

In Silico Strategies for Designing of Peptide Inhibitors of Oncogenic K-Ras G12V Mutant: Inhibiting Cancer Growth and Proliferation
 ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Structure Retrieval
2.2. Alanine Scanning Strategy
2.3. Residue Scan Strategy
2.4. Construction of Peptides Library
2.5. All-Atom Molecular Dynamics Simulation
2.6. Molecular Mechanics Generalized Born Surface Area (MMGBSA) Calculation
3. Results and Discussion
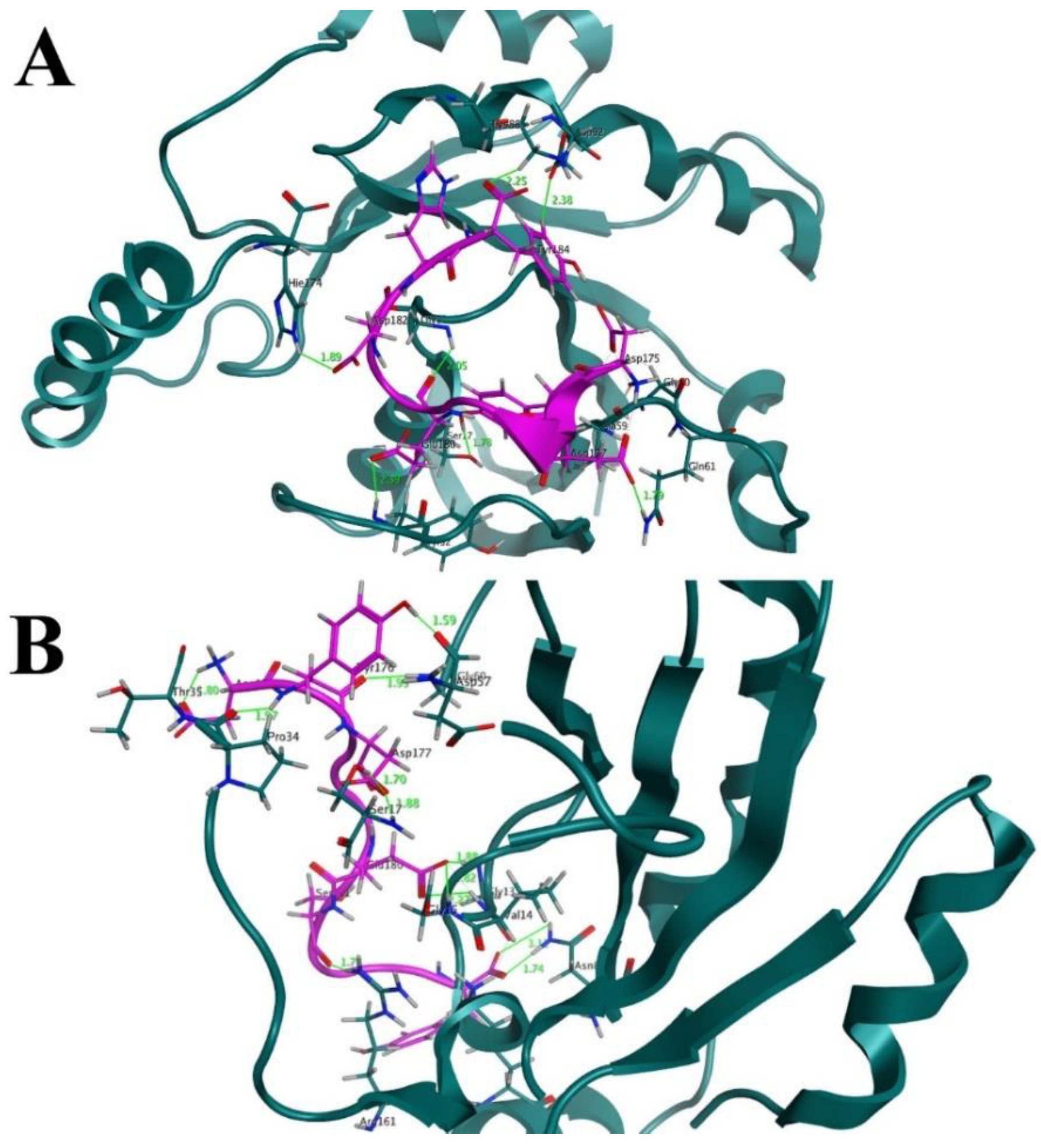
3.1. The Interface Analysis of the Mutant K-Ras/H-REV107 Complex Structure
3.2. Residue Scan to Design a Peptide Library
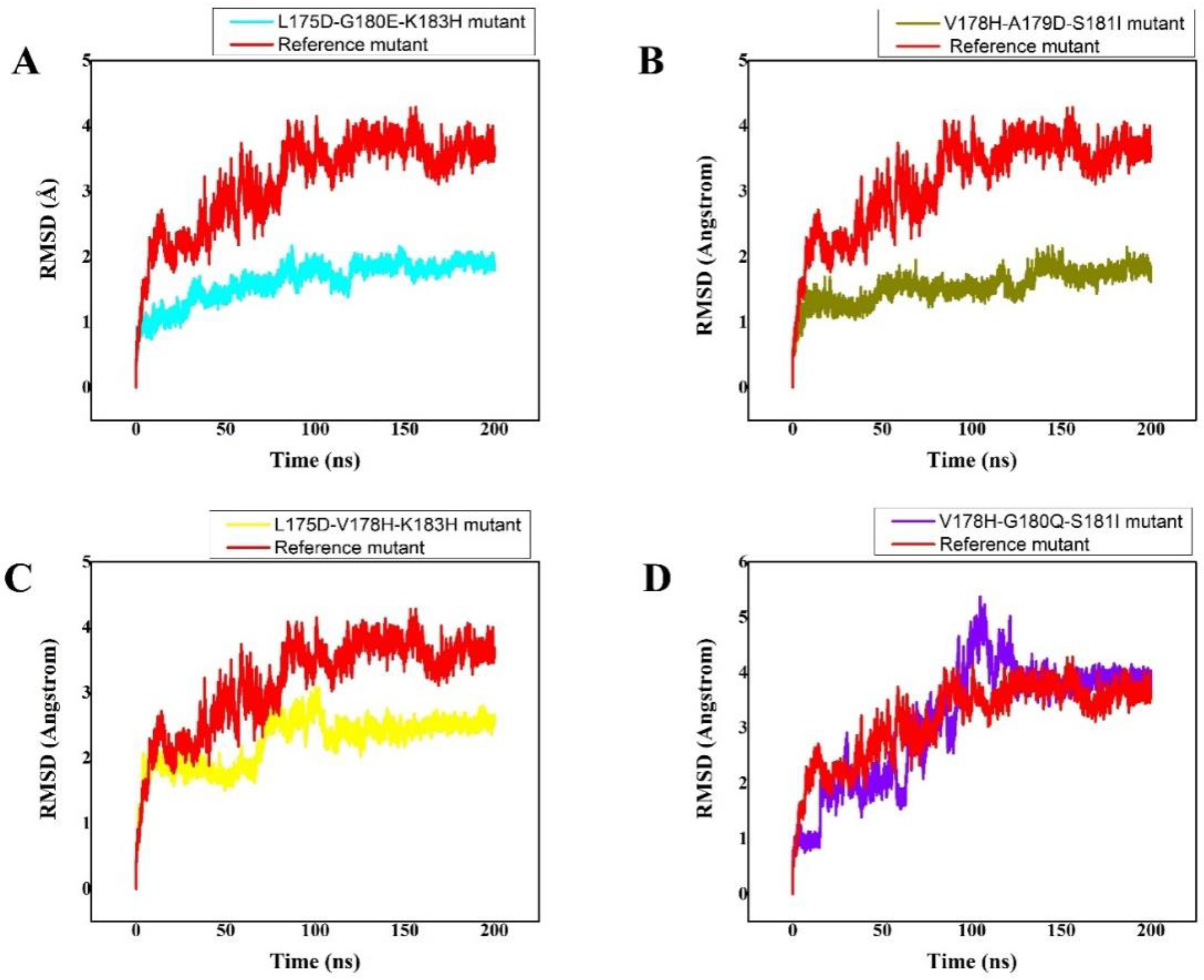
3.3. Molecular Dynamics Stability of K-RasPeptides Complexes
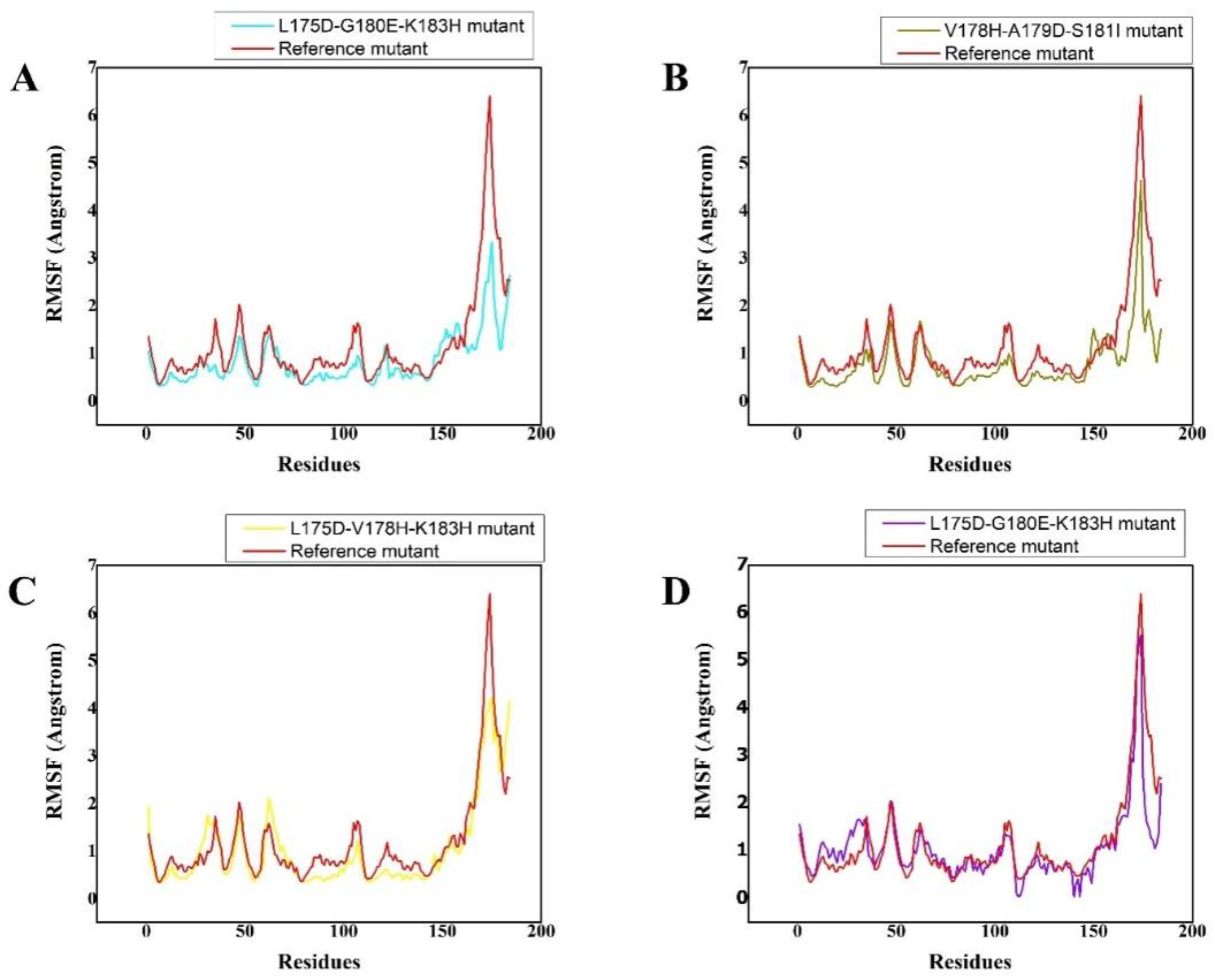
3.4. Residual Flexibility of the Complexes
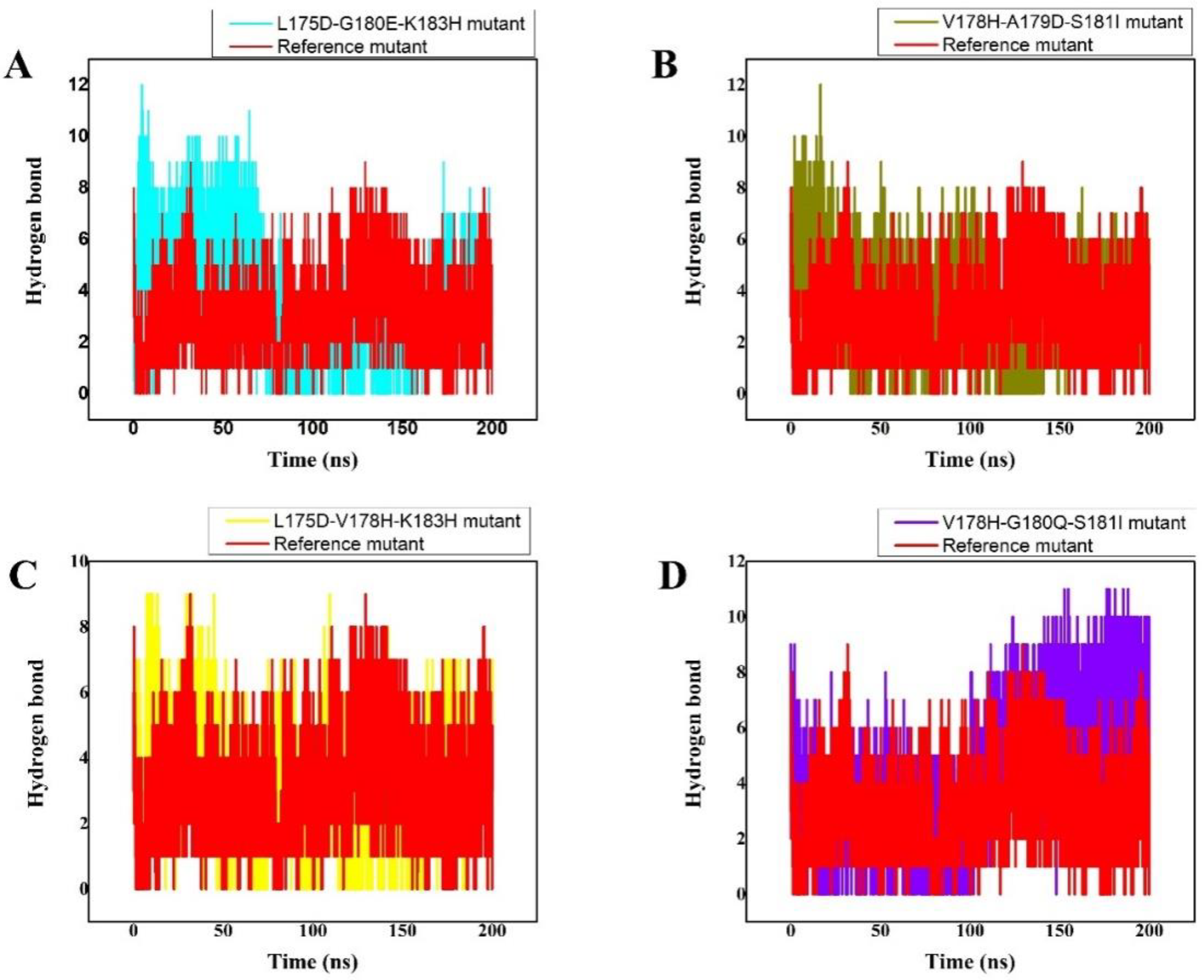
3.5. Hydrogen Bonds
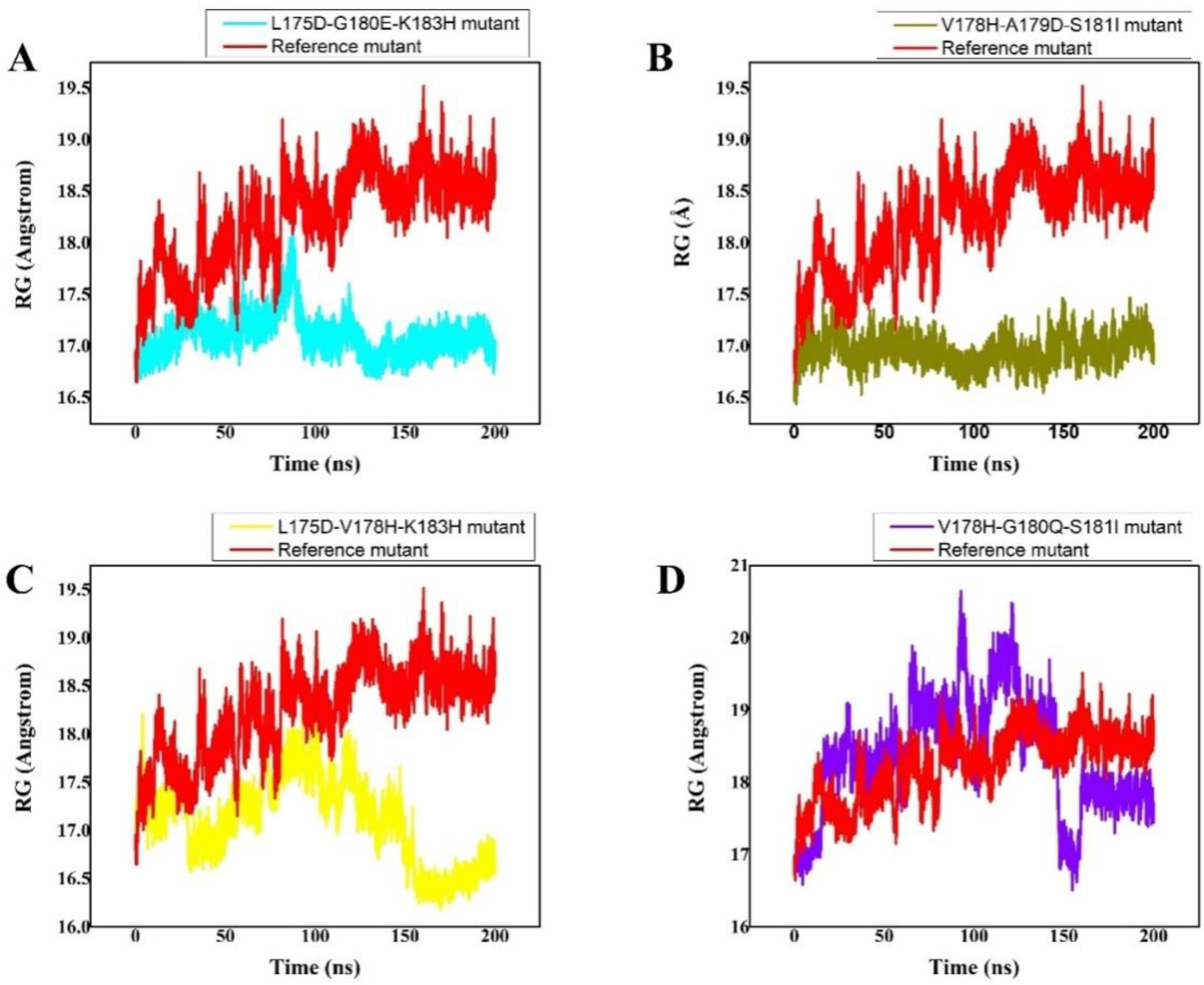
3.6. Radius of Gyration
3.7. Binding Free Energy Calculation
3.8. Interaction Analysis of the Designed Peptides with the K-Ras Protein
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.; Burkhart, D.L.; Haigis, K.M. Classification of KRAS-Activating Mutations and the Implications for Therapeutic Intervention. Cancer Discov. 2022, 12, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, T.J.; Bos, J.; Snel, B. Evolution of the Ras-like small GTPases and their regulators. Small GTPases 2011, 2, 4–16. