

HPV16 E7 Nucleotide Variants Found in Cancer-Free Subjects Affect E7 Protein Expression and Transformation
 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction and antiHPV16 E7 Antibodies
2.2. Mutant Construction
2.3. Cell Culture and Transfection
2.4. Western Blotting
2.5. Determination of Morphological Transformation
2.6. Anchorage-Independent Cell Growth
2.7. Wound Healing Assay
2.8. Statistical Analysis
3. Results
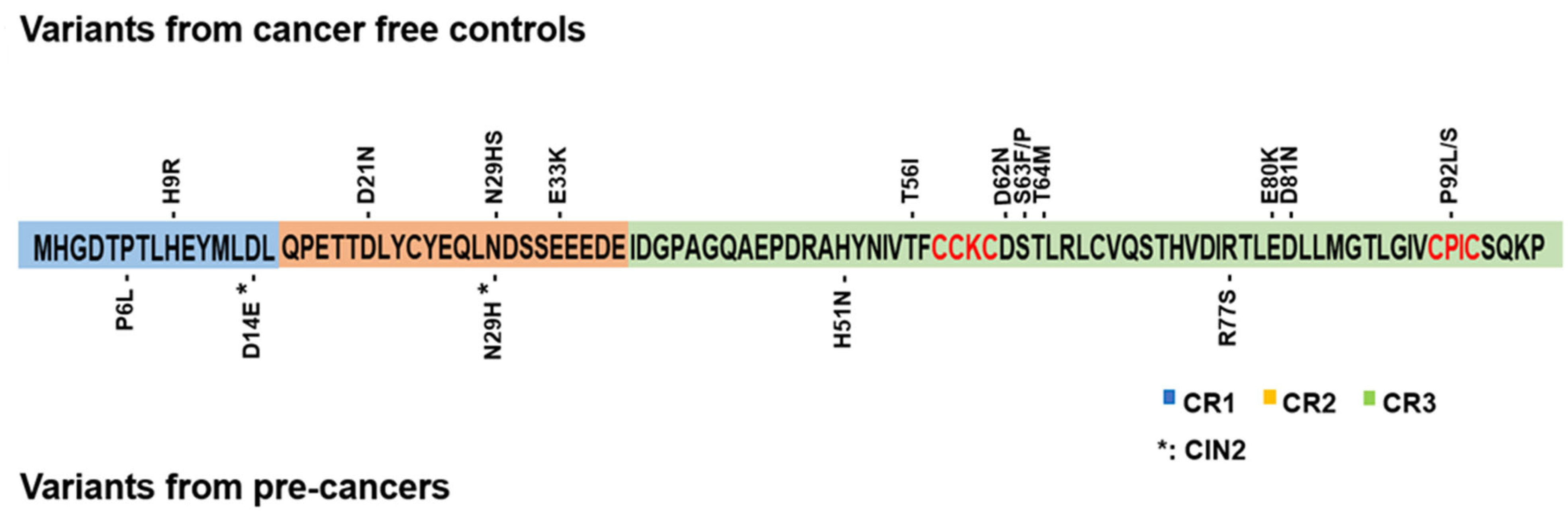
3.1. HPV16 E7 Mutagenesis and Protein Expression
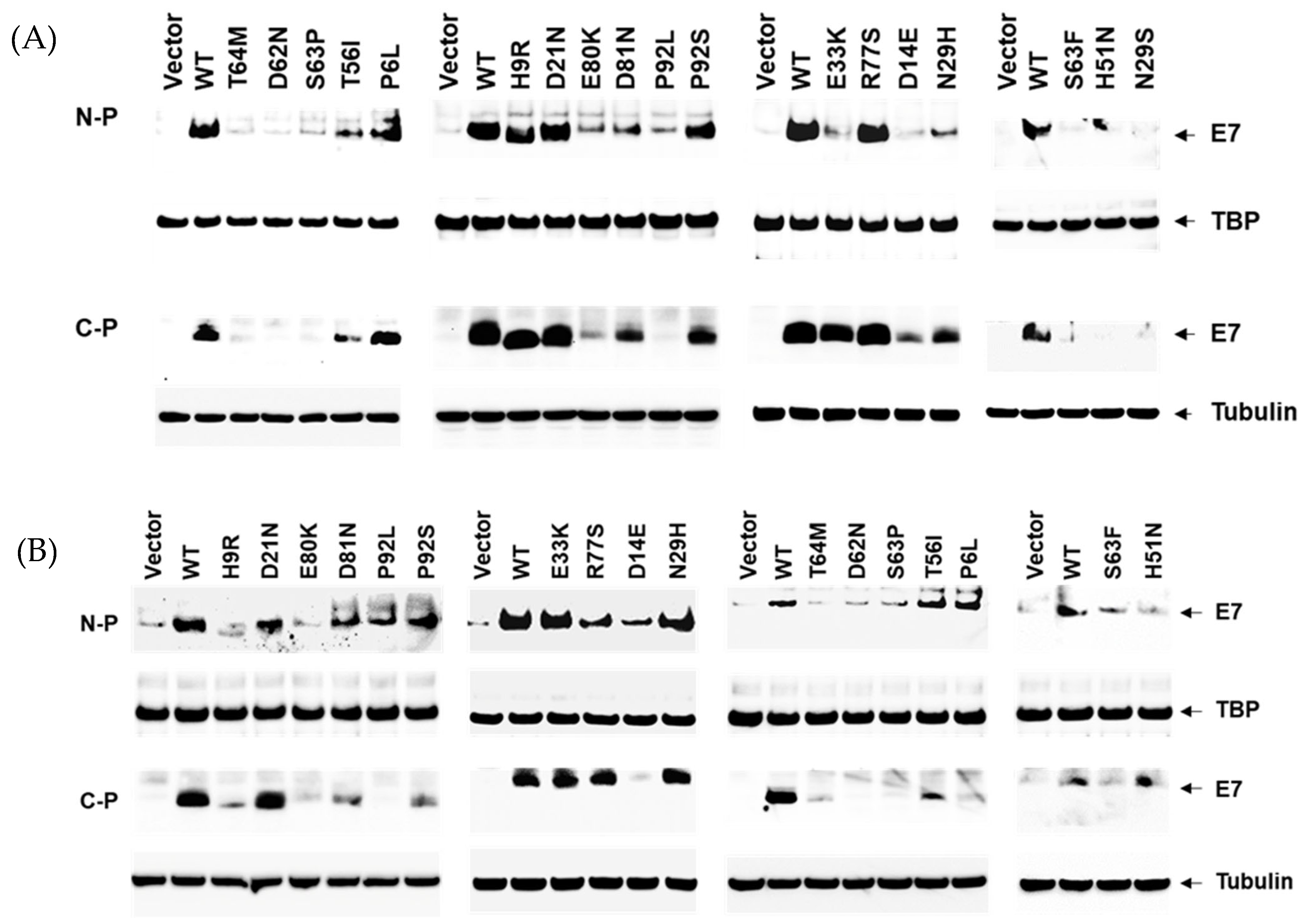
3.2. Reduced Protein Expression Levels of HPV16 E7 Variants
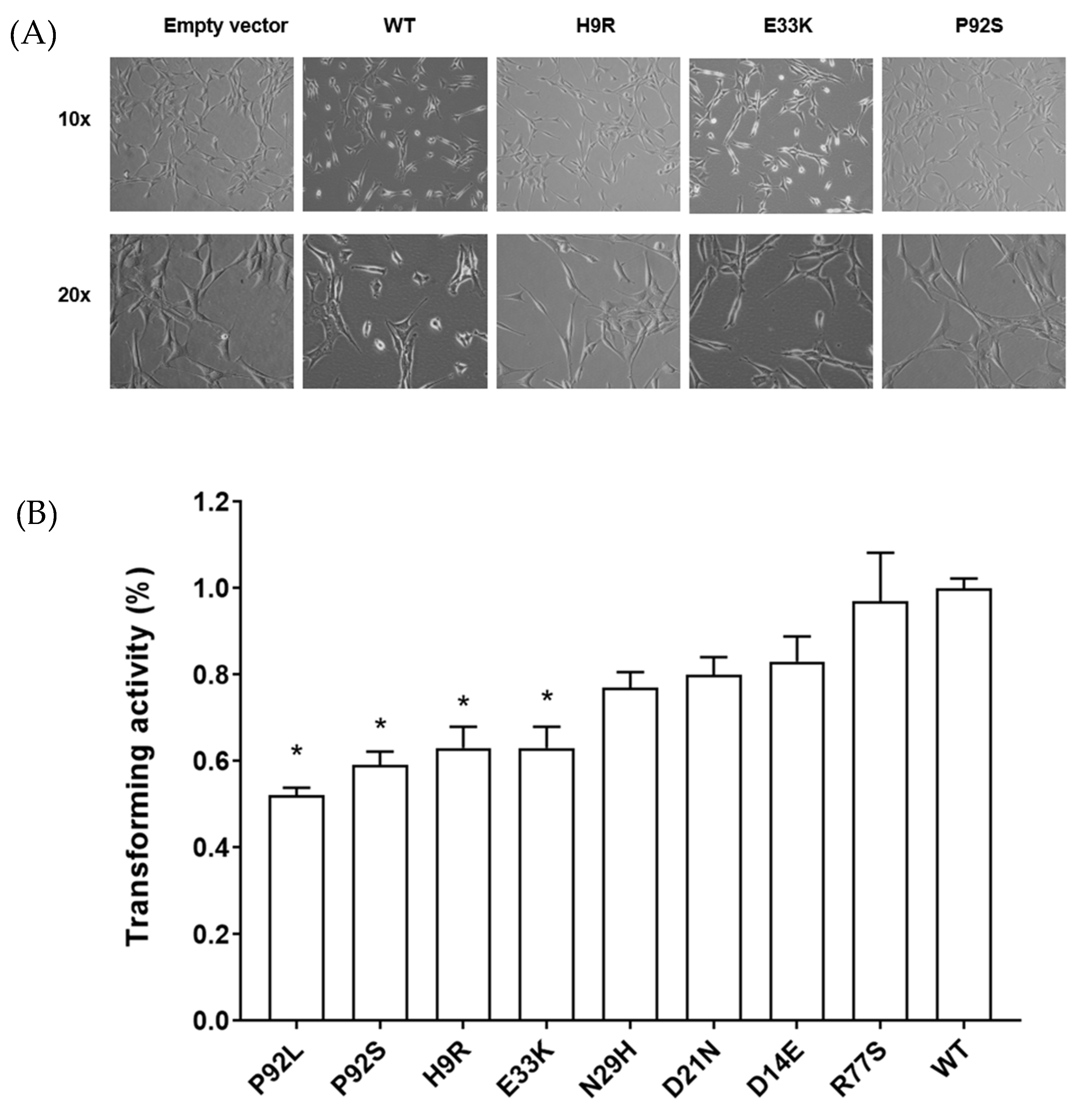
3.3. HPV16 E7 Variants Not Associated with Cancer Have a More Normal Cell Morphology and Reduced Anchorage-Independent Growth in NIH3T3 Cells
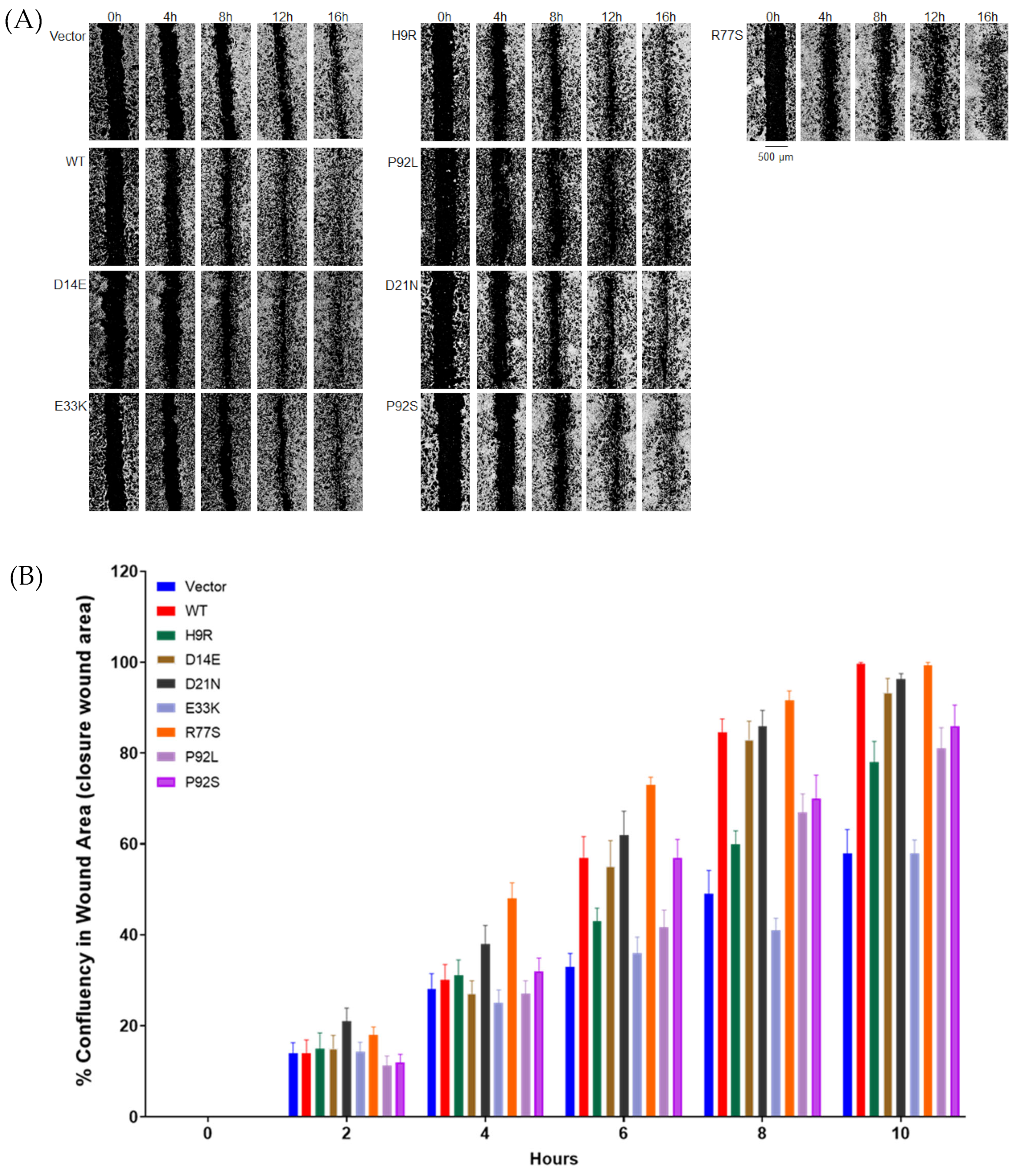
3.4. HPV16 E7 Variants Display Decreased NIH3T3 Cell Migration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schiffman, M.; Castle, P.E. The promise of global cervical-cancer prevention. N. Engl. J. Med. 2005, 353, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.M.; Mehta, K.P.; Cruz, L.; Meyers, C.; Laimins, L.A. The E7 open reading frame acts in cis and in trans to mediate differentiation-dependent activities in the human papillomavirus type 16 life cycle. J. Virol. 2011, 85, 8852–8862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stubenrauch, F.; Laimins, L.A. Human papillomavirus life cycle: Active and latent phases. Semin. Cancer Biol. 1999, 9, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Massimi, P.; Banks, L. Repression of p53 transcriptional activity by the HPV E7 proteins. Virology 1997, 227, 255–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Nielsen, S.J.