
Clinical and Molecular Features of KRAS-Mutated Lung Cancer Patients Treated with Immune Checkpoint Inhibitors
 ,
,  , , , ,
, , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Patients & Methods
2.1. Patients
2.2. Statistical Analysis
3. Results
3.1. Patient Characteristics
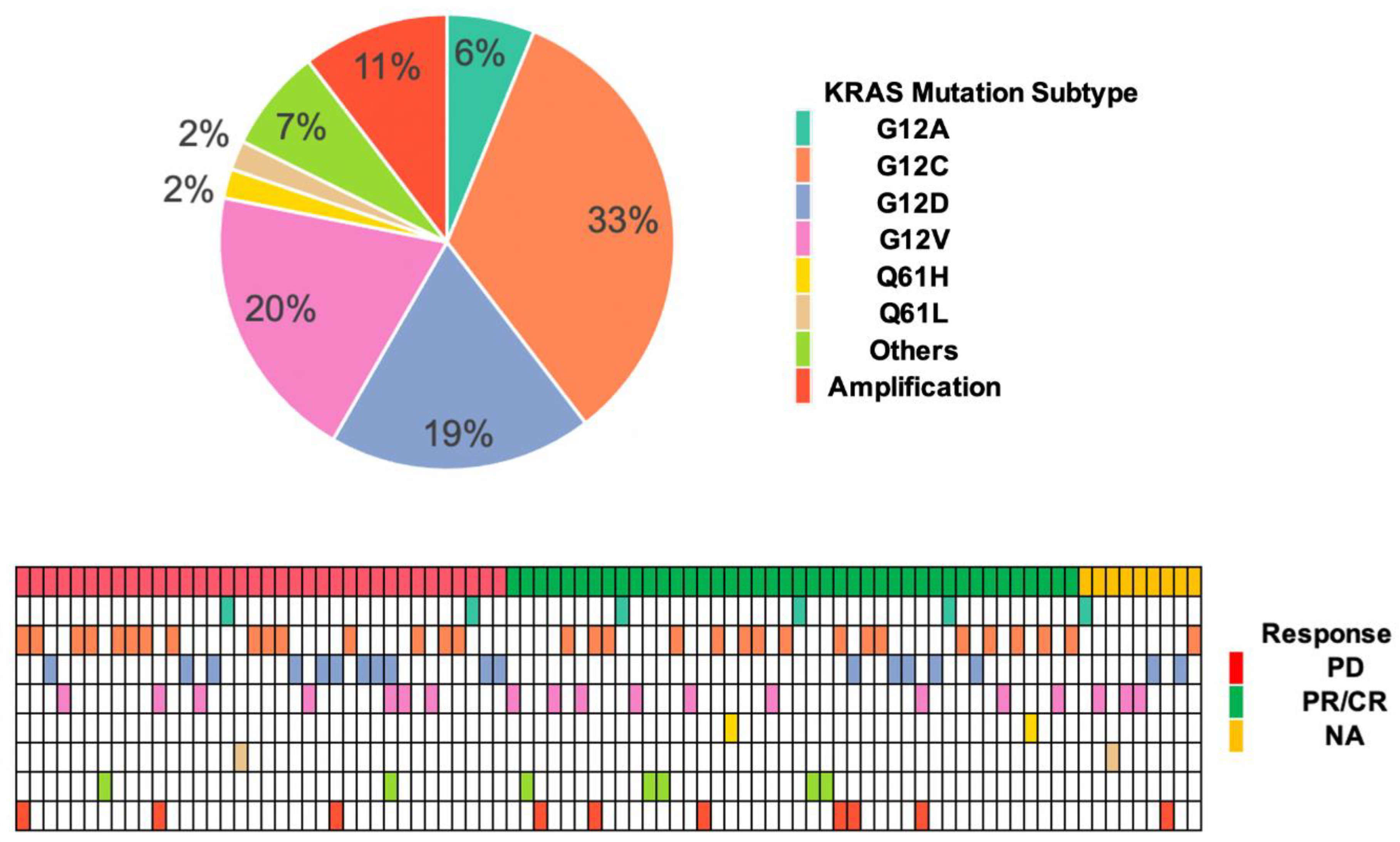
3.2. KRAS Mutation Subtypes with Responses and OS
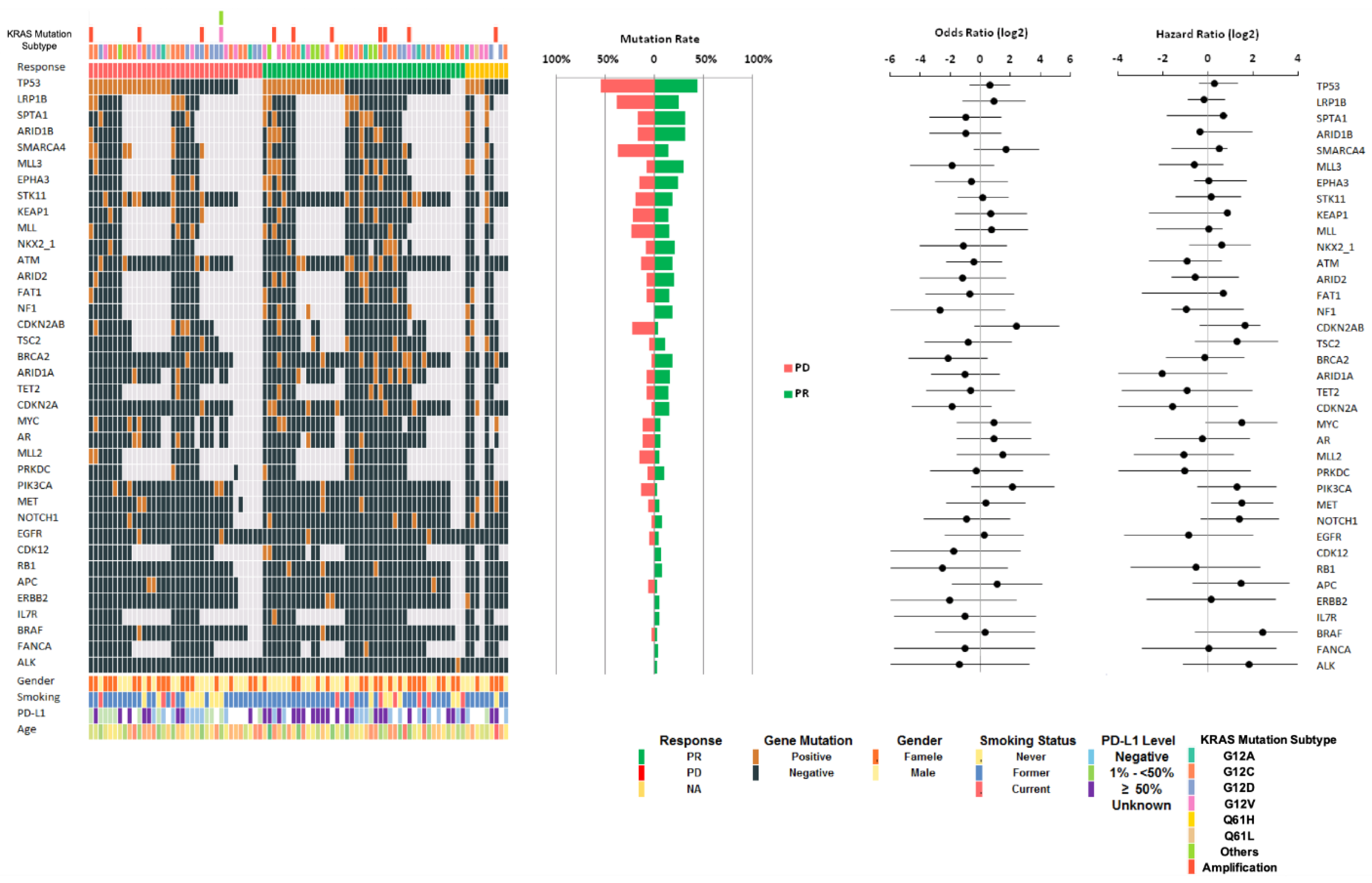
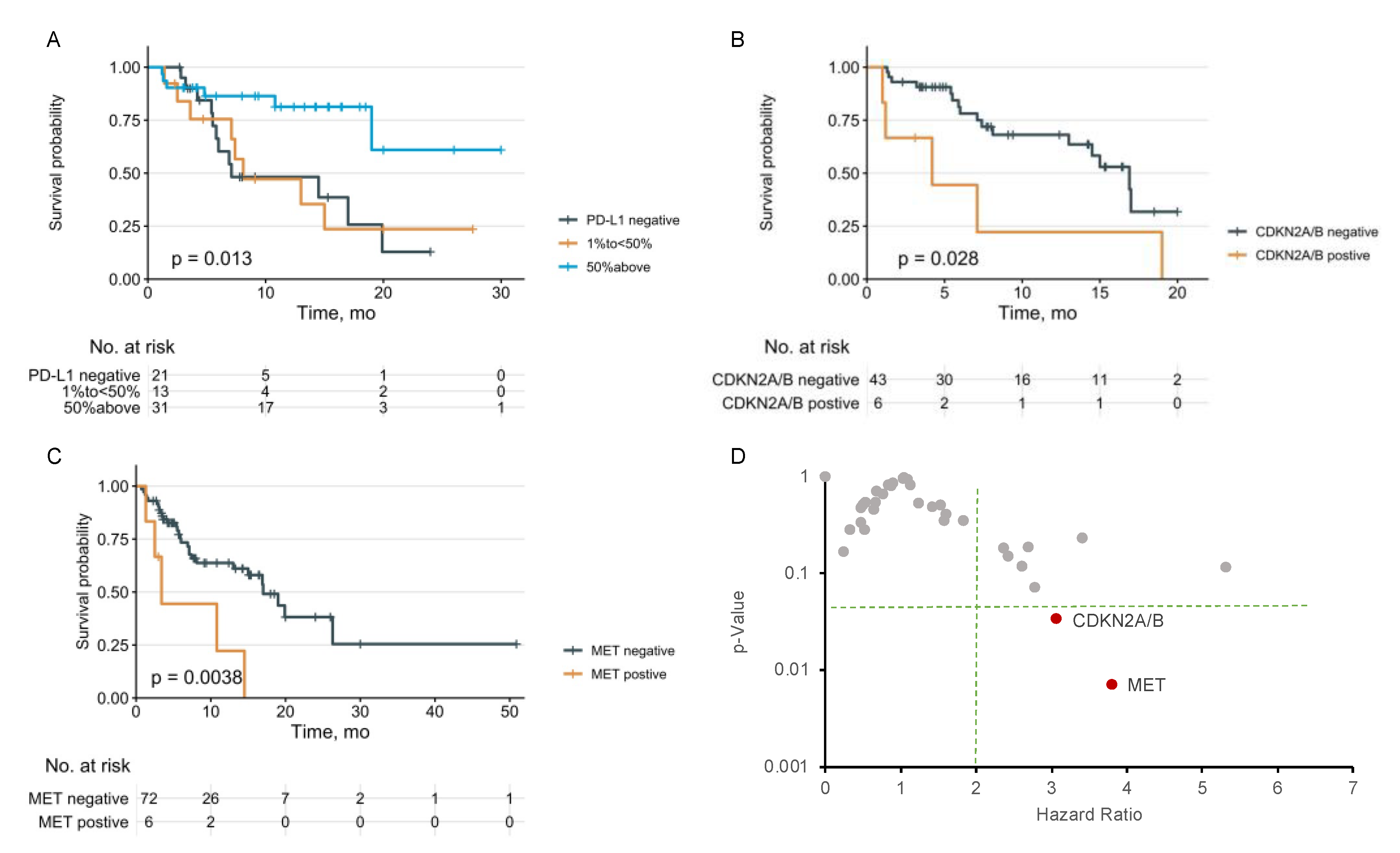
3.3. KRAS Comutations and OS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, M.L.; Sima, C.S.; Chaft, J.; Paik, P.K.; Pao, W.; Kris, M.G.; Ladanyi, M.; Riely, G.J. Association of KRAS and EGFR mutations with survival in patients with advanced lung adenocarcinomas. Cancer 2013, 119, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Slebos, R.J.; Kibbelaar, R.E.; Dalesio, O.; Kooistra, A.; Stam, J.; Meijer, C.J.; Wagenaar, S.S.; Vanderschueren, R.G.; van Zandwijk, N.; Mooi, W.J.; et al. K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N. Engl. J. Med. 1990, 323, 561–565. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stalnecker, C.A.; Der, C.J. RAS, wanted dead or alive: Advances in targeting RAS mutant cancers. Sci Signal 2020, 13, eaay6013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindan, R.; Fakih, M.; Price, T.; Falchook, G.; Desai, J.; Kuo, J.; Strickler, J.; Krauss, J.; Li, B.; Denlinger, C.; et al. Phase I study of AMG 510, a novel molecule targeting KRAS G12C mutant solid tumours. Ann. Oncol. 2019, 30, v159–v193. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Ou, S.I.; Jänne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients with Advanced KRASG12C Solid Tumors (KRYSTAL-1). J Clin Oncol 2022, Jco2102752. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R.; Pharaon, R.; Mambetsariev, I.; Nam, A.; Sattler, M. The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC). Cell. Rep. Med. 2021, 2, 100186. [Google Scholar] [CrossRef]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Lapinski, P.E.; King, P.D. Regulation of Ras signal transduction during T cell development and activation. Am. J. Clin. Exp. Immunol. 2012, 1, 147–153. [Google Scholar]
