
Modeling Therapy-Driven Evolution of Glioblastoma with Patient-Derived Xenografts
 , , , , , , , ,
, , , , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
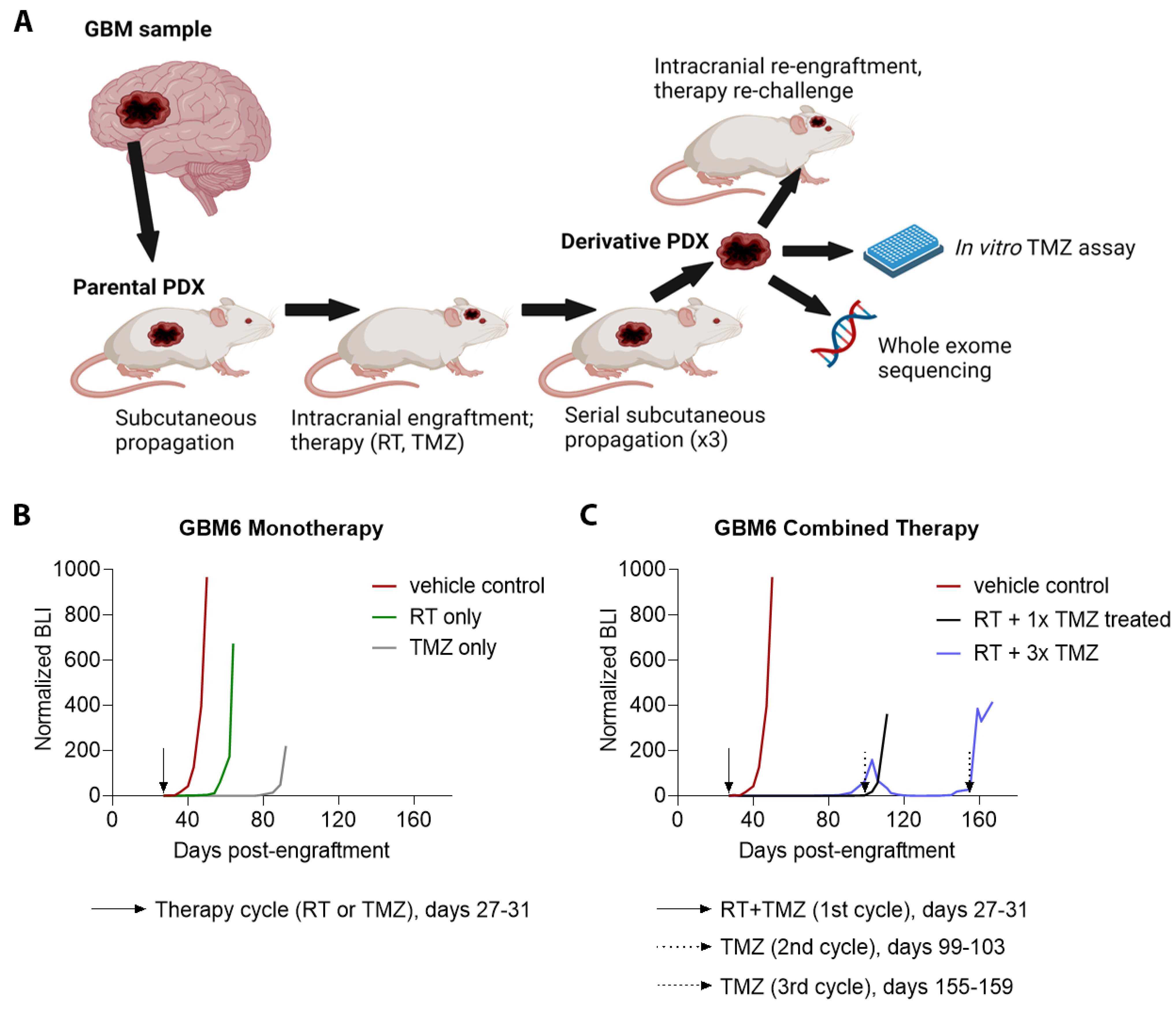
2.2. Intracranial Engraftment of PDX Cells
2.3. In Vivo Treatment of Parental PDX
2.4. Propagation of Derivative PDX
2.5. Cell Viability Assays
2.6. Testing Parental and Derivative PDX for Response to Therapy
2.7. Whole Exome Sequencing
2.8. Bioinformatics and Biostatistics Analyses
3. Results
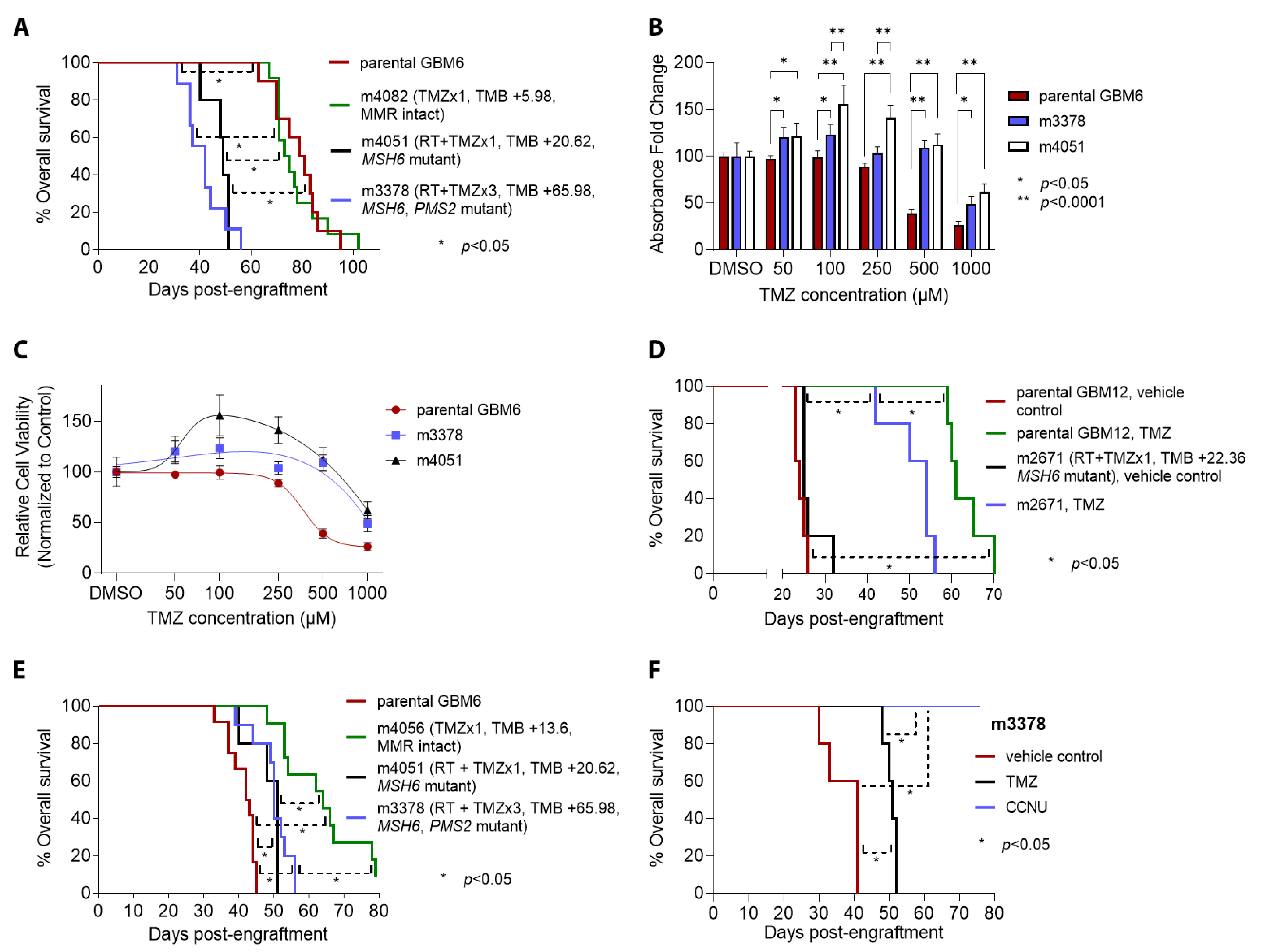
3.1. Establishment, Growth, and Primary Treatment of Parental PDX
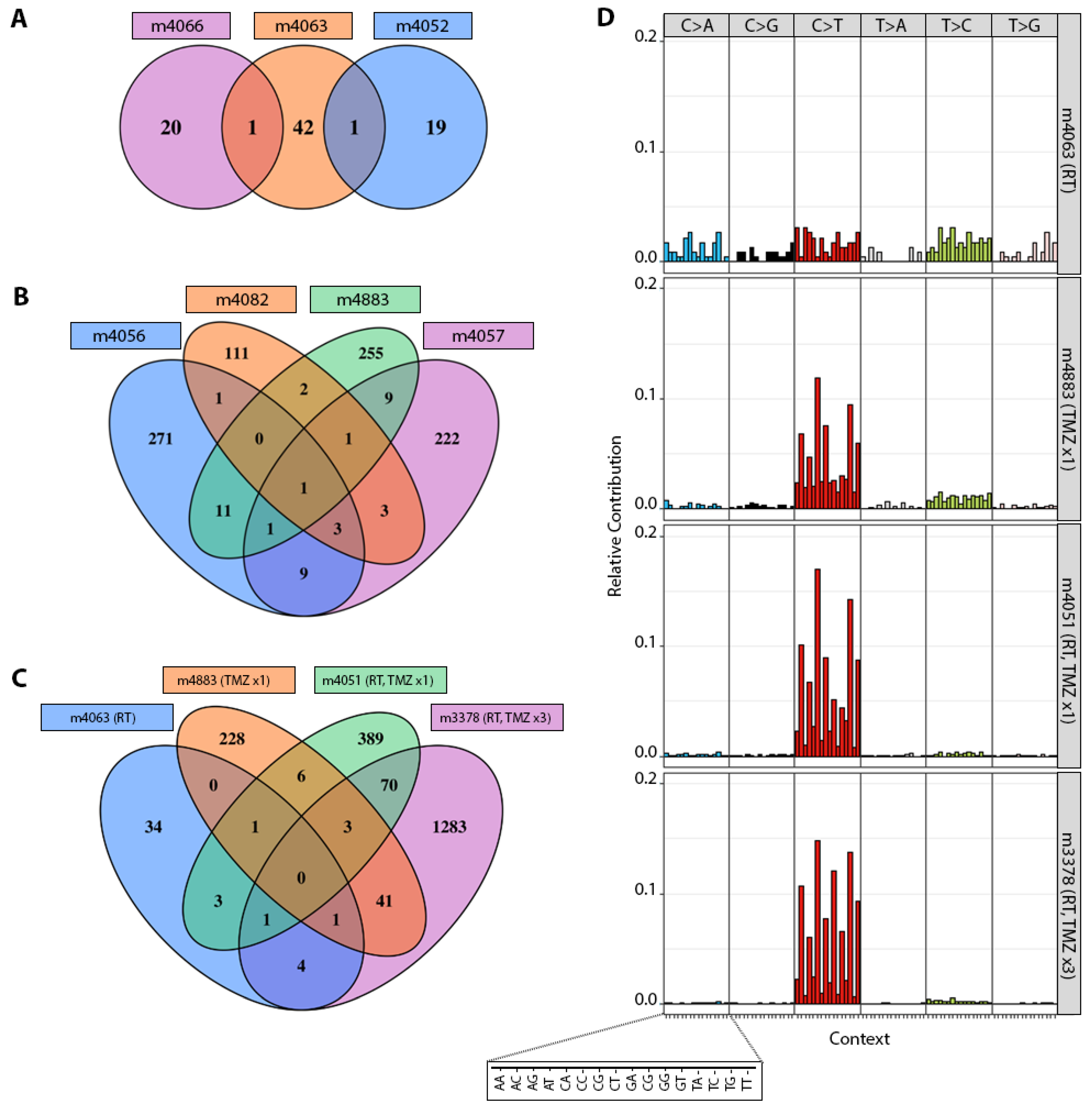
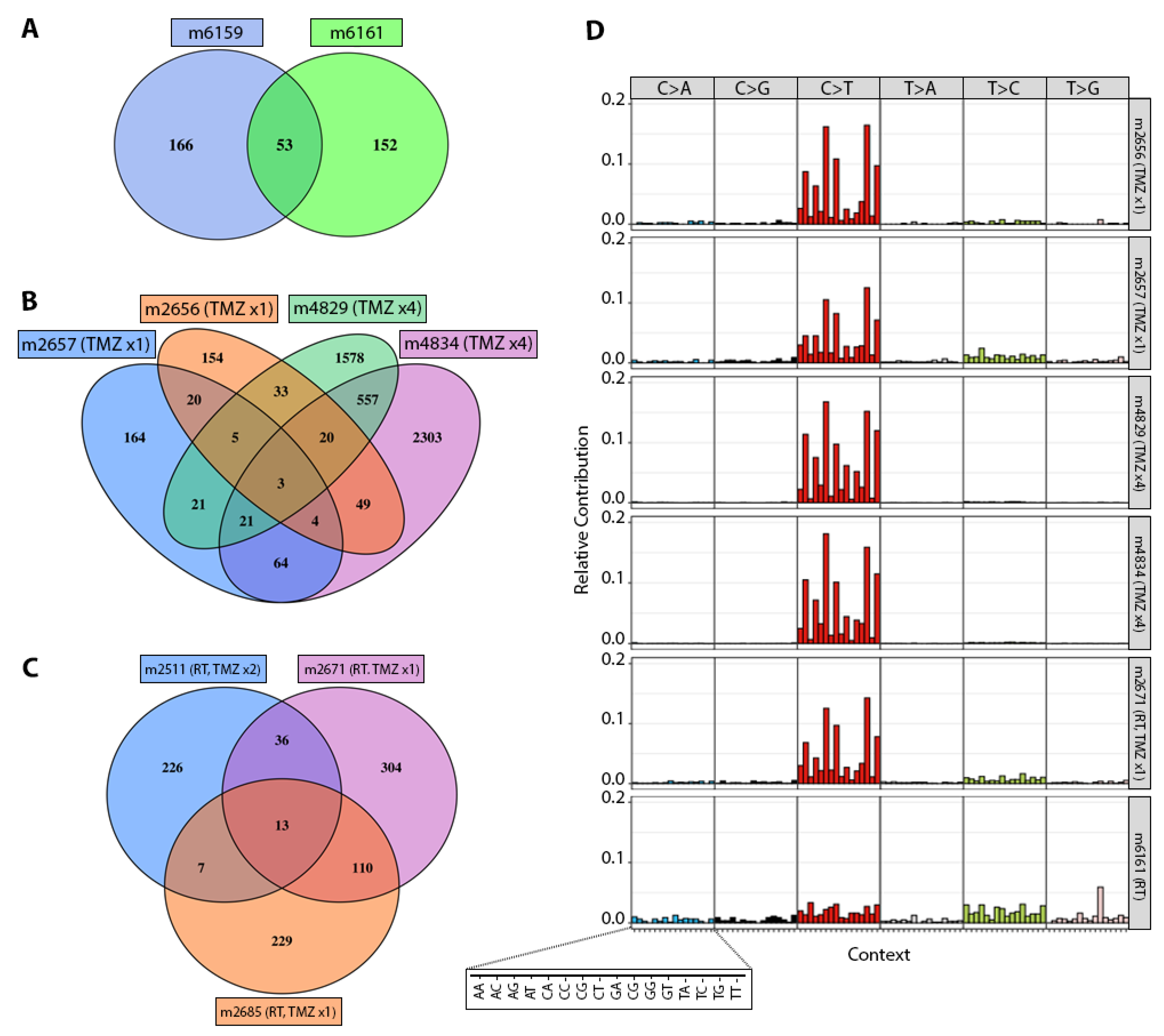
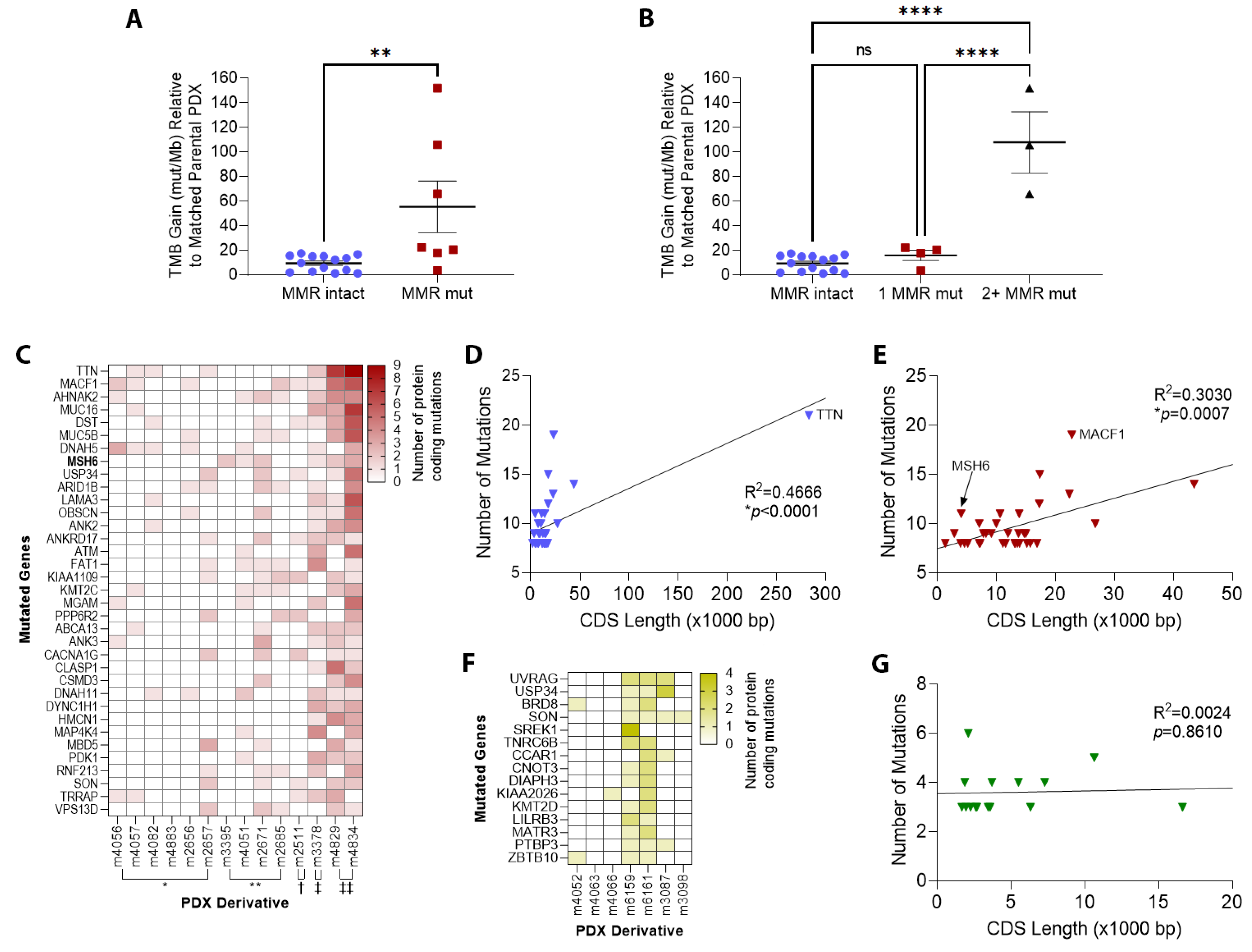
3.2. TMZ Treatment Results in Marked Increase in Tumor Mutation Burden (TMB) and a Distinct Mutation Profile
3.3. Patterns of Therapy Response in Post-Treatment GBM PDX
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 23, iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, M.T.C.; Sudlow, C.L.M.; Figueroa, J.D.; Brennan, P.M. Longer-term (≥2 years) survival in patients with glioblastoma in population-based studies pre- and post-2005: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 11622. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Xie, Q.; Mittal, S.; Berens, M.E. Targeting adaptive glioblastoma: An overview of proliferation and invasion. Neuro-Oncology 2014, 16, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.P.; Levine, K.K.; Betensky, R.A.; Codd, P.J.; Romany, C.A.; Reavie, L.B.; Batchelor, T.T.; Futreal, P.A.; Stratton, M.R.; Curry, W.T.; et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 