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999, 18, 2449–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodily, J.M.; Mehta, K.P.; Laimins, L.A. Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. 2011, 71, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- Berezutskaya, E.; Bagchi, S. The human papillomavirus E7 oncoprotein functionally interacts with the S4 subunit of the 26 S proteasome. J. Biol. Chem. 1997, 272, 30135–30140. [Google Scholar] [CrossRef] [Green Version]
- White, E.A.; Sowa, M.E.; Tan, M.J.; Jeudy, S.; Hayes, S.D.; Santha, S.; Munger, K.; Harper, J.W.; Howley, P.M. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc. Natl. Acad. Sci. USA 2012, 109, E260–E267. [Google Scholar] [CrossRef] [Green Version]
- Avvakumov, N.; Torchia, J.; Mymryk, J.S. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene 2003, 22, 3833–3841. [Google Scholar] [CrossRef]
- Helin, K.; Lees, J.A.; Vidal, M.; Dyson, N.; Harlow, E.; Fattaey, A. A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell 1992, 70, 337–350. [Google Scholar] [CrossRef]
- Dimova, D.K.; Dyson, N.J. The E2F transcriptional network: Old acquaintances with new faces. Oncogene 2005, 24, 2810–2826. [Google Scholar] [CrossRef] [Green Version]
- Pim, D.; Banks, L. Interaction of viral oncoproteins with cellular target molecules: Infection with high-risk vs. low-risk human papillomaviruses. APMIS 2010, 118, 471–493. [Google Scholar] [CrossRef]
- Phelps, W.C.; Yee, C.L.; Munger, K.; Howley, P.M. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell 1988, 53, 539–547. [Google Scholar] [CrossRef]
- Chemes, L.B.; Glavina, J.; Alonso, L.G.; Marino-Buslje, C.; De Prat-Gay, G.; Sanchez, I.E. Sequence evolution of the intrinsically disordered and globular domains of a model viral oncoprotein. PLoS ONE 2012, 7, e47661. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Clements, A.; Zhao, K.; Marmorstein, R. Structure of the human Papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J. Biol. Chem. 2006, 281, 578–586. [Google Scholar] [CrossRef] [Green Version]
