natl. Acad. Sci. USA 1980, 77, 4113–4117. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Ingram, L.; Tolosa, E.J.; Vera, R.E.; Li, Q.; Kim, S.; Ma, Y.; Spyropoulos, D.D.; Beharry, Z.; Huang, J.; et al. Gli Transcription Factors Mediate the Oncogenic Transformation of Prostate Basal Cells Induced by a Kras-Androgen Receptor Axis*. J. Biol. Chem. 2016, 291, 25749–25760. [Google Scholar] [CrossRef] [PubMed]
- Cardiff, R.D.; Anver, M.R.; Gusterson, B.A.; Hennighausen, L.; Jensen, R.A.; Merino, M.J.; Rehm, S.; Russo, J.; Tavassoli, F.A.; Wakefield, L.M.; et al. The mammary pathology of genetically engineered mice: The consensus report and recommendations from the Annapolis meeting. Oncogene 2000, 19, 968–988. [Google Scholar] [CrossRef] [PubMed]
- Cardiff, R.D.; Moghanaki, D.; Jensen, R.A. Genetically Engineered Mouse Models of Mammary Intraepithelial Neoplasia. J. Mammary Gland Biol. Neoplasia 2000, 5, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003, 30, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Fekete, J.T.; Győrffy, B. ROCplot.org: Validating predictive biomarkers of chemotherapy/hormonal therapy/anti-HER2 therapy using transcriptomic data of 3,104 breast cancer patients. Int. J. Cancer 2019, 145, 3140–3151. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.T.; Webster, M.A.; Schaller, M.; Parsons, T.J.; Cardiff, R.D.; Muller, W.J. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10578–10582. [Google Scholar] [CrossRef]
- Andrechek, E.R.; Hardy, W.R.; Siegel, P.M.; Rudnicki, M.A.; Cardiff, R.D.; Muller, W.J. Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 3444–3449. [Google Scholar] [CrossRef]
- Mihály, Z.; Győrffy, B. Improving Pathological Assessment of Breast Cancer by Employing Array-Based Transcriptome Analysis. Microarrays 2013, 2, 228–242. [Google Scholar] [CrossRef]
- Williams, J.M.; Daniel, C.W. Mammary ductal elongation: Differentiation of myoepithelium and basal lamina during branching morphogenesis. Dev. Biol. 1983, 97, 274–290. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, N.A.; Browne, B.C.; Chow, L.; Wang, Y.; Ginther, C.; Arboleda, J.; Duffy, M.J.; Crown, J.; O’Donovan, N.; Slamon, D.J. Activated Phosphoinositide 3-Kinase/AKT Signaling Confers Resistance to Trastuzumab but not Lapatinib. Mol. Cancer Ther. 2010, 9, 1489–1502. [Google Scholar] [CrossRef] [PubMed]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.B.; Mendrola, J.M.; Lemmon, M.A. ErbB3/HER3 does not homodimerize upon neuregulin binding at the cell surface. FEBS Lett. 2004, 569, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Bazley, L.A.; Gullick, W.J. The epidermal growth factor receptor family. Endocr. Relat. Cancer Endocr. Relat. Cancer 2005, 12, S17–S27. [Google Scholar] [CrossRef] [PubMed]
- Beigbeder, A.; Chartier, F.J.M.; Bisson, N. MPZL1 forms a signalling complex with GRB2 adaptor and PTPN11 phosphatase in HER2-positive breast cancer cells. Sci. Rep. 2017, 7, 11514. [Google Scholar] [CrossRef]
- Jin, Y.; Li, F.; Zheng, C.; Wang, Y.; Fang, Z.; Guo, C.; Wang, X.; Liu, H.; Deng, L.; Li, C.; et al. NEDD9 promotes lung cancer metastasis through epithelial–mesenchymal transition. Int. J. Cancer 2014, 134, 2294–2304. [Google Scholar] [CrossRef]
- Iida, J.; Dorchak, J.; Clancy, R.; Slavik, J.; Ellsworth, R.; Katagiri, Y.; Pugacheva, E.N.; van Kuppevelt, T.H.; Mural, R.J.; Cutler, M.L.; et al. Role for chondroitin sulfate glycosaminoglycan in NEDD9-mediated breast cancer cell growth. Exp. Cell Res. 2015, 330, 358–370. [Google Scholar] [CrossRef]
- Zelazny, E.; Li, B.; Anagnostopoulos, A.-M.; Coleman, A.; Perkins, A.S. Cooperating Oncogenic Events in Murine Mammary Tumorigenesis: Assessment of ErbB2, Mutant p53, and Mouse Mammary Tumor Virus. Exp. Mol. Pathol. 2001, 70, 183–193. [Google Scholar] [CrossRef]
