
Genomic Aberrations in Circulating Tumor DNAs from Palbociclib-Treated Metastatic Breast Cancer Patients Reveal a Novel Resistance Mechanism

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Plasma Separation
2.3. DNA Extraction
2.4. Targeted Gene Panel Sequencing
2.5. Somatic Mutation Calling
2.6. Statistical Analyses
3. Results
3.1. Patient Characteristics
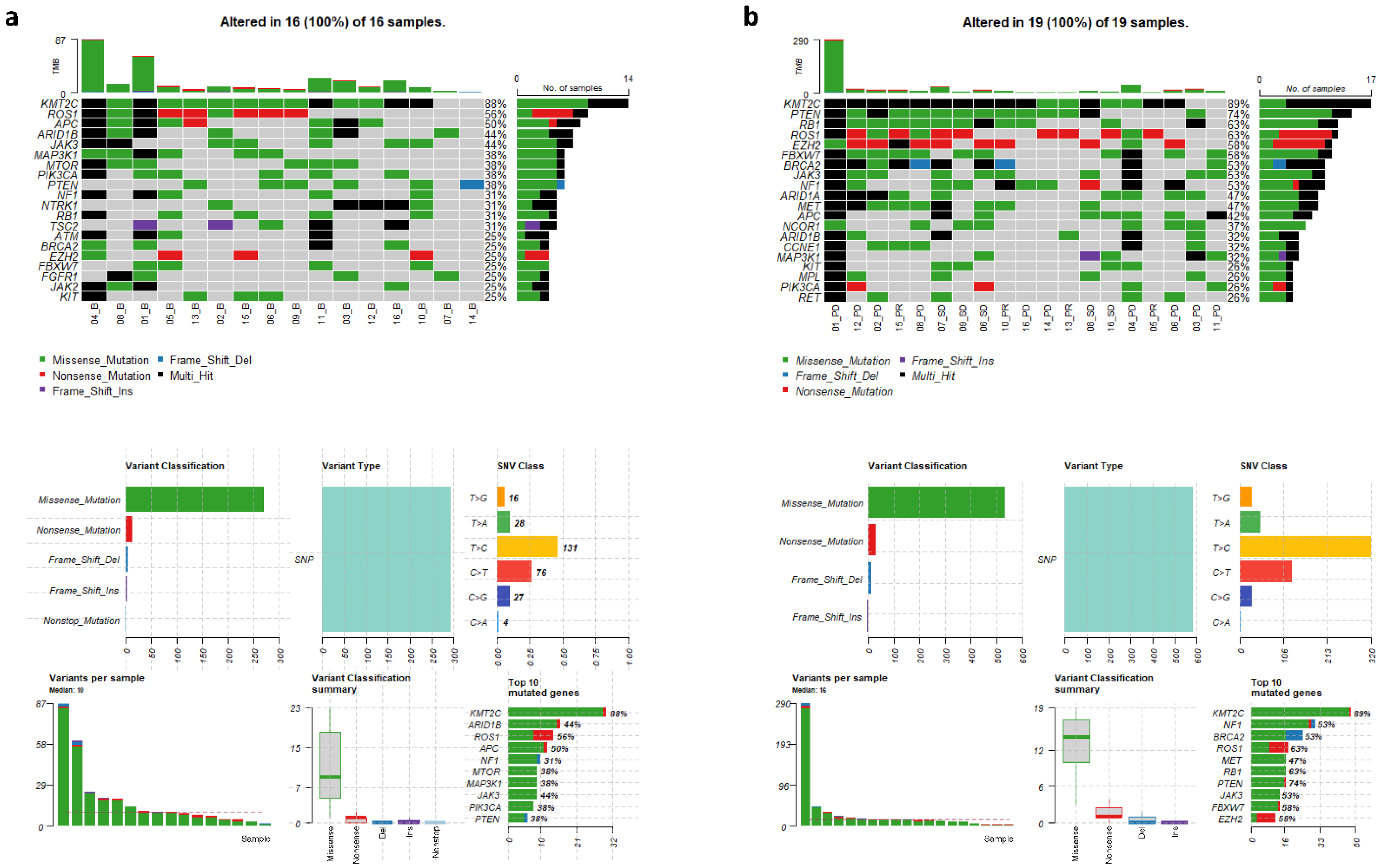
3.2. Genomic Variants in cfDNAs at Baseline and Follow-Up
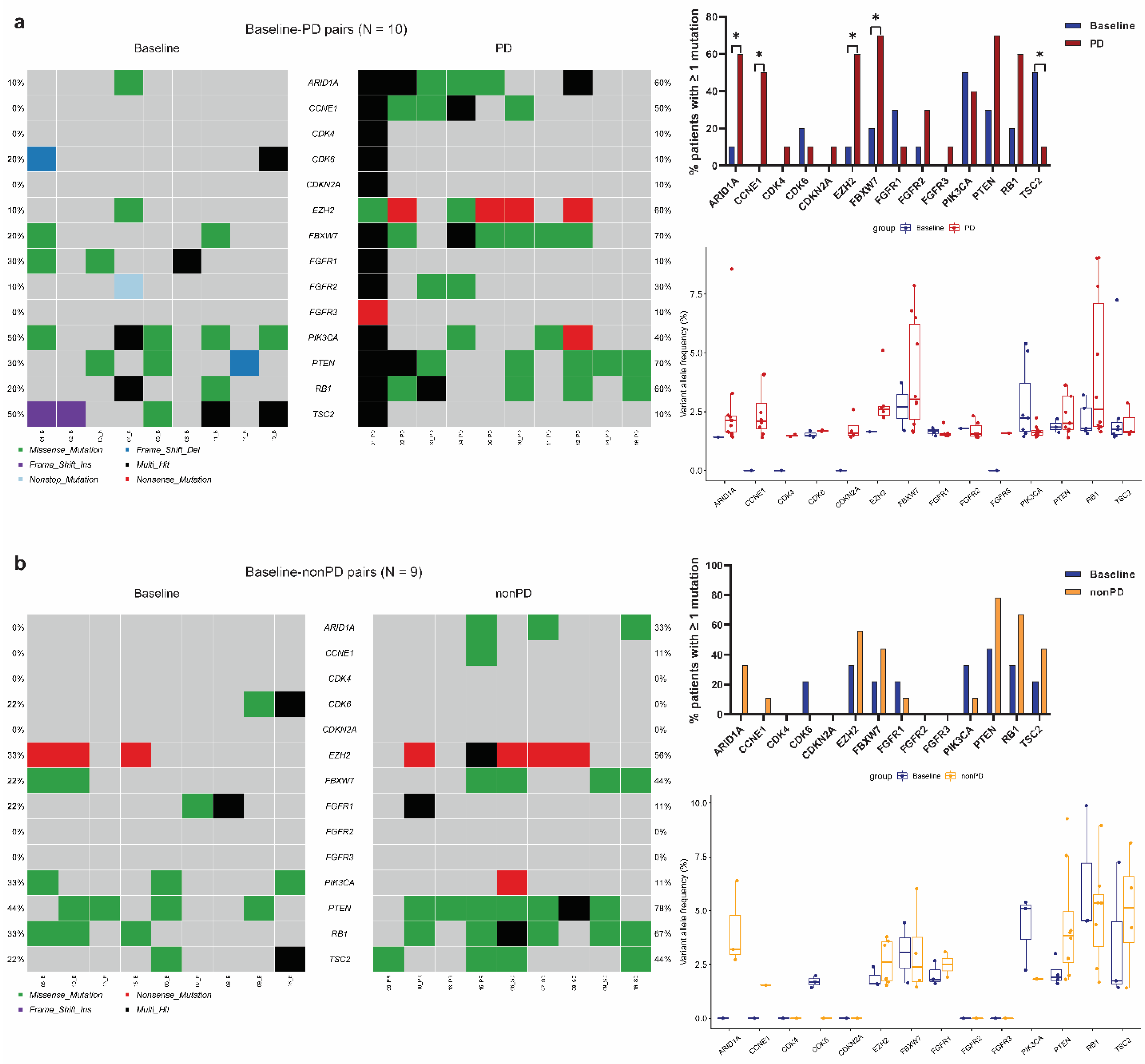
3.3. Identification of Significantly Mutated Genes at Progression by Paired Analyses
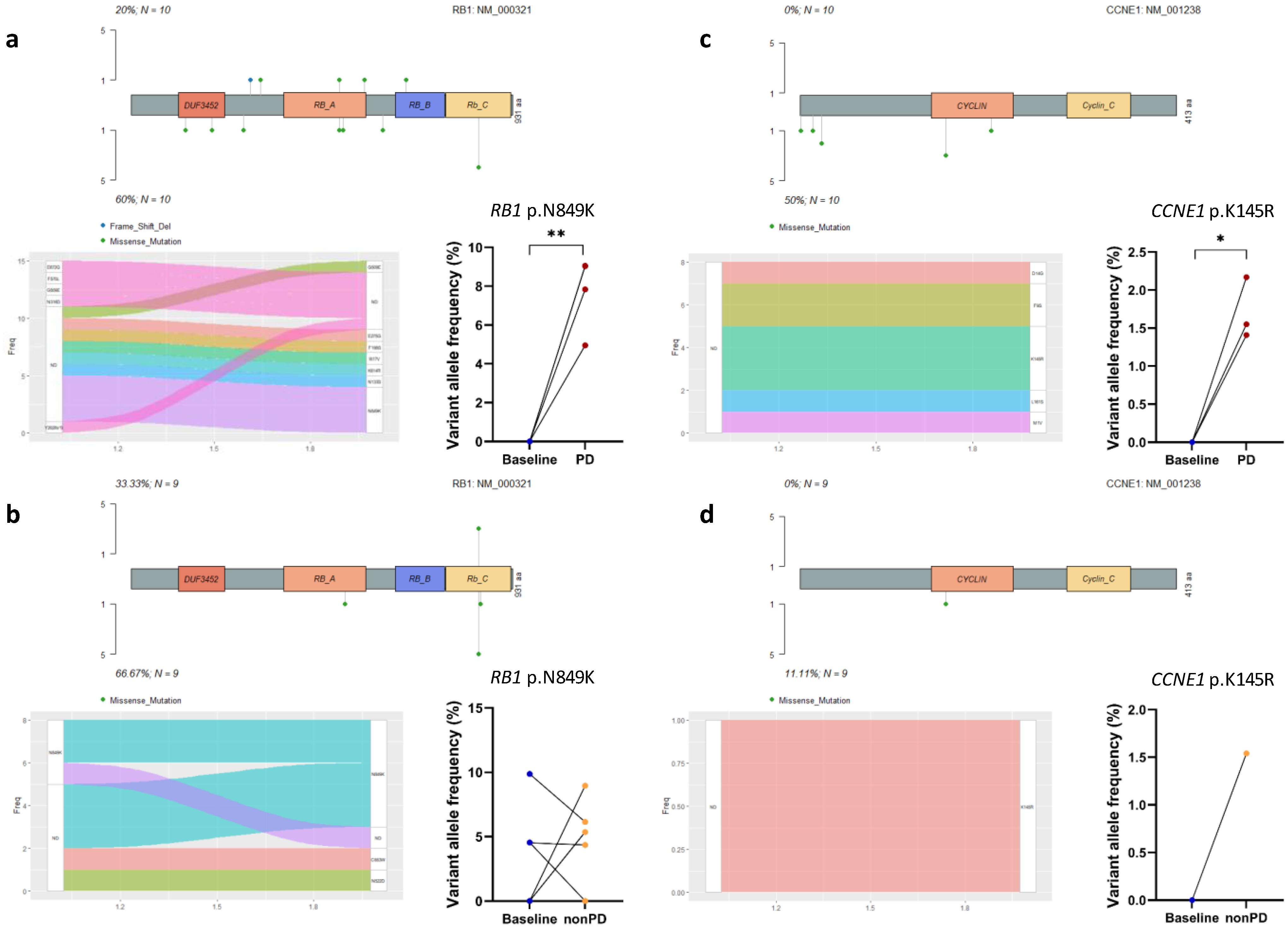
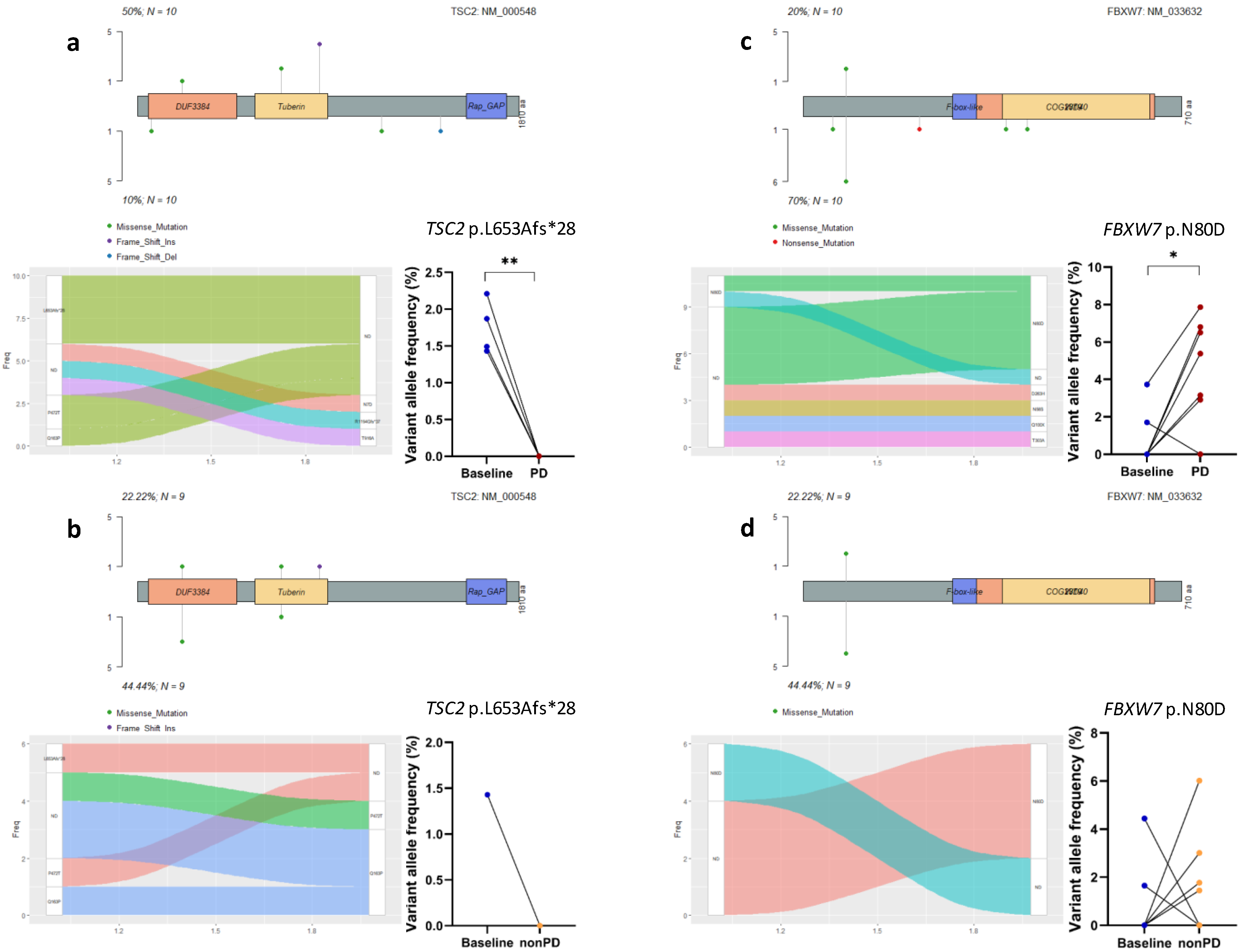
3.4. Acquired Mutations in Cell Cycle Pathway Genes
3.5. Acquired Mutations in Other Oncogenic Signaling Pathway Genes
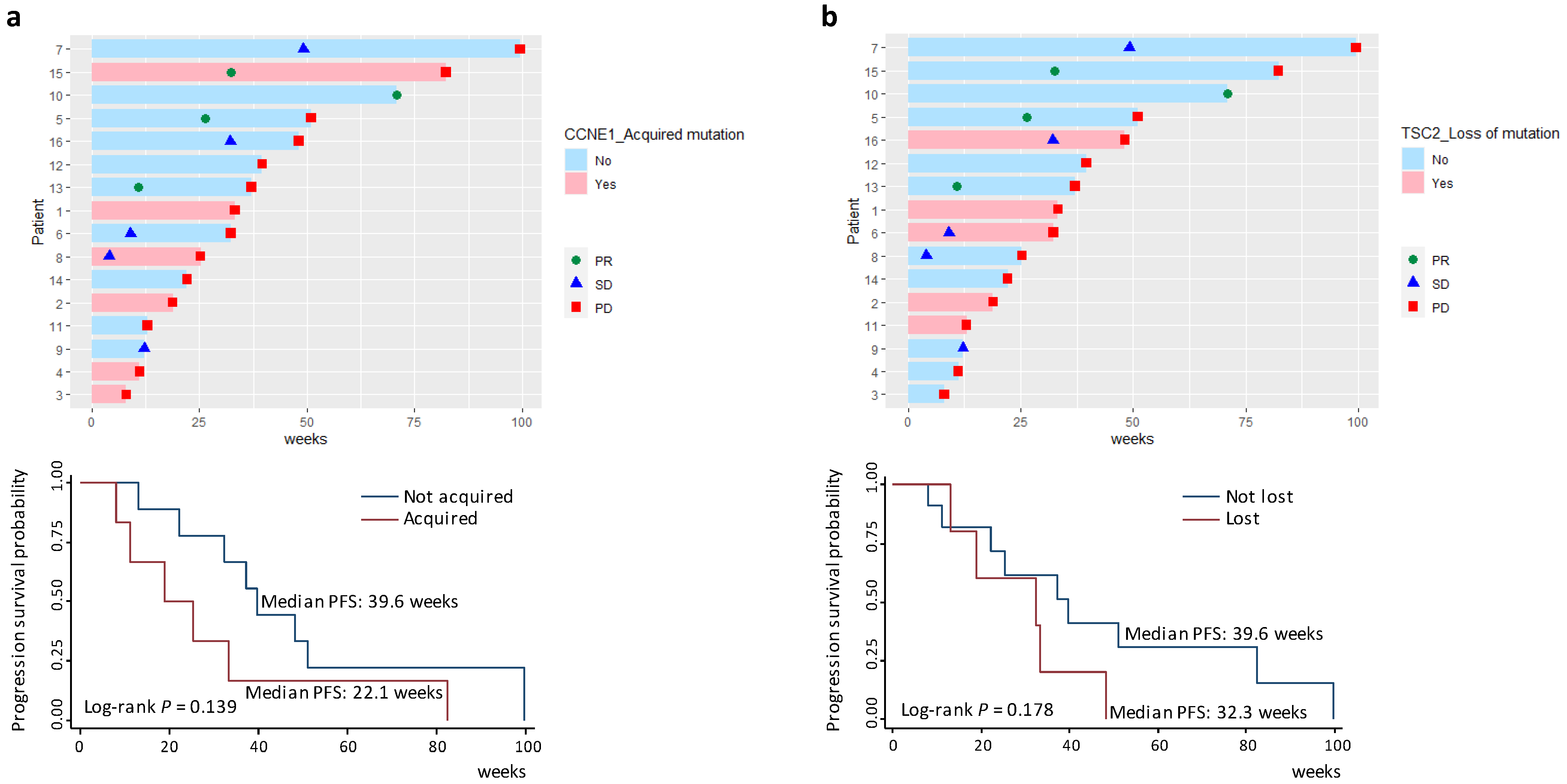
3.6. Survival Analyses of Acquired Gene Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Barrios, C.H. Optimal management of hormone receptor positive metastatic breast cancer in 2016. Ther. Adv. Med. Oncol. 2015, 7, 304–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, C.E. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr.-Relat. Cancer 2019, 26, R15–R30. [Google Scholar] [CrossRef] [Green Version]
- Gennari, A.; Andre, F.; Barrios, C.H.; Cortes, J.; de Azambuja, E.; DeMichele, A.; Dent, R.; Fenlon, D.; Gligorov, J.; Hurvitz, S.A.; et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 1475–1495. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.C.; Lee, S.S.; Abraham, J. Mechanisms of therapeutic CDK4/6 inhibition in breast cancer. Semin. Oncol. 2017, 44, 385–394. [Google Scholar] [CrossRef]