2007, 13, 2038–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCord, M.; Steffens, A.; Javier, R.; Kam, K.L.; McCortney, K.; Horbinski, C. The efficacy of DNA mismatch repair enzyme immunohistochemistry as a screening test for hypermutated gliomas. Acta Neuropathol. Commun. 2020, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Leelatian, N.; Hong, C.S.; Bindra, R.S. The Role of Mismatch Repair in Glioblastoma Multiforme Treatment Response and Resistance. Neurosurg. Clin. N. Am. 2021, 32, 171–180. [Google Scholar] [CrossRef]
- Poon, S.L.; McPherson, J.R.; Tan, P.; Teh, B.T.; Rozen, S.G. Mutation signatures of carcinogen exposure: Genome-wide detection and new opportunities for cancer prevention. Genome Med. 2014, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro-Oncology 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Daniel, P.; Sabri, S.; Chaddad, A.; Meehan, B.; Jean-Claude, B.; Rak, J.; Abdulkarim, B.S. Temozolomide Induced Hypermutation in Glioma: Evolutionary Mechanisms and Therapeutic Opportunities. Front. Oncol. 2019, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Lukas, R.V.; Rodon, J.; Becker, K.; Wong, E.T.; Shih, K.; Touat, M.; Fasso, M.; Osborne, S.; Molinero, L.; O’Hear, C.; et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J. Neurooncol. 2018, 140, 317–328. [Google Scholar] [CrossRef] [PubMed]