- Ohlenschlager, O.; Seiboth, T.; Zengerling, H.; Briese, L.; Marchanka, A.; Ramachandran, R.; Baum, M.; Korbas, M.; Meyer-Klaucke, W.; Durst, M.; et al. Solution structure of the partially folded high-risk human papilloma virus 45 oncoprotein E7. Oncogene 2006, 25, 5953–5959. [Google Scholar] [CrossRef] [Green Version]
- Kanda, T.; Furuno, A.; Yoshiike, K. Human papillomavirus type 16 open reading frame E7 encodes a transforming gene for rat 3Y1 cells. J. Virol. 1988, 62, 610–613. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Doniger, J.; DiPaolo, J.A.; Lowy, D.R. The E7 open reading frame of human papillomavirus type 16 encodes a transforming gene. Oncogene Res. 1988, 3, 167–175. [Google Scholar]
- Mirabello, L.; Yeager, M.; Yu, K.; Clifford, G.M.; Xiao, Y.; Zhu, B.; Cullen, M.; Boland, J.F.; Wentzensen, N.; Nelson, C.W.; et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 2017, 170, 1164–1174.e6. [Google Scholar] [CrossRef] [Green Version]
- Lou, H.; Villagran, G.; Boland, J.F.; Im, K.M.; Polo, S.; Zhou, W.; Odey, U.; Juarez-Torres, E.; Medina-Martinez, I.; Roman-Basaure, E.; et al. Genome Analysis of Latin American Cervical Cancer: Frequent Activation of the PIK3CA Pathway. Clin. Cancer Res. 2015, 21, 5360–5370. [Google Scholar] [CrossRef]
- Lou, H.; Li, H.; Yeager, M.; Im, K.; Gold, B.; Schneider, T.D.; Fraumeni, J.F., Jr.; Chanock, S.J.; Anderson, S.K.; Dean, M. Promoter variants in the MSMB gene associated with prostate cancer regulate MSMB/NCOA4 fusion transcripts. Hum. Genet. 2012, 131, 1453–1466. [Google Scholar] [CrossRef] [Green Version]