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Buday, L.; Downward, J. Many faces of Ras activation. Biochim. Biophys. Acta BBA Rev. Cancer 2008, 1786, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.T.; Ovais, M.; Iqbal, J.; Ali, A.; Ayaz, M.; Abbas, M.; Ahmad, I.; Devkota, H.P. Microbes-mediated synthesis strategies of metal nanoparticles and their potential role in cancer therapeutics. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar]
- Hofmann, M.H.; Gerlach, D.; Misale, S.; Petronczki, M.; Kraut, N. Expanding the Reach of Precision Oncology by Drugging All KRAS MutantsDrugging All KRAS Mutants. Cancer Discov. 2022, 12, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Timar, J.; Kashofer, K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metast. Rev. 2020, 39, 1029–1038. [Google Scholar] [CrossRef]
- Song, Y.; Bi, Z.; Liu, Y.; Qin, F.; Wei, Y.; Wei, X. Targeting RAS-RAF-MEK-ERK signaling pathway in human cancer: Current status in clinical trials. Genes Dis. 2022, in press. [Google Scholar] [CrossRef]
- Fu, X.-H.; Chen, Z.-T.; Wang, W.-H.; Fan, X.-J.; Huang, Y.; Wu, X.-B.; Huang, J.-L.; Wang, J.-X.; Lin, H.-J.; Tan, X.-L. KRAS G12V mutation is an adverse prognostic factor of Chinese gastric cancer patients. J. Cancer 2019, 10, 821. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, L.; Tan, S.; Kim, G.P.; Dou, R.; Chen, D.; Cai, Y.; Fu, X.; Wang, L.; Zhu, J. KRAS as a predictor of poor prognosis and benefit from postoperative FOLFOX chemotherapy in patients with stage II and III colorectal cancer. Mol. Oncol. 2015, 9, 1341–1347. [Google Scholar] [CrossRef]