- McCord, M.; Lukas, R.V.; Amidei, C.; Demars, N.; Gelb, A.; Buck, J.; Sachdev, S.; Feldman, A.; Tate, M.; Dixit, K.; et al. Disappearance of MMR-deficient subclones after controlled IL-12 and PD-1 inhibition in a glioma patient. Neurooncol. Adv. 2021, 3, vdab045. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.; Meehan, B.; Sabri, S.; Jamali, F.; Sarkaria, J.N.; Choi, D.; Garnier, D.; Kitange, G.; Glennon, K.I.; Paccard, A.; et al. Detection of temozolomide-induced hypermutation and response to PD-1 checkpoint inhibitor in recurrent glioblastoma. Neurooncol. Adv. 2022, 4, vdac076. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, R.; Samstein, R.M.; Lee, K.W.; Havel, J.J.; Wang, H.; Krishna, C.; Sabio, E.Y.; Makarov, V.; Kuo, F.; Blecua, P.; et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019, 364, 485–491. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Kurz, S.C.; Cabrera, L.P.; Hastie, D.; Huang, R.; Unadkat, P.; Rinne, M.; Nayak, L.; Lee, E.Q.; Reardon, D.A.; Wen, P.Y. PD-1 inhibition has only limited clinical benefit in patients with recurrent high-grade glioma. Neurology 2018, 91, e1355–e1359. [Google Scholar] [CrossRef]
- McGrail, D.J.; Pilie, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Ueno, N.T.; et al. Validation of cancer-type dependent benefit from immune checkpoint blockade in TMB-H tumors identified by the FoundationOne CDx assay. Ann. Oncol. 2022, 33, 1204–1206. [Google Scholar] [CrossRef]
- Giannini, C.; Sarkaria, J.N.; Saito, A.; Uhm, J.H.; Galanis, E.; Carlson, B.L.; Schroeder, M.A.; James, C.D. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro-Oncology 2005, 7, 164–176. [Google Scholar] [CrossRef] [Green Version]