- Psyrri, A.; DeFilippis, R.A.; Edwards, A.P.; Yates, K.E.; Manuelidis, L.; DiMaio, D. Role of the retinoblastoma pathway in senescence triggered by repression of the human papillomavirus E7 protein in cervical carcinoma cells. Cancer Res. 2004, 64, 3079–3086. [Google Scholar] [CrossRef] [Green Version]
- Baker, C.C.; Phelps, W.C.; Lindgren, V.; Braun, M.J.; Gonda, M.A.; Howley, P.M. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 1987, 61, 962–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrede, D.; Tidy, J.A.; Crook, T.; Lane, D.; Vousden, K.H. Expression of RB and p53 proteins in HPV-positive and HPV-negative cervical carcinoma cell lines. Mol. Carcinog. 1991, 4, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.I.; Francis, D.A.; Karpova, A.Y.; Dowhanick, J.J.; Benson, J.D.; Howley, P.M. Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21(CIP)-dependent pathways. EMBO J. 2000, 19, 5762–5771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenison, S.A.; Yu, X.P.; Valentine, J.M.; Koutsky, L.A.; Christiansen, A.E.; Beckmann, A.M.; Galloway, D.A. Evidence of prevalent genital-type human papillomavirus infections in adults and children. J. Infect. Dis. 1990, 162, 60–69. [Google Scholar] [CrossRef]
- Tindle, R.W.; Smith, J.A.; Geysen, H.M.; Selvey, L.A.; Frazer, I.H. Identification of B epitopes in human papillomavirus type 16 E7 open reading frame protein. J. Gen. Virol. 1990, 71 Pt 6, 1347–1354. [Google Scholar] [CrossRef]
- Greenfield, I.; Nickerson, J.; Penman, S.; Stanley, M. Human papillomavirus 16 E7 protein is associated with the nuclear matrix. Proc. Natl. Acad. Sci. USA 1991, 88, 11217–11221. [Google Scholar] [CrossRef] [Green Version]