- Briere, D.M.; Li, S.; Calinisan, A.; Sudhakar, N.; Aranda, R.; Hargis, L.; Peng, D.H.; Deng, J.; Engstrom, L.D.; Hallin, J.; et al. The KRAS(G12C) Inhibitor MRTX849 Reconditions the Tumor Immune Microenvironment and Sensitizes Tumors to Checkpoint Inhibitor Therapy. Mol. Cancer Ther. 2021, 20, 975–985. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Govindan, R.; Anders, R.A.; Antonia, S.J.; Sagorsky, S.; Davies, M.J.; Dubinett, S.M.; Ferris, A.; Gandhi, L.; Garon, E.B.; et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2018, 6, 75. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Mansfield, A.S.; Szczesna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gumus, M.; Mazieres, J.; Hermes, B.; Cay Senler, F.; Csoszi, T.; Fulop, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. New Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghaei, H.; Langer, C.J.; Gadgeel, S.; Papadimitrakopoulou, V.A.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. 24-Month Overall Survival from KEYNOTE-021 Cohort G: Pemetrexed and Carboplatin with or without Pembrolizumab as First-Line Therapy for Advanced Nonsquamous Non-Small Cell Lung Cancer. J. Thorac.Oncol.: Off. Publ. Int. Assoc. Study Lung Cancer 2018, 14, 124–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalvala, A.; Wallet, P.; Yang, L.; Wang, C.; Li, H.; Nam, A.; Nathan, A.; Mambetsariev, I.; Poroyko, V.; Gao, H.; et al. Phenotypic Switching of Naïve T Cells to Immune-Suppressive Treg-Like Cells by Mutant KRAS. J. Clin. Med. 2019, 8, 1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Best, S.A.; Ding, S.; Kersbergen, A.; Dong, X.; Song, J.Y.; Xie, Y.; Reljic, B.; Li, K.; Vince, J.E.; Rathi, V.; et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat. Commun. 2019, 10, 4190. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Yonesaka, K.; Teramura, T.; Takehara, T.; Kato, R.; Sakai, H.; Haratani, K.; Tanizaki, J.; Kawakami, H.; Hayashi, H.; et al. KRAS Inhibitor Resistance in MET-Amplified KRAS (G12C) Non-Small Cell Lung Cancer Induced By RAS- and Non-RAS-Mediated Cell Signaling Mechanisms. Clin. Cancer Res. 2021, 27, 5697–5707. [Google Scholar] [CrossRef]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef]