- O’Leary, B.; Cutts, R.J.; Liu, Y.; Hrebien, S.; Huang, X.; Fenwick, K.; Andre, F.; Loibl, S.; Loi, S.; Garcia-Murillas, I.; et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 2018, 8, 1390–1403. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Liu, Y.; Zhu, Z.; Martin, M.; Rugo, H.S.; Dieras, V.; Im, S.A.; Gelmon, K.A.; Harbeck, N.; Lu, D.R.; et al. Biomarker Analyses of Response to Cyclin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naive Metastatic Breast Cancer. Clin. Cancer Res. 2020, 26, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Ignatiadis, M. Promises and Pitfalls of Using Liquid Biopsy for Precision Medicine. Cancer Res. 2019, 79, 2798–2804. [Google Scholar] [CrossRef] [Green Version]
- Meador, C.B.; Lovly, C.M. Liquid biopsies reveal the dynamic nature of resistance mechanisms in solid tumors. Nat. Med. 2015, 21, 663–665. [Google Scholar] [CrossRef]
- Cescon, D.W.; Bratman, S.V.; Chan, S.M.; Siu, L.L. Circulating tumor DNA and liquid biopsy in oncology. Nat. Cancer 2020, 1, 276–290. [Google Scholar] [CrossRef]
- Wang, C.; Mu, Z.; Chervoneva, I.; Austin, L.; Ye, Z.; Rossi, G.; Palazzo, J.P.; Sun, C.; Abu-Khalaf, M.; Myers, R.E.; et al. Longitudinally collected CTCs and CTC-clusters and clinical outcomes of metastatic breast cancer. Breast Cancer Res. Treat. 2017, 161, 83–94. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Wei, Q.; Zhan, X.; Zhong, X.; Liu, Y.; Han, Y.; Chen, W.; Li, B. A Bayesian framework for de novo mutation calling in parents-offspring trios. Bioinformatics 2015, 31, 1375–1381. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, W.; O’Connor, T.D.; Jun, G.; Kang, H.M.; Abecasis, G.; Leal, S.M.; Gabriel, S.; Rieder, M.J.; Altshuler, D.; Shendure, J.; et al. Analysis of 6515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013, 493, 216–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Chong, W.; Wei, Q.; Zhang, Z.; Wang, C.; Ye, Z.; Abu-Khalaf, M.M.; Silver, D.P.; Stapp, R.T.; Jiang, W.; et al. Whole-exome sequencing identifies somatic mutations and intratumor heterogeneity in inflammatory breast cancer. NPJ Breast Cancer 2021, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 2019, 10, 1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliaccio, I.; Leo, A.; Galardi, F.; Guarducci, C.; Fusco, G.M.; Benelli, M.; Di Leo, A.; Biganzoli, L.; Malorni, L. Circulating Biomarkers of CDK4/6 Inhibitors Response in Hormone Receptor Positive and HER2 Negative Breast Cancer. Cancers 2021, 13, 2640. [Google Scholar] [CrossRef]
- Lloyd, M.R.; Spring, L.M.; Bardia, A.; Wander, S.A. Mechanisms of Resistance to CDK4/6 Blockade in Advanced Hormone Receptor-Positive, HER2-Negative Breast Cancer and Emerging Therapeutic Opportunities. Clin. Cancer Res. 2021, 28, 821–830. [Google Scholar] [CrossRef]
- Caldon, C.E.; Musgrove, E.A. Distinct and redundant functions of cyclin E1 and cyclin E2 in development and cancer. Cell Div. 2010, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.N.; Roberts, C.W. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih Ie, M.; Conejo-Garcia, J.R.; et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Guarducci, C.; Bonechi, M.; Benelli, M.; Biagioni, C.; Boccalini, G.; Romagnoli, D.; Verardo, R.; Schiff, R.; Osborne, C.K.; De Angelis, C.; et al. Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. NPJ Breast Cancer 2018, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, B.; Cutts, R.J.; Huang, X.; Hrebien, S.; Liu, Y.; Andre, F.; Loibl, S.; Loi, S.; Garcia-Murillas, I.; Cristofanilli, M.; et al. Circulating Tumor DNA Markers for Early Progression on Fulvestrant With or Without Palbociclib in ER+ Advanced Breast Cancer. J. Natl. Cancer Inst. 2021, 113, 309–317. [Google Scholar] [CrossRef]
- Turner, N.C.; Liu, Y.; Zhu, Z.; Loi, S.; Colleoni, M.; Loibl, S.; DeMichele, A.; Harbeck, N.; Andre, F.; Bayar, M.A.; et al. Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2019, 37, 1169–1178. [Google Scholar] [CrossRef]
- Ma, C.X.; Gao, F.; Luo, J.; Northfelt, D.W.; Goetz, M.; Forero, A.; Hoog, J.; Naughton, M.; Ademuyiwa, F.; Suresh, R.; et al. NeoPalAna: Neoadjuvant Palbociclib, a Cyclin-Dependent Kinase 4/6 Inhibitor, and Anastrozole for Clinical Stage 2 or 3 Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2017, 23, 4055–4065. [Google Scholar] [CrossRef] [Green Version]
- Wander, S.A.; Han, H.S.; Zangardi, M.L.; Niemierko, A.; Mariotti, V.; Kim, L.S.L.; Xi, J.; Pandey, A.; Dunne, S.; Nasrazadani, A.; et al. Clinical Outcomes With Abemaciclib After Prior CDK4/6 Inhibitor Progression in Breast Cancer: A Multicenter Experience. J. Natl. Compr. Canc Netw 2021, 1, 1–8. [Google Scholar] [CrossRef]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Miricescu, D.; Totan, A.; Stanescu, S., II; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]
