
The Genomics of Hairy Cell Leukaemia and Splenic Diffuse Red Pulp Lymphoma



1
Department of Haematology, Royal Bournemouth and Christchurch NHS Trust, Bournemouth BH7 7DW, UK
2
Institute of Applied Biosciences, Centre for Research and Technology-Hellas, 57001 Thessaloniki, Greece
3
Cancer Genomics Group, Southampton General Hospital, Tremona Road, Southampton SO16 6YD, UK
*
Author to whom correspondence should be addressed.
Cancers 2022, 14(3), 697; https://doi.org/10.3390/cancers14030697
Submission received: 29 December 2021
/
Revised: 24 January 2022
/
Accepted: 25 January 2022
/
Published: 29 January 2022
(This article belongs to the Special Issue Genomics of Rare Hematologic Cancers)
Abstract
:Simple Summary
Hairy cell leukaemia is a rare chronic lymphoid malignancy with distinctive clinical and laboratory features which include an enlarged spleen, low blood counts, and infiltration of the spleen and bone marrow, with lymphocytes that have a villous or hairy cytoplasmic border. Historically it has been responsive to a range of treatment modalities including splenectomy, alpha interferon, and more recently chemotherapy, but none are curative. This review describes the chromosome abnormalities, genomic mutations, DNA methylation patterns, and immunoglobulin gene usage in this disease. We then discuss how the discovery of a specific mutation in a single gene (BRAF), present in almost all cases but not in hairy cell variant or splenic lymphoma with villous lymphocytes, two other splenic lymphomas with similar features, has provided new insights into its biology, a new diagnostic test, and a new therapeutic target.
Abstract
Classical hairy cell leukaemia (HCLc), its variant form (HCLv), and splenic diffuse red pulp lymphoma (SDRPL) constitute a subset of relatively indolent B cell tumours, with low incidence rates of high-grade transformations, which primarily involve the spleen and bone marrow and are usually associated with circulating tumour cells characterised by villous or irregular cytoplasmic borders. The primary aim of this review is to summarise their cytogenetic, genomic, immunogenetic, and epigenetic features, with a particular focus on the clonal BRAFV600E mutation, present in most cases currently diagnosed with HCLc. We then reflect on their cell of origin and pathogenesis as well as present the clinical implications of improved biological understanding, extending from diagnosis to prognosis assessment and therapy response.
1. Introduction
The 2017 WHO classification of haematological malignancies recognises classical hairy cell leukaemia (HCLc) as a discrete entity and its variant form (HCLv) and splenic diffuse red pulp lymphoma (SDRPL) as provisional entities [1]. HCLc is a rare chronic lymphoproliferative disorder, with an incidence of 0.4/100,000. It is approximately four times more common in men than women and typically presents in middle age, with fatigue, infections, and abdominal discomfort due to splenomegaly. A complete blood count frequently shows cytopenia with almost universal monocytopenia, and a blood film usually reveals small numbers of medium-sized lymphoid cells with a ‘kidney-shaped nucleus’, infrequent nucleoli, weakly basophilic cytoplasm, and hairy projections. A modest lymphocytosis is seen in 5–10% of cases. Splenic histology shows diffuse infiltration of the red pulp with atrophy of the white pulp, a pattern also seen in HCLv and SDRPL, in contrast to the predominant white pulp involvement found in Splenic Marginal Zone Lymphoma (SMZL), which is the subject of a separate review in this series. Although originally believed to represent a tumour of haemopoietic progenitor reticuloendothelial cells [2], immunophenotyping identified HCLc as a tumour of mature B cells, typically expressing CD19, CD20, CD200, Tbet, PD1, and four markers of diagnostic value: CD11c and CD103, components of integrin receptors, and CD25 and CD123, components of interleukin receptors, of which at least three are expressed in all cases. Hairy cells do not express CD5 or CD27. Immunohistochemistry of bone marrow trephines additionally shows the expression of cyclin D1, CD72 (DBA-44), and Annexin A1, a specific marker for HCLc among B-cell malignancies, while bone marrow aspiration is usually unsuccessful due to the presence of reticulin fibrosis [3,4].