- Takahashi, N.; Yamada, Y.; Taniguchi, H.; Fukahori, M.; Sasaki, Y.; Shoji, H.; Honma, Y.; Iwasa, S.; Takashima, A.; Kato, K. Clinicopathological features and prognostic roles of KRAS, BRAF, PIK3CA and NRAS mutations in advanced gastric cancer. BMC Res. Notes 2014, 7, 271. [Google Scholar] [CrossRef] [Green Version]
- Alamo, P.; Gallardo, A.; Di Nicolantonio, F.; Pavón, M.A.; Casanova, I.; Trias, M.; Mangues, M.A.; Lopez-Pousa, A.; Villaverde, A.; Vázquez, E. Higher metastatic efficiency of KRas G12V than KRas G13D in a colorectal cancer model. FASEB J. 2015, 29, 464–476. [Google Scholar] [CrossRef] [Green Version]
- Fiala, O.; Buchler, T.; Mohelnikova-Duchonova, B.; Melichar, B.; Matejka, V.M.; Holubec, L.; Kulhankova, J.; Bortlicek, Z.; Bartouskova, M.; Liska, V. G12V and G12A KRAS mutations are associated with poor outcome in patients with metastatic colorectal cancer treated with bevacizumab. Tumor Biol. 2016, 37, 6823–6830. [Google Scholar] [CrossRef] [PubMed]
- Olmedillas López, S.; García-Olmo, D.C.; García-Arranz, M.; Guadalajara, H.; Pastor, C.; García-Olmo, D. KRAS G12V mutation detection by droplet digital PCR in circulating cell-free DNA of colorectal cancer patients. Int. J. Mol. Sci. 2016, 17, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raub, C.B.; Lee, C.