- Costa, C.; Wang, Y.; Ly, A.; Hosono, Y.; Murchie, E.; Walmsley, C.S.; Huynh, T.; Healy, C.; Peterson, R.; Yanase, S.; et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kalpha Inhibitors in Breast Cancer. Cancer Discov. 2020, 10, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Romero-Pozuelo, J.; Figlia, G.; Kaya, O.; Martin-Villalba, A.; Teleman, A.A. Cdk4 and Cdk6 Couple the Cell-Cycle Machinery to Cell Growth via mTORC1. Cell Rep. 2020, 31, 107504. [Google Scholar] [CrossRef]
- Maskey, R.S.; Wang, F.; Lehman, E.; Wang, Y.; Emmanuel, N.; Zhong, W.; Jin, G.; Abraham, R.T.; Arndt, K.T.; Myers, J.S.; et al. Sustained mTORC1 activity during palbociclib-induced growth arrest triggers senescence in ER+ breast cancer cells. Cell Cycle 2021, 20, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, I.; Bonechi, M.; McCartney, A.; Guarducci, C.; Benelli, M.; Biganzoli, L.; Di Leo, A.; Malorni, L. CDK4/6 inhibitors: A focus on biomarkers of response and post-treatment therapeutic strategies in hormone receptor-positive HER2-negative breast cancer. Cancer Treat. Rev. 2021, 93, 102136. [Google Scholar] [CrossRef] [PubMed]
- Sailo, B.L.; Banik, K.; Girisa, S.; Bordoloi, D.; Fan, L.; Halim, C.E.; Wang, H.; Kumar, A.P.; Zheng, D.; Mao, X.; et al. FBXW7 in Cancer: What Has Been Unraveled Thus Far? Cancers 2019, 11, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohmaier, H.; Spruck, C.H.; Kaiser, P.; Won, K.A.; Sangfelt, O.; Reed, S.I. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 2001, 413, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Wang, Y.; Zhang, P.; Lu, J.; Mao, J.H. Evaluating the prognostic significance of FBXW7 expression level in human breast cancer by a meta-analysis of transcriptional profiles. J. Cancer Sci. 2012, 4, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Gala, K.; Li, Q.; Sinha, A.; Razavi, P.; Dorso, M.; Sanchez-Vega, F.; Chung, Y.R.; Hendrickson, R.; Hsieh, J.J.; Berger, M.; et al. KMT2C mediates the estrogen dependence of breast cancer through regulation of ERalpha enhancer function. Oncogene 2018, 37, 4692–4710. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Rampias, T.; Karagiannis, D.; Avgeris, M.; Polyzos, A.; Kokkalis, A.; Kanaki, Z.; Kousidou, E.; Tzetis, M.; Kanavakis, E.; Stravodimos, K.; et al. The lysine-specific methyltransferase KMT2C/MLL3 regulates DNA repair components in cancer. EMBO Rep. 2019, 20, e46821. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, F.; Rypens, C.; Finetti, P.; Guille, A.; Adélaïde, J.; Monneur, A.; Carbuccia, N.; Garnier, S.; Dirix, P.; Gonçalves, A.; et al. NOTCH and DNA repair pathways are more frequently targeted by genomic alterations in inflammatory than in non-inflammatory breast cancers. Mol. Oncol. 2020, 14, 504–519. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Chhangawala, S.; Cocco, E.; Razavi, P.; Cai, Y.; Otto, J.E.; Ferrando, L.; Selenica, P.; Ladewig, E.; Chan, C.; et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat. Genet. 