- Sarkaria, J.N.; Yang, L.; Grogan, P.T.; Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Galanis, E.; Giannini, C.; Wu, W.; Dinca, E.B.; et al. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol. Cancer Ther. 2007, 6, 1167–1174. [Google Scholar] [CrossRef] [Green Version]
- Michaud, K.; Solomon, D.A.; Oermann, E.; Kim, J.S.; Zhong, W.Z.; Prados, M.D.; Ozawa, T.; James, C.D.; Waldman, T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010, 70, 3228–3238. [Google Scholar] [CrossRef] [PubMed]
- Supino, R. MTT assays. In In Vitro Toxicity Testing Protocols; O’Hare, S.A.C., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 1995; Volume 43, pp. 137–149. [Google Scholar]
- Obenchain, V.; Lawrence, M.; Carey, V.; Gogarten, S.; Shannon, P.; Morgan, M. VariantAnnotation: A Bioconductor package for exploration and annotation of genetic variants. Bioinformatics 2014, 30, 2076–2078. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Bang, M.L.; Centner, T.; Fornoff, F.; Geach, A.J.; Gotthardt, M.; McNabb, M.; Witt, C.C.; Labeit, D.; Gregorio, C.C.; Granzier, H.; et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 2001, 89, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. [Google Scholar] [CrossRef]
- Taal, W.; Oosterkamp, H.M.; Walenkamp, A.M.; Dubbink, H.J.; Beerepoot, L.V.; Hanse, M.C.; Buter, J.; Honkoop, A.H.; Boerman, D.; de Vos, F.Y.; et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): A randomised controlled phase 2 trial. Lancet Oncol. 2014, 15, 943–953. [Google Scholar] [CrossRef]