HCLv has an incidence of 0.04/100,000, a median age at presentation of 70 years, and a male-to-female ratio of 1.5–2. It was first described in 1980 in two patients with bulky splenomegaly, a marked leucocytosis with villous lymphocytes, and splenic histology showing red pulp involvement similar to that seen in HCLc, together with a number of distinctive features not found in HCLc. These include the absence of monocytopenia, larger tumour cells with prominent nucleoli, and bone marrow that is easy to aspirate due to absence or minimal marrow reticulin. Immunophenotyping shows the expression of CD19, CD20, and of the four archetypal HCLc markers, only CD11c and CD103 are commonly expressed, while CD25, CD123, and CD200 are negative or only weakly expressed. Immunohistochemistry shows the expression of CD72 but not annexin A1, and cyclin D1 is negative or weak [5,6].
In 2008, the term SDRPL was introduced to describe a further type of splenic lymphoma with circulating villous lymphocytes [7]. The frequency of SDRPL has not been established in the general population but represented 9% of splenic B-cell lymphomas seen in a 12-year period reviewed at the Spanish National Cancer Research Centre [8]. The clinical and laboratory features overlap those seen in HCLv, but the tumour cells generally lack a prominent nucleolus, and patients pursue a more indolent clinical course. Key demographic, morphological, phenotypic, and clinical differences between the three disorders are shown in Table 1.
2. Hairy Cell Leukaemia
2.1. BRAF V600E Mutations
The whole-exome sequencing of a single case of HCLc led to the discovery of a single somatic, point mutation in the DNA sequence of v-Raf murine sarcoma viral oncogene homolog B (BRAF), a kinase-encoding proto-oncogene. The same mutation was subsequently found in all 47 additional cases studied. The mutation replaces thymine (T) with adenine (A) in exon 15 of BRAF at position 1799 of the gene-coding sequence located in chromosome 7q34. In turn, this produces an amino acid change from valine (V) to glutamate (E) at position 600 (V600E) of the protein sequence, ultimately leading to aberrant activation of the BRAF oncogenic kinase and, thus, of the downstream MEK–ERK signalling pathway, such that ERK phosphorylation (pERK), detectable by immunohistochemistry, is a ubiquitous finding in BRAF-V600E positive HCLc [9,10]. The BRAF-V600E mutation in HCL is clonal and heterozygous, except in a minority of patients who lose the wild-type allele as a result of a concomitant 7q deletion [11]. Details of the RAS–RAF–MEK–ERK pathway and the BRAF protein with the site of the BRAFV600E mutation are shown in Figure 1 and Figure 2, respectively, and described in the accompanying legends.
BRAF mutations are found in a wide range of both solid and haematopoietic tumours, with a particularly high incidence in benign melanocytic nevi, malignant melanoma, papillary carcinoma of the thyroid [16], and the primary histiocytic disorders, Langerhans cell histiocytosis (LCH) and Erdheim–Chester disease (ECD) [17,18]. The clinical and biological consequences of BRAF mutations are highly variable and include the induction of a senescent phenotype, oncogenic transformation, and the emergence of secondary histiocytic sarcomas in a variety of acute or chronic, B or T cell, leukaemia, or lymphomas [19,20,21]. This variability may reflect the acquisition of additional genomic abnormalities such as the inactivation of cell cycle inhibitors, and the differentiation stage, transcriptomic and epigenetic features of the cell type in which the BRAF mutation arises [22,23].
2.1.1. Haematopoietic Stem Cell Origin of BRAFV600E Mutation in HCLc
To identify the cell population from which the BRAFV600E mutation arises, immunophenotypically distinct CD34+, CD38− lineage-negative cells which encompass haemopoietic stem cells (HSCs) and their immediate multipotent progenitors [24,25], CD34+, CD38+ pro-B cells, myeloid progenitor cells, and HCLc cells were isolated with >97% purity from the bone marrow of 14 HCLc patients and age-matched controls [26].