2020, 52, 198–207. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Age (yr) | Race | ER/PR /HER2 | Tumor Differentiate | IBC | Lines of Previous Therapies * | Regimen | Number of Blood Samples | Response at EOT | TTP (wk) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | F/U | ||||||||||
| 1 | 54.56 | White | +/+/+ | Poorly | Yes | 6 | Palbociclib + TDM-1 | 1 | 1 (PD) | PD | 33.29 |
| 2 | 66.05 | Black | +/+/− | Poorly | No | 4 | Palbociclib + letrozole | 1 | 1 (PD) | PD | 18.86 |
| 3 | 33.03 | White | +/+/− | Poorly | Yes | 0 | Palbociclib + fulvestrant | 1 | 1 (PD) | PD | 8.00 |
| 4 | 42.79 | White | +/+/− | Moderately | No | 1 | Palbociclib + fulvestrant | 1 | 1 (PD) | PD | 11.14 |
| 5 | 39.42 | White | +/+/− | Poorly | No | 2 | Palbociclib + letrozole | 1 | 1 (PR) | PD | 51.00 |
| 6 | 58.14 | White | +/+/− | Moderately | No | 1 | Palbociclib + fulvestrant | 1 | 1 (SD), 1 (PD) | PD | 32.29 |
| 7 | 66.25 | White | +/+/− | Moderately | Yes | 3 | Palbociclib + fulvestrant | 1 | 1 (SD) | PD | 99.57 |
| 8 | 39.20 | White | +/−/− | Poorly | Yes | 8 | Palbociclib + letrozole | 1 | 1 (SD), 1 (PD) | PD | 25.29 |
| 9 | 54.87 | White | +/+/− | Unknown | Yes | 1 | Palbociclib + fulvestrant | 1 | 1 (SD) | SD (Side effects) | (12.14) |
| 10 | 32.56 | Black | +/+/− | Poorly | Yes | 1 | Palbociclib + letrozole | 1 | 1 (PR) | PR# (LTFU) | (70.86) |
| 11 | 51.12 | White | +/−/− | Poorly | Yes | 3 | Palbociclib + letrozole | 1 | 1 (PD) | PD | 13.00 |
| 12 | 74.81 | White | +/+/− | Moderately | No | 4 | Palbociclib + letrozole | 1 | 1 (PD) | PD | 39.57 |