- Jeanson, A.; Tomasini, P.; Souquet-Bressand, M.; Brandone, N.; Boucekine, M.; Grangeon, M.; Chaleat, S.; Khobta, N.; Milia, J.; Mhanna, L.; et al. Efficacy of Immune Checkpoint Inhibitors in KRAS-Mutant Non-Small Cell Lung Cancer (NSCLC). J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, 1095–1101. [Google Scholar] [CrossRef]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y.; et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients with Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Biton, J.; Mansuet-Lupo, A.; Pecuchet, N.; Alifano, M.; Ouakrim, H.; Arrondeau, J.; Boudou-Rouquette, P.; Goldwasser, F.; Leroy, K.; Goc, J.; et al. TP53, STK11, and EGFR Mutations Predict Tumor Immune Profile and the Response to Anti-PD-1 in Lung Adenocarcinoma. Clin. Cancer Res. 2018, 24, 5710–5723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Xie, Z.; Liu, S.Y.; Tu, H.Y.; Chen, H.J.; Sun, Y.L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [Green Version]
- Blons, H.; Garinet, S.; Laurent-Puig, P.; Oudart, J.B. Molecular markers and prediction of response to immunotherapy in non-small cell lung cancer, an update. J. Thorac. Dis. 2019, 11, S25–S36. [Google Scholar] [CrossRef]
- Amanam, I.; Mambetsariev, I.; Gupta, R.; Achuthan, S.; Wang, Y.; Pharaon, R.; Massarelli, E.; Koczywas, M.; Reckamp, K.; Salgia, R. Role of immunotherapy and co-mutations on KRAS-mutant non-small cell lung cancer survival. J. Thorac. Dis. 2020, 12, 5086–5095. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Zhao, D.; Mambetsariev, I.; Li, H.; Chen, C.; Fricke, J.; Fann, P.; Kulkarni, P.; Xing, Y.; Lee, P.P.; Bild, A.; et al. Association of molecular characteristics with survival in advanced non-small cell lung cancer patients treated with checkpoint inhibitors. Lung Cancer 2020, 146, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Gutiontov, S.I.; Turchan, W.T.; Spurr, L.F.; Rouhani, S.J.; Chervin, C.S.; Steinhardt, G.; Lager, A.M.; Wanjari, P.; Malik, R.; Connell, P.P.; et al. CDKN2A loss-of-function predicts immunotherapy resistance in non-small cell lung cancer. Sci. Rep. 2021, 11, 20059. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R. MET in Lung Cancer: Biomarker Selection Based on Scientific Rationale. Mol. Cancer Ther. 2017, 16, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabari, J.K.; Leonardi, G.C.; Shu, C.A.; Umeton, R.; Montecalvo, J.; Ni, A.; Chen, R.; Dienstag, J.; Mrad, C.; Bergagnini, I.; et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann.Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 2085–2091. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann.Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Galimi, F.; Cottone, E.; Vigna, E.; Arena, N.; Boccaccio, C.; Giordano, S.; Naldini, L.; Comoglio, P.M. Hepatocyte growth factor is a regulator of monocyte-macrophage function. J. Immunol. 2001, 166, 1241–1247. [Google Scholar] [CrossRef] [Green Version]
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Thompson, A.A.; Wauters, E.; Walmsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015, 522, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Titmarsh, H.F.; O’Connor, R.; Dhaliwal, K.; Akram, A.R. The Emerging Role of the c-MET-HGF Axis in Non-small Lung Cancer Tumor Immunology and Immunotherapy. Front. Oncol. 2020, 10, 54. [Google Scholar] [CrossRef] [Green Version]
- Papaccio, F.; Della Corte, C.M.; Viscardi, G.; Di Liello, R.; Esposito, G.; Sparano, F.; Ciardiello, F.; Morgillo, F. HGF/MET and the Immune System: Relevance for Cancer Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3595. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Nagelberg, A.; Chow, J.L.; Chen, Y.T.; Michalchuk, Q.; Somwar, R.; Lockwood, W.W. MET Exon 14 Splice-Site Mutations Preferentially Activate KRAS Signaling to Drive Tumourigenesis. Cancers 2022, 14, 1378. [Google Scholar] [CrossRef]