HCLc patients were characterised by an expansion of HSCs and a marked decrease in the frequency of granulocyte-macrophage progenitor cells, consistent with the neutropenia and monocytopenia characteristics of HCLc. The BRAFV600E mutation was identified in the HSC, pro-B cell, and HCL cell populations, and quantitative sequencing analysis revealed a mean BRAFV600E-mutant allele frequency of 4.97% in the HSCs. Furthermore, in one patient who also had chronic lymphocytic leukaemia (CLL), the BRAFV600E mutation was present in both tumour cell populations, consistent with the mutation arising in a common precursor. To identify additional co-occurring genetic lesions that might cooperate with the BRAFV600E mutation to promote haematopoietic transformation, targeted mutational analysis was performed on HCL cells from three patients in whom the BRAFV600E mutation had been detected in HSCs. An additional ARID1A or KMT2C mutation was present in the leukemic cells but not the HSCs of 2/3 cases. HCLc patients treated with vemurafenib showed restoration of normal myelopoiesis, demonstrating that the impaired myeloid differentiation in HCL is dependent on mutant BRAFV600E signalling. This raises the question as to whether the clinical response to BRAF inhibitors may be mediated through their effects on mature leukemic cells, as well as through targeted inhibition of signalling and survival in mutant HSPCs.
2.1.2. Biological Consequences of the BRAFV600E Mutation in HCLc
The biology of HCLc reflects both cell-intrinsic factors as well as interactions with antigen(s), the extracellular matrix, the multiple cell types, and their secreted products present in the tissue microenvironment (TME) [30,31]. To ascertain the contribution of mutant BRAF to the HCLc phenotype, hairy cells from 26 patients were exposed in vitro to the specific BRAF inhibitors, vemurafenib or dabrafenib, or the MEK inhibitor trametinib. This resulted in the silencing of a gene expression signature which is specific to HCLc among B-cell tumours, with downregulation of genes including CCND1, CD25, and feedback inhibitors of ERK signalling such as members of the dual-specificity phosphatase (DSP) gene family. Additionally, BRAF or MEK inhibition caused loss of the hairy morphology and induced apoptosis which could be partially abrogated by co-culture with a bone marrow stromal cell line. BRAF and MEK inhibitors did not elicit any of the above-described biological effects in leukemic cells from four cases with HCLv, although the MAP2K1 mutation status of these cases was not documented [32,33,34].
2.1.3. Incidence of BRAFV600E in HCLc
The initial description of the BRAFV600E mutation in HCLc identified the mutation in all 48 patients tested [9], and several subsequent studies also found an incidence of 100% [11,35,36,37]. In contrast, other studies of patients reported having the typical clinical, morphological, and immunophenotypic features of HCLc have included a varying percentage of cases lacking the BRAFV600E mutation, with by far the highest incidence (21–25%) found among cases with relapsed/refractory disease [38,39]. Alternate methods of MAPK pathway activation have been discovered in some of these cases, including rare BRAF exon 11 mutations [40] and a single case with a t(7;14) (q34;q32) translocation resulting in an IGH-BRAF fusion [41]. The translocation juxtaposes the IGM switch region with exons 10–18 of BRAF, including the protein kinase domain, while removing the N-terminal auto-inhibitory Ras binding domain which spans exons 3–5. This results in ERK phosphorylation of tumour cells, indicating upregulation of MAPK signalling. Of potential clinical relevance, this patient would be expected to respond to MEK, but not BRAFV600E, mutation-specific inhibitors.
In a study of targeted gene sequencing in 20 HCLc cases, 2 lacked a BRAF mutation, of which 1 had a MAP2K1 mutation, while no genomic abnormality was detected in the other case [42]. Among 53 cases with relapsed/refractory disease, BRAF was wild type in 11 (21%), including all 5 cases with clonotypic B-cell receptor immunoglobulin (BcR IG) utilising the IGHV4-34 gene [38]. In a follow-up study of 27 HCLc cases, 7 were IGHV4-34 positive, and activating MAP2K1 mutations were identified in 5 IGHV4-34-positive but only 1 IGHV4-34-negative case [43].