-C.; Shibata, D.; Taylor, C.; Kartalov, E. HistoMosaic Detecting KRAS G12V Mutation Across Colorectal Cancer Tissue Slices through in Situ PCR. Anal. Chem. 2016, 88, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.; Falcoz, P.-E.; Schaeffer, M.; Guenot, D.; Romain, B.; Olland, A.; Reeb, J.; Santelmo, N.; Chenard, M.-P.; Legrain, M. Prognostic value of the KRAS G12V mutation in 841 surgically resected Caucasian lung adenocarcinoma cases. Br. J. Cancer 2015, 113, 1206–1215. [Google Scholar] [CrossRef] [Green Version]
- Chaft, J.E.; Litvak, A.; Arcila, M.E.; Patel, P.; D’Angelo, S.P.; Krug, L.M.; Rusch, V.; Mattson, A.; Coeshott, C.; Park, B. Phase II study of the GI-4000 KRAS vaccine after curative therapy in patients with stage I-III lung adenocarcinoma harboring a KRAS G12C, G12D, or G12V mutation. Clin. Lung Cancer 2014, 15, 405–410. [Google Scholar] [CrossRef]
- Provost, E.; Bailey, J.M.; Aldrugh, S.; Liu, S.; Iacobuzio-Donahue, C.; Leach, S.D. The tumor suppressor rpl36 restrains KRASG12V-induced pancreatic cancer. Zebrafish 2014, 11, 551–559. [Google Scholar] [CrossRef] [Green Version]
- Margonis, G.A.; Kim, Y.; Spolverato, G.; Ejaz, A.; Gupta, R.; Cosgrove, D.; Anders, R.; Karagkounis, G.; Choti, M.A.; Pawlik, T.M. Association between specific mutations in KRAS codon 12 and colorectal liver metastasis. JAMA