- Huh, K.W.; DeMasi, J.; Ogawa, H.; Nakatani, Y.; Howley, P.M.; Munger, K. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc. Natl. Acad. Sci. USA 2005, 102, 11492–11497. [Google Scholar] [CrossRef] [Green Version]
- Ressler, S.; Scheiden, R.; Dreier, K.; Laich, A.; Muller-Holzner, E.; Pircher, H.; Morandell, D.; Stein, I.; Viertler, H.P.; Santer, F.R.; et al. High-risk human papillomavirus E7 oncoprotein detection in cervical squamous cell carcinoma. Clin. Cancer Res. 2007, 13, 7067–7072. [Google Scholar] [CrossRef]
- Nguyen, C.L.; Munger, K. Human papillomavirus E7 protein deregulates mitosis via an association with nuclear mitotic apparatus protein 1. J. Virol. 2009, 83, 1700–1707. [Google Scholar] [CrossRef] [Green Version]
- Zatsepina, O.; Braspenning, J.; Robberson, D.; Hajibagheri, M.A.; Blight, K.J.; Ely, S.; Hibma, M.; Spitkovsky, D.; Trendelenburg, M.; Crawford, L.; et al. The human papillomavirus type 16 E7 protein is associated with the nucleolus in mammalian and yeast cells. Oncogene 1997, 14, 1137–1145. [Google Scholar] [CrossRef] [Green Version]
- Fujinaga, Y.; Okazawa, K.; Nishikawa, A.; Yamakawa, Y.; Fukushima, M.; Kato, I.; Fujinaga, K. Sequence variation of human papillomavirus type 16 E7 in preinvasive and invasive cervical neoplasias. Virus Genes 1994, 9, 85–92. [Google Scholar] [CrossRef]
- Edmonds, C.; Vousden, K.H. A point mutational analysis of human papillomavirus type 16 E7 protein. J. Virol. 1989, 63, 2650–2656. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L.; Akagi, K.; Xiao, W.; Jiang, B.; Pickard, R.K.L.; Li, J.; Swanson, B.J.; Agrawal, A.D.; Zucker, M.; Stache-Crain, B.; et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res. 2019, 29, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Lee, E.E.; Kim, J.; Yang, R.; Chamseddin, B.; Ni, C.; Gusho, E.; Xie, Y.; Chiang, C.M.; Buszczak, M.; et al. Transforming activity of an oncoprotein-encoding circular RNA from human papillomavirus. Nat. Commun. 2019, 10, 2300. [Google Scholar] [CrossRef] [Green Version]