2.2. Other Genomic Abnormalities
The most frequent cytogenetic and genomic abnormalities and immunogenetic features found in BRAFV600E mutated HCLc, BRAF WT HCLc, HCLv, and SDRPL are summarised in Table 2.
2.2.1. Cytogenetic and DNA Copy Number Aberrations in HCL
Chromosome banding analysis (CBA) using a variety of B-cell mitogens identified clonal abnormalities in 70–80% of evaluable metaphases, but the nature and frequency of recurring abnormalities differed among studies. Abnormalities of chromosome 5, most commonly trisomy 5, or pericentric inversions and interstitial deletions involving band 5q13 were the most frequent abnormality in one study, detected in 12/30 cases [44]. Subsequent studies employing comparative genomic hybridisation also showed a varying incidence of copy number abnormalities (CNAs) but confirmed recurrent gains of 5q13-q31 and loss of 7q [45,46,47,48].
Two deep-targeted sequencing studies have enabled copy number analysis of regions sequenced by the panels. Among 53 BRAFV600E mutated cases of whom 22 were treatment naïve, recurrent abnormalities included deletions of 7q and of 13q14.3, encompassing RB1 and the miR-15a and miR-16-1 microRNA cluster at 13q [11]. A second study of 20 cases sampled at diagnosis found loss of MAPK15 in 7 (35%) of patients. The MAPK15 gene, located on chromosome 8, encodes extracellular regulated kinase 8 (ERK8), a member of the MAPK family. The presence of a MAPK15 CNA had no impact on treatment-free survival (TFS) and overall survival (OS), but progression-free survival (PFS) was significantly longer in cases with a MAPK15 deletion [42].
2.2.2. Somatic Genomic Mutations
Current information on the nature and incidence of somatic mutations other than BRAF in HCLc is based on limited data—namely, whole-exome sequencing (WES) of 9 cases [9,49,50] and targeted sequencing using a large panel of cancer-related genes in 73 cases [11,26,42], together with targeted sequencing of specific genes: CDKN1B, MAP2K1, and KLF2. Mutations in these genes are described in more detail below. Recurring low-frequency mutations also involve chromatin modifiers, discussed in the next section on epigenetic abnormalities and genes involved in Notch signalling (NOTCH1 and NOTCH2), and DNA repair (RAD50).
- CDKN1B
CDKN1B maps to 12p13 and encodes p27Kip1(p27), an intrinsically unstructured protein which regulates the transition from the G1 to the S phase of the cell cycle (Figure 3) and also has CDK-independent functions [51].
Low expression of p27 was demonstrated in all 58 cases of HCL studied and was associated with post-transcriptional downregulation, although the precise cause was not ascertained [53]. Subsequent studies in melanoma revealed a direct role for the BRAFV600E mutation: Expression of mutant BRAF was sufficient to upregulate cyclin D1 and downregulate p27 in human melanocytes [54], while in melanoma cells, mutant B-RAF controls p27Kip1 expression via mRNA abundance and proteasomal degradation [55]. A further potential mechanism for low p27 in HCLc emerged from microarray expression profiling of HCLc, compared with normal and other malignant B cells, which identified overexpression of miR-221/miR-222c which negatively regulates the expression of p27 [56,57].
More recently mutations of CDKN1B were identified in 13 of 81 (16%) patients with HCLc. All harboured at least one CDKN1B nonsense or splice site variant, except for one case in which a missense mutation was identified. Three patients had more than one mutation. implying selective pressure to inactivate CDKN1B. Overall, 11/13 CDKN1B mutations had allele frequencies very similar to those of the BRAF mutant clone, suggesting that CDKN1B mutations are early lesions that may contribute to HCLc pathogenesis by impairing cell cycle control and/or circumventing oncogene-induced senescence. CDKN1B mutations did not impact treatment response to PNAs [49].