Surg. 2015, 150, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Koltun, E.; Cregg, J.; Rice, M.A.; Whalen, D.M.; Freilich, R.; Jiang, J.; Hansen, R.; Bermingham, A.; Knox, J.E.; Dinglasan, J. First-in-class, orally bioavailable KRASG12V (ON) tri-complex inhibitors, as single agents and in combinations, drive profound anti-tumor activity in preclinical models of KRASG12V mutant cancers. Cancer Res. 2021, 81, 1260. [Google Scholar] [CrossRef]
- Luo, J. KRAS mutation in pancreatic cancer. Semin. Oncol. 2021, 48, 10–18. [Google Scholar] [CrossRef]
- Wang, X.; Ni, D.; Liu, Y.; Lu, S. Rational design of peptide-based inhibitors disrupting protein-protein interactions. Front. Chem. 2021, 294. [Google Scholar] [CrossRef]
- Hurd, C.A.; Mott, H.R.; Owen, D. Therapeutic peptides targeting the Ras superfamily. Pept. Sci. 2020, 112, e24165. [Google Scholar] [CrossRef]
- Levinson, A.M.; McGee, J.H.; Roberts, A.G.; Creech, G.S.; Wang, T.; Peterson, M.T.; Hendrickson, R.C.; Verdine, G.L.; Danishefsky, S.J. Total chemical synthesis and folding of all-L and all-D variants of oncogenic KRas (G12V). J. Am. Chem. Soc. 2017, 139, 7632–7639. [Google Scholar] [CrossRef] [PubMed]
- McGee, J.H.; Shim, S.Y.; Lee, S.-J.; Swanson, P.K.; Jiang, S.Y.; Durney, M.A.; Verdine, G.L. Exceptionally high-affinity Ras binders that remodel its effector domain. J. Biol. Chem. 2018, 293, 3265–3280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef]