| 13 | 45.15 | White | +/+/− | Poorly | Yes | 1 | Palbociclib + fulvestrant | 1 | 1 (PR) | PD | 37.14 |
| 14 | 64.12 | White | +/−/− | Moderately | No | 4 | Palbociclib + fulvestrant | 1 | 1 (PD) | PD | 22.14 |
| 15 | 58.62 | White | +/+/− | Poorly | Yes | 1 | Palbociclib + fulvestrant | 1 | 1 (PR) | PD | 82.29 |
| 16 | 63.78 | White | +/−/− | Moderately | No | 0 | Palbociclib + letrozole | 1 | 1 (SD), 1 (PD) | PD | 48.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu-Khalaf, M.; Wang, C.; Zhang, Z.; Luo, R.; Chong, W.; Silver, D.P.; Fellin, F.; Jaslow, R.; Lopez, A.; Cescon, T.; et al. Genomic Aberrations in Circulating Tumor DNAs from Palbociclib-Treated Metastatic Breast Cancer Patients Reveal a Novel Resistance Mechanism. Cancers 2022, 14, 2872. https://doi.org/10.3390/cancers14122872
Abu-Khalaf M, Wang C, Zhang Z, Luo R, Chong W, Silver DP, Fellin F, Jaslow R, Lopez A, Cescon T, et al. Genomic Aberrations in Circulating Tumor DNAs from Palbociclib-Treated Metastatic Breast Cancer Patients Reveal a Novel Resistance Mechanism. Cancers. 2022; 14(12):2872. https://doi.org/10.3390/cancers14122872
Chicago/Turabian StyleAbu-Khalaf, Maysa, Chun Wang, Zhenchao Zhang, Rui Luo, Weelic Chong, Daniel P. Silver, Frederick Fellin, Rebecca Jaslow, AnaMaria Lopez, Terrence Cescon, and et al. 2022. "Genomic Aberrations in Circulating Tumor DNAs from Palbociclib-Treated Metastatic Breast Cancer Patients Reveal a Novel Resistance Mechanism" Cancers 14, no. 12: 2872. https://doi.org/10.3390/cancers14122872