- KLF2
The Krüppel-like factor 2 (KLF2) zinc-finger gene, located at chromosome 19p13.1, encodes a transcription factor widely expressed in haemopoietic, endothelial, and lung cells. In B cells, KLF2 regulates the expression of genes involved in cell homing, NF-κB signalling, and cell cycle control. B-cell-specific Klf2-deficient mice show a dramatic increase in cells with a marginal zone-like phenotype [58,59]. The KLF2 protein comprises activating and inhibitory domains, two nuclear localisation sequences (NLSs), and three zinc finger motifs (ZnFs). KLF2 mutations are present in 20–40% of SMZL cases [60,61] and in three studies were also found in 9/74 (12%) cases of HCLc cases [42,62,63]. Mutations may occur in the activation, inhibitory, zinc finger or nuclear localisation domains, are predominantly truncating or missense, and reduce the transcriptional activity of KLF2, partly by displacement from the nucleus if mutations involve the NLS (Figure 4).
2.3. Germline Variants
Familial HCLc exhibits similar clinical features to sporadic HCLc but is rare, with fewer than 20 families reported in the literature. Four multiplex HCLc pedigrees were recently screened for shared germline variants, conferring HCLc susceptibility. Although there was only limited overlap between the pedigrees on a variant or gene level, several functional pathways such as neutrophil-mediated immunity and G-protein-coupled receptor signalling were shared in 3/4 and MAPK and RAS signalling in 2/4 pedigrees, respectively [66].
2.4. Epigenetic Abnormalities
The epigenome comprises chemical modifications to DNA and DNA-associated proteins which modify gene expression mediated through DNA methylation, histone tail modifications, chromatin accessibility, and DNA architecture and are critical for cellular differentiation and response to environmental stimuli. Post-translational modifications (PTM) of histone proteins regulate the accessibility of DNA to transcription factors and DNA repair enzymes by a variety of mechanisms which include recruiting additional chromatin-modifying factors, reducing the positive charge of histones, and altering the positioning of nucleosomes [67]. Mutations of genes that encode chromatin modifiers may also target nonhistone proteins.
2.4.1. Mutations in Chromatin Modifiers
Mutations in genes involved in transcriptional regulation were found in 26/74 (35%) of HCLc cases [11,26,42]. The most frequently mutated gene was the histone methyltransferase KMT2C (MLL3) in which loss-of-function mutations throughout the coding region were identified in 15% (8 of 53) of cases [11]. KMT2C is a member of the KMT2 gene family which promotes methylation of H3K4 at enhancers and super-enhancers and transcription of genes related to cell differentiation or tumour suppression [68]. Other recurring mutations involve CREBBP and EP300, two interacting histone acetylation genes, and BRD4, CEBPA, RUNX1, and MED12. The transcriptional and phenotypic consequences of these mutations are highly context dependent, and their functional consequences in HCLc are unknown.
2.4.2. DNA Methylation Profile
DNA methylation profiling was analysed with the low-resolution Infinium HumanMethylation27 array in 11 cases of BRAFV600E mutated HCLc, together with cases of CLL, SMZL, and normal B-cell subsets. HCLc had a distinct global methylation profile which, nevertheless, was more closely related to SMZL than to CLL and to normal post germinal centre (GC) memory B cells and marginal zone B cells than to pre-GC and GC B cells. When probes inside or outside cytosine guanine dinucleotide islands (CGIs) were analysed separately, the CGI-only methylation profile clustered all HCLc samples in an independent branch, separately from post-GC B cells but together with two of seven SMZL cases.
An integrated analysis of the HCL methylation profile and the previously published gene expression profile showed an inverse correlation between gene expression and methylation, alluding to a role for DNA promoter methylation in the regulation of specific gene expression. Independent supervised analyses were then performed to compare HCL methylation with that of post-GC B cells, SMZL, and CLL. Differential methylation changes observed in HCLc that were also reflected in gene expression patterns were consistent with constitutive activation of the RAS–RAF–MEK–ERK pathway and also affected pathways involved in the homing, migration, and survival of HCL cells [69].