- Prat, A.; Karginova, O.; Parker, J.S.; Fan, C.; He, X.; Bixby, L.; Harrell, J.C.; Roman, E.; Adamo, B.; Troester, M.; et al. Characterization of cell lines derived from breast cancers and normal mammary tissues for the study of the intrinsic molecular subtypes. Breast Cancer Res. Treat. 2013, 142, 237–255. [Google Scholar] [CrossRef]
- Gabbasov, R.; Xiao, F.; Howe, C.G.; Bickel, L.E.; O’Brien, S.W.; Benrubi, D.; Do, T.-V.; Zhou, Y.; Nicolas, E.; Cai, K.Q.; et al. NEDD9 promotes oncogenic signaling, a stem/mesenchymal gene signature, and aggressive ovarian cancer growth in mice. Oncogene 2018, 37, 4854–4870. [Google Scholar] [CrossRef]
- Yue, D.; Liu, S.; Zhang, T.; Wang, Y.; Qin, G.; Chen, X.; Zhang, H.; Wang, D.; Huang, D.; Wang, F.; et al. NEDD9 promotes cancer stemness by recruiting myeloid-derived suppressor cells via CXCL8 in esophageal squamous cell carcinoma. Cancer Biol. Med. 2021, 18, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Masciale, V.; Banchelli, F.; Grisendi, G.; D’Amico, R.; Maiorana, A.; Stefani, A.; Morandi, U.; Stella, F.; Dominici, M.; Aramini, B. The Influence of Cancer Stem Cells on the Risk of Relapse in Adenocarcinoma and Squamous Cell Carcinoma of the Lung: A Prospective Cohort Study. Stem Cells Transl. Med. 2022, 11, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Marzagalli, M.; Fontana, F.; Raimondi, M.; Limonta, P. Cancer Stem Cells—Key Players in Tumor Relapse. Cancers 2021, 13, 376. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wei, F.; Su, Y.; Wang, Y.; Shen, Y.; Fang, Y.; Ding, J.; Chen, Y. Histone deacetylase inhibitors promote breast cancer metastasis by elevating NEDD9 expression. Signal Transduct. Target. Ther. 2023, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Kozyreva, V.K.; Kiseleva, A.A.; Ice, R.J.; Jones, B.C.; Loskutov, Y.V.; Matalkah, F.; Smolkin, M.B.; Marinak, K.; Livengood, R.H.; Salkeni, M.A.; et al. Combination of Eribulin and Aurora A Inhibitor MLN8237 Prevents Metastatic Colonization and Induces Cytotoxic Autophagy in Breast Cancer. Mol. Cancer Ther. 2016, 15, 1809–1822. [Google Scholar] [CrossRef]
- Mihály, Z.; Kormos, M.; Lánczky, A.; Dank, M.; Budczies, J.; Szász, M.A.; Győrffy, B. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res. Treat. 2013, 140, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Tortora, G. Mechanisms of Resistance to HER2 Target Therapy. JNCI Monogr. 2011, 2011, 95–98. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | MIN+, N (%) | MIN−, N(%) |
|---|---|---|
| MMTV-Cre | 1 (10%) | 9 (90%) |
| MMTV-Cre-Erbb2 | 3 (30%) | 7 (70%) |
| MMTV-Cre-NEDD9 | 5 (50%) | 5 (50%) |
| MMTV-Cre-Erbb2-NEDD9 | 8 (80%) | 2 (20%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purazo, M.L.; Ice, R.J.; Shimpi, R.; Hoenerhoff, M.; Pugacheva, E.N. NEDD9 Overexpression Causes Hyperproliferation of Luminal Cells and Cooperates with HER2 Oncogene in Tumor Initiation: A Novel Prognostic Marker in Breast Cancer. Cancers 2023, 15, 1119. https://doi.org/10.3390/cancers15041119
Purazo ML, Ice RJ, Shimpi R, Hoenerhoff M, Pugacheva EN. NEDD9 Overexpression Causes Hyperproliferation of Luminal Cells and Cooperates with HER2 Oncogene in Tumor Initiation: A Novel Prognostic Marker in Breast Cancer. Cancers. 2023; 15(4):1119. https://doi.org/10.3390/cancers15041119
Chicago/Turabian StylePurazo, Marc L., Ryan J. Ice, Rahul Shimpi, Mark Hoenerhoff, and Elena N. Pugacheva. 2023. "NEDD9 Overexpression Causes Hyperproliferation of Luminal Cells and Cooperates with HER2 Oncogene in Tumor Initiation: A Novel Prognostic Marker in Breast Cancer" Cancers 15, no. 4: 1119. https://doi.org/10.3390/cancers15041119