- Han, C.W.; Jeong, M.S.; Ha, S.C.; Jang, S.B. A H-REV107 peptide inhibits tumor growth and interacts directly with oncogenic KRAS mutants. Cancers 2020, 12, 1412. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Umbreen, S.; Hameed, A.; Fatima, R.; Zahoor, U.; Babar, Z.; Waseem, M.; Hussain, Z.; Rizwan, M.; Zaman, N. In silico mutagenesis-based remodelling of SARS-CoV-1 Peptide (ATLQAIAS) to Inhibit SARS-CoV-2: Structural-dynamics and free energy calculations. Interdiscip. Sci. Comput. Life Sci. 2021, 13, 521–534. [Google Scholar] [CrossRef]
- Ghufran, M.; Rehman, A.U.; Shah, M.; Ayaz, M.; Ng, H.L.; Wadood, A. In-silico design of peptide inhibitors of K-Ras target in cancer disease. J. Biomol. Struct. Dyn. 2020, 38, 5488–5499. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Junaid, M.; Shah, M.; Khan, A.; Li, C.-D.; Khan, M.T.; Kaushik, A.C.; Ali, A.; Mehmood, A.; Nangraj, A.S.; Choi, S. Structural-dynamic insights into the H. pylori cytotoxin-associated gene A (CagA) and its abrogation to interact with the tumor suppressor protein ASPP2 using decoy peptides. J. Biomol. Struct. Dyn. 2018, 37, 4035–4050. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Zwanzig, R. Nonlinear generalized Langevin equations. J. Stat. Phys. 1973, 9, 215–220. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Niida, A.; Sasaki, S.; Yonemori, K.; Sameshima, T.; Yaguchi, M.; Asami, T.; Sakamoto, K.; Kamaura, M. Investigation of the structural requirements of K-Ras (G12D) selective inhibitory peptide KRpep-2d using alanine scans and cysteine bridging. Bioorg. Med. Chem. Lett. 2017, 27, 2757–2761. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Fu, H.; Lee Fryar, K.; Landua, J.; Trevino, S.R.; Schell, D.; Thurlkill, R.L.; Imura, S.; Scholtz, J.M.; Gajiwala, K. Contribution of hydrogen bonds to protein stability. Protein Sci. 2014, 23, 652–661. [Google Scholar] [CrossRef]
- Islam, M.S.; Al-Majid, A.M.; Sholkamy, E.N.; Yousuf, S.; Ayaz, M.; Nawaz, A.; Wadood, A.; Rehman, A.U.; Verma, V.P.; Bari, A. Synthesis, molecular docking and enzyme inhibitory approaches of some new chalcones engrafted pyrazole as potential antialzheimer, antidiabetic and antioxidant agents. J. Mol. Struct. 2022, 1269, 133843. [Google Scholar] [CrossRef]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure of biomolecules through molecular dynamics simulations. Procedia Comput. Sci. 2019, 156, 69–78. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Peptide Sequence | Affinity (kcal/mol) | dAffinity (kcal/mol) |
|---|---|---|---|
| 1 | DYDVAESDHY | −11.4881 | −1.1435 |
| 2 | LYDHDGIDKY | −11.0227 | −1.2644 |
| 3 | DYDHAGSDHY | −10.9856 | −1.0907 |
| 4 | LYDHAQIDKY | −10.6353 | −0.4426 |
| WT | LYDVAGSDKY | −9.5301 | 0 |
| No# | Peptide Sequence | VDWAALS | ESURF | EGB | EEL | Delta Total Binding Free Energies |