- Bahcall, M.; Awad, M.M.; Sholl, L.M.; Wilson, F.H.; Xu, M.; Wang, S.; Palakurthi, S.; Choi, J.; Ivanova, E.V.; Leonardi, G.C.; et al. Amplification of Wild-type KRAS Imparts Resistance to Crizotinib in MET Exon 14 Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 5963–5976. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.; Hensing, T.; Malik, R.; Salgia, R. Prognostic and Predictive Value in KRAS in Non-Small-Cell Lung Cancer: A Review. JAMA Oncol. 2016, 2, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Burkhart, D.L.; Haigis, K.M. Classification of KRAS-Activating Mutations and the Implications for Therapeutic Intervention. Cancer Discov. 2022, 12, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, G.A.; Der, C.J. RAS Mutations Are Not Created Equal. Cancer Discov. 2019, 9, 696–698. [Google Scholar] [CrossRef] [Green Version]
- Ricciuti, B.; Son, J.; Okoro, J.J.; Mira, A.; Patrucco, E.; Eum, Y.; Wang, X.; Paranal, R.; Wang, H.; Lin, M.; et al. Comparative Analysis and Isoform-Specific Therapeutic Vulnerabilities of KRAS Mutations in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2022, 28, 1640–1650. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Chhoeu, C.; Li, J.; Price, K.S.; Kiedrowski, L.A.; Hutchins, J.L.; Hardin, A.I.; Wei, Z.; Hong, F.; Bahcall, M.; et al. Silent mutations reveal therapeutic vulnerability in RAS Q61 cancers. Nature 2022, 603, 335–342. [Google Scholar] [CrossRef]
- Cui, W.; Franchini, F.; Alexander, M.; Officer, A.; Wong, H.L.; IJzerman, M.J.; Desai, J.; Solomon, B.J. Assessing the significance of KRAS G12C mutation: Clinicopathologic features, treatments, and survival outcomes in a real-world KRAS mutant non-small cell lung cancer cohort. J. Clin. Oncol. 2020, 38, e19324. [Google Scholar] [CrossRef]
- Tamiya, Y.; Zenke, Y.; Matsumoto, S.; Furuya, N.; Sakamoto, T.; Kato, T.; Nishino, K.; Shingyoji, M.; Miyamoto, S.; Shirakawa, C.; et al. Therapeutic impact of mutation subtypes and concomitant STK11 mutations in KRAS–mutated non-small cell lung cancer (NSCLC): A result of nationwide genomic screening project (LC-SCRUM-Japan). J. Clin. Oncol. 2020, 38, 9589. [Google Scholar] [CrossRef]