- Androphy, E.J.; Schiller, J.T.; Lowy, D.R. Identification of the protein encoded by the E6 transforming gene of bovine papillomavirus. Science 1985, 230, 442–445. [Google Scholar] [CrossRef]
- Firzlaff, J.M.; Galloway, D.A.; Eisenman, R.N.; Luscher, B. The E7 protein of human papillomavirus type 16 is phosphorylated by casein kinase II. New Biol. 1989, 1, 44–53. [Google Scholar]
- Knapp, A.A.; McManus, P.M.; Bockstall, K.; Moroianu, J. Identification of the nuclear localization and export signals of high risk HPV16 E7 oncoprotein. Virology 2009, 383, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Zhou, J.; Wang, F.; Shi, H.; Li, Y.; Li, B. HPV-16 E6/E7 promotes cell migration and invasion in cervical cancer via regulating cadherin switch in vitro and in vivo. Arch. Gynecol. Obstet. 2015, 292, 1345–1354. [Google Scholar] [CrossRef]
- Chuerduangphui, J.; Pientong, C.; Chaiyarit, P.; Patarapadungkit, N.; Chotiyano, A.; Kongyingyoes, B.; Promthet, S.; Swangphon, P.; Wongjampa, W.; Ekalaksananan, T. Effect of human papillomavirus 16 oncoproteins on oncostatin M upregulation in oral squamous cell carcinoma. Med. Oncol. 2016, 33, 83. [Google Scholar] [CrossRef]
- Todorovic, B.; Massimi, P.; Hung, K.; Shaw, G.S.; Banks, L.; Mymryk, J.S. Systematic analysis of the amino acid residues of human papillomavirus type 16 E7 conserved region 3 involved in dimerization and transformation. J. Virol. 2011, 85, 10048–10057. [Google Scholar] [CrossRef] [Green Version]
- Caldeira, S.; De Villiers, E.M.; Tommasino, M. Human papillomavirus E7 proteins stimulate proliferation independently of their ability to associate with retinoblastoma protein. Oncogene 2000, 19, 821–826. [Google Scholar] [CrossRef] [Green Version]