- Burnet, N.G.; Jefferies, S.J.; Benson, R.J.; Hunt, D.P.; Treasure, F.P. Years of life lost (YLL) from cancer is an important measure of population burden—And should be considered when allocating research funds. Br. J. Cancer 2005, 92, 241–245. [Google Scholar] [CrossRef]
- Higuchi, F.; Nagashima, H.; Ning, J.; Koerner, M.V.A.; Wakimoto, H.; Cahill, D.P. Restoration of Temozolomide Sensitivity by PARP Inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin. Cancer Res. 2020, 26, 1690–1699. [Google Scholar] [CrossRef]
- Kamoun, W.S.; Ley, C.D.; Farrar, C.T.; Duyverman, A.M.; Lahdenranta, J.; Lacorre, D.A.; Batchelor, T.T.; di Tomaso, E.; Duda, D.G.; Munn, L.L.; et al. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J. Clin. Oncol. 2009, 27, 2542–2552. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Mulholland, P.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218. [Google Scholar] [CrossRef] [PubMed]
- Stefanik, D.F.; Fellows, W.K.; Rizkalla, L.R.; Rizkalla, W.M.; Stefanik, P.P.; Deleo, A.B.; Welch, W.C. Monoclonal antibodies to vascular endothelial growth factor (VEGF) and the VEGF receptor, FLT-1, inhibit the growth of C6 glioma in a mouse xenograft. J. Neurooncol. 2001, 55, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.P.; Codd, P.J.; Batchelor, T.T.; Curry, W.T.; Louis, D.N. MSH6 inactivation and emergent temozolomide resistance in human glioblastomas. Clin. Neurosurg. 2008, 55, 165–171. [Google Scholar]