- Arbour, K.C.; Rizvi, H.; Plodkowski, A.J.; Hellmann, M.D.; Knezevic, A.; Heller, G.; Yu, H.A.; Ladanyi, M.; Kris, M.G.; Arcila, M.E.; et al. Treatment Outcomes and Clinical Characteristics of Patients with KRAS-G12C-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 2209–2215. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | No. of Patients (n = 87) | CR/PR (n = 42) | PD (n = 36) | p-Value b |
|---|---|---|---|---|
| Age, years, at ICIs | ||||
| <70 | 49 (56.3%) | 24 (57%) | 20 (56%) | 1 |
| ≥70 | 38 (43.7%) | 18 (43%) | 16 (44%) | |
| Sex | ||||
| Women | 42 (48.3%) | 20 (47%) | 18 (50%) | 0.82 |
| Men | 45 (51.7%) | 22 (53%) | 18 (50%) | |
| Smoking status | ||||
| Current | 10 (11.5%) | 7 (17%) | 3 (8%) | 0.33 |
| Former | 61 (70.1%) | 29 (69%) | 24 (67%) | |
| Never | 16 (18.4%) | 6 (14%) | 9 (25%) | |
| Histology | ||||
| Lung adenocarcinoma | 83 (95.4%) | 40 (95.2%) | 34 (94%) | 0.99 |
| Lung squamous | 2 (2.3%) | 1 (2.4%) | 1 (3%) | |
| Others a | 2 (2.3%) | 1 (2.4%) | 1 (3%) | |
| TP53 | ||||
| Positive | 38 (48.1%) c | 17 (44%) | 17 (55%) | 0.49 |
| Negative | 41 (51.9%) c | 22 (56%) | 14 (45%) | |
| Total tested | 79 | 39 | 31 | |
| PD-L1 | ||||
| Negative | 21 (32.3%) c | 11 (32%) | 8 (30%) | 0.001 |
| 1–<50% | 13 (20%) c | 1 (6%) | 11 (40%) | |
| ≥50% | 31 (47.7%) c | 21 (62%) | 8 (30%) | |
| Total tested | 65 | 33 | 27 |
| KRAS GAs | No. (n = 87) | CR/PR (n = 42) | PD (n = 36) | OR (95% CI) | OR p-Value | OS Association HR (95% CI) | HR p-Value |
|---|---|---|---|---|---|---|---|
| G12C | 32 (36.8%) | 16 | 15 | 0.86 (0.35–2.15) | 0.748 | 1.00 (0.52–1.93) | 0.997 |
| G12V | 19 (21.9%) | 9 | 7 | 1.13 (0.37–3.53) | 0.829 | 1.94 (0.95–3.96) | 0.068 |
| G12D | 18 (20.7%) | 5 | 11 | 0.31 (0.09–0.95) | 0.048 * | 0.53 (0.21–1.36) | 0.185 |
| G12A | 6 (6.9%) | 3 | 2 | 1.31 (0.21–10.37) | 0.776 | 1.09 (0.38–3.09) | 0.875 |
| G12R | 3 (3.5%) | 1 | 1 | 0.85 (0.03–22.12) | 0.912 | 0 (0–inf) | 0.997 |
| Q61H | 2 (2.3%) | 2 | 0 | NA | 0.992 | 0.34 (0.04–2.67) | 0.306 |
| Q61L a | 2 (2.3%) | 0 | 1 | NA | 0.991 | 7.75 (1.71–35) | 0.008 ** |
| G12S | 1 (1.1%) | 1 | 0 | NA | 0.992 | 0 (0–inf) | 0.997 |
| G13D | 1 (1.1%) | 1 | 0 | NA | 0.992 | 0 (0–inf) | 0.996 |