- Lam, E.W.; Morris, J.D.; Davies, R.; Crook, T.; Watson, R.J.; Vousden, K.H. HPV16 E7 oncoprotein deregulates B-myb expression: Correlation with targeting of p107/E2F complexes. EMBO J. 1994, 13, 871–878. [Google Scholar] [CrossRef]
- Storey, A.; Pim, D.; Murray, A.; Osborn, K.; Banks, L.; Crawford, L. Comparison of the in vitro transforming activities of human papillomavirus types. EMBO J. 1988, 7, 1815–1820. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variants | Original Codon | Mutant Codon | Nucleotide Change | Control (No.) | Precancer (No.) | E7 * |
|---|---|---|---|---|---|---|
| Wild type | 1 | |||||
| Control | ||||||
| H9R | CAT | CGT | A > G | 1 | 0 | 0.8 |
| D14E | GAT | GAA | T > A | 0 | 1 (CIN2 **) | 0.11 |
| D21N | GAT | AAT | G > A | 1 | 0 | 0.92 |
| N29H | AAT | CAT | A > C | 0 | 1 (CIN2 **) | 0.3 |
| N29S | AAT | AGT | A > G | 2 | 0 | 0.018 |
| E33K | GAG | AAG | G > A | 2 | 0 | 0.2 |
| T56I | ACC | ATC | C > T | 1 | 0 | 0.55 |
| D62N | GAC | AAC | G > A | 1 | 0 | 0.11 |
| S63F | TCT | TTT | C > T | 1 | 0 | 0.1 |
| S63P | TCT | CCT | T > C | 1 | 0 | 0.3 |
| T64M | ACG | ATG | C > T | 1 | 0 | 0.11 |
| E80K | GAA | AAA | G > A | 1 | 0 | 0.26 |
| D81N | GAC | AAC | G > A | 1 | 0 | 0.42 |
| P92L | CCC | CTC | C > T | 2 | 0 | 0.1 |
| P92S | CCC | TCC | C > T | 2 | 0 | 0.68 |
| Precancer | ||||||
| P6L | CCT | CTT | C > T | 1 | 1 (CIN3 **) | 1.1 |
| H51N | CAT | AAT | C > A | 6 | 3 (CIN3/AIS ***) | 0.46 |
| R77S | CGT | AGT | C > A | 1 | 1 (AIS) | 0.99 |
| Construct | Cervical Disease | HPV16 Clearance | Function Domain | Nuclear Protein | Transforming Activity (% Growth in Soft Agar) | Wound Healing |
|---|---|---|---|---|---|---|
| Vector | + | |||||