- Vaubel, R.A.; Tian, S.; Remonde, D.; Schroeder, M.A.; Mladek, A.C.; Kitange, G.J.; Caron, A.; Kollmeyer, T.M.; Grove, R.; Peng, S.; et al. Genomic and Phenotypic Characterization of a Broad Panel of Patient-Derived Xenografts Reflects the Diversity of Glioblastoma. Clin. Cancer Res. 2020, 26, 1094–1104. [Google Scholar] [CrossRef]
- Chakravarti, A.; Erkkinen, M.G.; Nestler, U.; Stupp, R.; Mehta, M.; Aldape, K.; Gilbert, M.R.; Black, P.M.; Loeffler, J.S. Temozolomide-mediated radiation enhancement in glioblastoma: A report on underlying mechanisms. Clin. Cancer Res. 2006, 12, 4738–4746. [Google Scholar] [CrossRef] [Green Version]
- Kil, W.J.; Cerna, D.; Burgan, W.E.; Beam, K.; Carter, D.; Steeg, P.S.; Tofilon, P.J.; Camphausen, K. In vitro and in vivo radiosensitization induced by the DNA methylating agent temozolomide. Clin. Cancer Res. 2008, 14, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Aquilina, G.; Ceccotti, S.; Martinelli, S.; Hampson, R.; Bignami, M. N-(2-chloroethyl)-N′-cyclohexyl-N-nitrosourea sensitivity in mismatch repair-defective human cells. Cancer Res. 1998, 58, 135–141. [Google Scholar]
- Alnahhas, I.; Rayi, A.; Ong, S.; Giglio, P.; Puduvalli, V. Management of gliomas in patients with Lynch syndrome. Neuro-Oncology 2021, 23, 167–168. [Google Scholar] [CrossRef]
- Ollier, E.; Mazzocco, P.; Ricard, D.; Kaloshi, G.; Idbaih, A.; Alentorn, A.; Psimaras, D.; Honnorat, J.; Delattre, J.Y.; Grenier, E.; et al. Analysis of temozolomide resistance in low-grade gliomas using a mechanistic mathematical model. Fundam. Clin. Pharmacol. 