| G13R | 1 (1.1%) | 0 | 1 | NA | 0.991 | 0 (0–inf) | 0.997 |
| K117N | 1 (1.1%) | 1 | 0 | NA | 0.992 | 2.41 (0.33–17.82) | 0.338 |
| KRAS a amp | 10 (11.5%) | 6 | 3 | 1.83 (0.45–9.24) | 0.417 | 0.59 (0.18–1.92) | 0.381 |
| Other b | 1 (1.1%) | 0 | 1 | NA | 0.991 | 0 (0–inf) | 0.997 |
| Risk Factors | HR (95% CI) | p-Values 1 |
|---|---|---|
| PD-L1 | ||
| Negative or less than 50% | Reference | |
| 50% above | 0.12 (0.03–0.57) | 0.007 ** |
| CDKN2A/B Loss | ||
| Negative | Reference | |
| Positive | 9.44 (1.90–46.93) | 0.006 ** |
| MET Mutation | ||
| Negative | Reference | |
| Positive | 3.46 (0.55–21.91) | 0.186 |
| KRAS G12V Mutation | ||
| Negative | Reference | |
| Positive | 4.13 (0.98–17.50) | 0.053 |
| KRAS G12D Mutation | ||
| Negative | Reference | |
| Positive | 0.09 (0.01–0.68) | 0.02 * |
| Age | ||
| <70 | Reference | |
| >=70 | 1.26 (0.35–5.25) | 0.66 |
| Sex | ||
| Female | Reference | |
| Male | 1.53 (0.45–5.14) | 0.49 |
| Smoking Status | ||
| Never | Reference | |
| Current | 0.38 (0.04–3.25) | 0.38 |
| Former | 0.33 (0.07–1.47) | 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, D.; Li, H.; Mambetsariev, I.; Mirzapoiazova, T.; Chen, C.; Fricke, J.; Kulkarni, P.; Villaflor, V.; Arvanitis, L.; Hamilton, S.; et al. Clinical and Molecular Features of KRAS-Mutated Lung Cancer Patients Treated with Immune Checkpoint Inhibitors. Cancers 2022, 14, 4933. https://doi.org/10.3390/cancers14194933
Zhao D, Li H, Mambetsariev I, Mirzapoiazova T, Chen C, Fricke J, Kulkarni P, Villaflor V, Arvanitis L, Hamilton S, et al. Clinical and Molecular Features of KRAS-Mutated Lung Cancer Patients Treated with Immune Checkpoint Inhibitors. Cancers. 2022; 14(19):4933. https://doi.org/10.3390/cancers14194933
Chicago/Turabian StyleZhao, Dan, Haiqing Li, Isa Mambetsariev, Tamara Mirzapoiazova, Chen Chen, Jeremy Fricke, Prakash Kulkarni, Victoria Villaflor, Leonidas Arvanitis, Stanley Hamilton, and et al. 2022. "Clinical and Molecular Features of KRAS-Mutated Lung Cancer Patients Treated with Immune Checkpoint Inhibitors" Cancers 14, no. 19: 4933. https://doi.org/10.3390/cancers14194933