|---|---|---|---|---|---|---|
| 1 | DYDVAESDHY | −86.0276 | −11.5218 | 77.7663 | −39.5265 | −59.3096 |
| 2 | LYDHDGIDKY | −81.6682 | −10.6545 | −35.5413 | 75.1207 | −52.7433 |
| 3 | DYDHAGSDHY | −77.1634 | −9.6102 | −103.8830 | 147.1721 | −43.4844 |
| 4 | LYDHAQIDKY | −62.7196 | −8.9737 | −159.3055 | 196.6481 | −34.3507 |
| WT | LYDVAGSDKY | −62.9248 | −9.7345 | 54.2766 | −12.8694 | −31.2521 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghufran, M.; Khan, H.A.; Ullah, M.; Ghufran, S.; Ayaz, M.; Siddiq, M.; Hassan, S.S.u.; Bungau, S. In Silico Strategies for Designing of Peptide Inhibitors of Oncogenic K-Ras G12V Mutant: Inhibiting Cancer Growth and Proliferation. Cancers 2022, 14, 4884. https://doi.org/10.3390/cancers14194884
Ghufran M, Khan HA, Ullah M, Ghufran S, Ayaz M, Siddiq M, Hassan SSu, Bungau S. In Silico Strategies for Designing of Peptide Inhibitors of Oncogenic K-Ras G12V Mutant: Inhibiting Cancer Growth and Proliferation. Cancers. 2022; 14(19):4884. https://doi.org/10.3390/cancers14194884
Chicago/Turabian StyleGhufran, Mehreen, Haider Ali Khan, Mehran Ullah, Sabreen Ghufran, Muhammad Ayaz, Muhammad Siddiq, Syed Shams ul Hassan, and Simona Bungau. 2022. "In Silico Strategies for Designing of Peptide Inhibitors of Oncogenic K-Ras G12V Mutant: Inhibiting Cancer Growth and Proliferation" Cancers 14, no. 19: 4884. https://doi.org/10.3390/cancers14194884