2017, 31, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Barthel, F.P.; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; Anderson, K.J.; Abiola, O.; Aldape, K.; Alfaro, K.D.; et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019, 576, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G. Glioma through the looking GLASS: Molecular evolution of diffuse gliomas and the Glioma Longitudinal Analysis Consortium. Neuro-Oncology 2018, 20, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Varn, F.S.; Johnson, K.C.; Martinek, J.; Huse, J.T.; Nasrallah, M.P.; Wesseling, P.; Cooper, L.A.D.; Malta, T.M.; Wade, T.E.; Sabedot, T.S.; et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 2022, 185, 2184–2199.e2116. [Google Scholar] [CrossRef] [PubMed]
- Yeo, A.T.; Rawal, S.; Delcuze, B.; Christofides, A.; Atayde, A.; Strauss, L.; Balaj, L.; Rogers, V.A.; Uhlmann, E.J.; Varma, H.; et al. Single-cell RNA sequencing reveals evolution of immune landscape during glioblastoma progression. Nat. Immunol. 2022, 23, 971–984. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parental PDX | ID * | Therapy ** | Genes Mutated | TMB Gain *** | MSH6 † | MSH2 † | MLH1 † | PMS2 † |
|---|---|---|---|---|---|---|---|---|
| GBM6 | m4052 | RT | 20 | 2.10 | 0 | 0 | 0 | 0 |
| m4063 | 44 | 2.78 | 0 | 0 | 0 | 0 | ||
| m4066 | 21 | 1.24 | 0 | 0 | 0 | 0 | ||
| GBM12 | m6159 | 219 | 15.12 | 0 | 0 | 0 | 0 | |
| m6161 | 205 | 15.63 | 0 | 0 | 0 | 0 | ||
| GBM43 | m3087 | 48 | 4.02 | 0 | 0 | 0 | 0 | |
| m3098 | 27 | 1.30 | 0 | 0 | 0 | 0 | ||
| GBM6 | m4056 | TMZ ×1 | 297 | 13.60 | 0 | 0 | 0 | 0 |
| m4057 | 249 | 9.80 | 0 | 0 | 0 | 0 | ||