| D81N | Control | CR3 | ++ | NA | NA | |
| E80K | Control | CR3 | ++ | NA | NA | |
| P92S | Control | CR3 | ++ | ++ | ++ | |
| S63F | Control | CR3 | + | NA | NA | |
| T56I | Control | CR3 | ++ | NA | NA | |
| T64M | Control | CR3 | + | NA | NA | |
| D21N | Control | yes | CR2 | +++ | +++ | +++ |
| D62N | Control | CR3 | + | NA | NA | |
| P92L | Control | yes | CR3 | + | ++ | ++ |
| S63P | Control | yes | CR3 | + | NA | NA |
| E33K | Control | yes | CR2 | + | ++ | + |
| H9R | Control | CR1 | +++ | ++ | ++ | |
| N29S | Control, CIN2 | yes (both) | CR2 | + | NA | NA |
| N29H | CIN2 | CR2 | ++ | +++ | NA | |
| D14E | CIN2 | CR1 | ++ | +++ | +++ | |
| H51N * | CIN3/AIS | CR3 | ++ | NA | NA | |
| P6L | CIN3 | CR1 | ++++ | NA | NA | |
| R77S | AIS | CR3 | ++++ | ++++ | ++++ | |
| Wild type | ++++ | ++++ | ++++ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lou, H.; Boland, J.F.; Li, H.; Burk, R.; Yeager, M.; Anderson, S.K.; Wentzensen, N.; Schiffman, M.; Mirabello, L.; Dean, M. HPV16 E7 Nucleotide Variants Found in Cancer-Free Subjects Affect E7 Protein Expression and Transformation. Cancers 2022, 14, 4895. https://doi.org/10.3390/cancers14194895
Lou H, Boland JF, Li H, Burk R, Yeager M, Anderson SK, Wentzensen N, Schiffman M, Mirabello L, Dean M. HPV16 E7 Nucleotide Variants Found in Cancer-Free Subjects Affect E7 Protein Expression and Transformation. Cancers. 2022; 14(19):4895. https://doi.org/10.3390/cancers14194895
Chicago/Turabian StyleLou, Hong, Joseph F. Boland, Hongchuan Li, Robert Burk, Meredith Yeager, Stephen K. Anderson, Nicolas Wentzensen, Mark Schiffman, Lisa Mirabello, and Michael Dean. 2022. "HPV16 E7 Nucleotide Variants Found in Cancer-Free Subjects Affect E7 Protein Expression and Transformation" Cancers 14, no. 19: 4895. https://doi.org/10.3390/cancers14194895