| m4082 | 122 | 5.98 | 0 | 0 | 0 | 0 | ||
| m4883 | 280 | 17.59 | 0 | 0 | 0 | 0 | ||
| GBM12 | m2656 | 288 | 12.38 | 0 | 0 | 0 | 0 | |
| m2657 | 302 | 16.67 | 0 | 0 | 0 | 0 | ||
| GBM6 | m3395 | RT+TMZ ×1 | 78 | 3.69 | 2 | 0 | 0 | 0 |
| m4051 | 473 | 20.62 | 1 | 0 | 0 | 0 | ||
| GBM12 | m2671 | 463 | 22.36 | 2 | 0 | 0 | 0 | |
| m2685 | 359 | 15.28 | 0 | 0 | 0 | 0 | ||
| m2511 | RT+TMZ ×2 | 282 | 17.86 | 0 | 0 | 1 | 0 | |
| GBM6 | m3378 | RT+TMZ ×3 | 1403 | 65.98 | 1 | 0 | 0 | 1 |
| GBM12 | m4829 | TMZ ×4 | 2238 | 105.78 | 2 | 1 | 0 | 0 |
| m4834 | 3021 | 151.85 | 3 | 1 | 0 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCord, M.; Bartom, E.; Burdett, K.; Baran, A.; Eckerdt, F.D.; Balyasnikova, I.V.; McCortney, K.; Sears, T.; Cheng, S.-Y.; Sarkaria, J.N.; et al. Modeling Therapy-Driven Evolution of Glioblastoma with Patient-Derived Xenografts. Cancers 2022, 14, 5494. https://doi.org/10.3390/cancers14225494
McCord M, Bartom E, Burdett K, Baran A, Eckerdt FD, Balyasnikova IV, McCortney K, Sears T, Cheng S-Y, Sarkaria JN, et al. Modeling Therapy-Driven Evolution of Glioblastoma with Patient-Derived Xenografts. Cancers. 2022; 14(22):5494. https://doi.org/10.3390/cancers14225494
Chicago/Turabian StyleMcCord, Matthew, Elizabeth Bartom, Kirsten Burdett, Aneta Baran, Frank D. Eckerdt, Irina V. Balyasnikova, Kathleen McCortney, Thomas Sears, Shi-Yuan Cheng, Jann N. Sarkaria, and et al. 2022. "Modeling Therapy-Driven Evolution of Glioblastoma with Patient-Derived Xenografts" Cancers 14, no. 22: 5494. https://doi.org/10.3390/cancers14225494