2.5. Immunogenetic Features
The analysis of immunoglobulin gene repertoires in B-cell malignancies has provided key insights into their ontogeny, including their cell(s) of origin, the role and nature of antigenic stimulation in tumour development and evolution; moreover, in some diseases, especially CLL, IGHV gene SHM status has prognostic and predictive value [70].
While the majority of cases of HCLc have mutated IGHV genes, 17–20% are unmutated using a 98% cut-off and <5% have completely unmutated IGHV genes, with 100% identity to the germline. Compared with the normal B-cell repertoire [71], there is biased usage of the IGHV3-21, IGHV3-30, IGHV3-33, and IGHV4-34 genes, each found in 7–10% of cases, with preferential use of IGHV3-30 and IGHV4-34, especially among the unmutated cases. Biased usage of IGHD genes has also been documented [72,73,74,75].
While kappa is the most frequently used immunoglobulin light chain in the normal B-cell repertoire and in other B-cell tumours, HCLc is associated with preferential use of lambda light chains, resulting in an inverted Igκ:Igλ ratio (0.7:1). The explanation for this is unclear but may derive from secondary IG light chain gene rearrangements as part of receptor editing, a physiological process leading to the pairing of the authentic heavy chain with a novel light chain as a means to alleviate intense autoreactivity.
While there is much less diversity within the light chain, compared with the heavy chain repertoire, there is evidence of biased usage in HCLc, as virtually all cases that express lambda light chains utilise the IGLJ3 gene. In addition, the variable lambda complementarity determining region 3 (VL CDR3) of lambda-expressing cases frequently share structural features, while restricted pairings exist between certain conserved lambda light chains and heavy chains encoded by the IGHV3-21/30/33 genes [72].
All of the above features support a role for antigen selective pressure in tumour ontogeny. Moreover, the presence of intra-clonal diversification within the clonotypic IG genes indicates that ongoing SHM occurs post-transformation likely in a context of continuous interactions with antigen(s).
An unusual feature of HCLc is the expression of multiple IGH isotypes on the cell surface, documented in 40% to over 80% of cases. Single-cell analysis has confirmed that this phenomenon is attributable to the expression of multiple isotypes in individual cells rather than to clonal heterogeneity [76]. Heavy-chain isotype switching is mediated through class-switch DNA recombination (CSR) which occurs between two switch (S) regions located 5′ of each IGHC gene. The intervening DNA segments are extruded via a cohesion-driven process and form extrachromosomal DNA switch circles. Deleted circle transcripts are not seen in HCLc cases expressing multiple isotypes, suggesting an arrest of CSR prior to deletional switching but where multiple isotypes can still be generated. CSR requires the upregulation of AID, enhanced chromatin accessibility mediated by histone modifications, and upregulation of factors such as IL-4, TGFβ, or IFNγ whose transcription is dependent on microenvironmental stimuli which determine the choice of specific isotypes. No genomic differences have been reported between cases that express either a single or multiple H chain isotypes, and the cause of the aberrant CSR remains uncertain [77,78].
The expression of multiple CH isotypes, including IgM with IgG or IgA, has also been reported in HCLv and SDRPL, although it has not been demonstrated if they are expressed in single cells. If so, this might point to a microenvironmental factor.
A further anomalous immunogenetic feature of HCLc is an increased incidence of cells expressing both IG kappa and lambda light chains. Dual expression of IG K and L light chains is rare in health, documented in only 0.2–0.5% of B cells from five normal controls [79]. Immunophenotypic analysis of 105 HCLc cases identified 3 (2.86%) that co-expressed surface kappa/lambda in virtually all cells. The immunogenetic analysis identified an additional case with a functional IGK/IGL transcript that also expressed multiple IGH isotypes and a RAG1 transcript. An increased incidence of dual light chain expressing cells is also seen in SLE [80] and in mouse models of autoimmunity [81,82,83]. The functional consequences of dual kappa and lambda light-chain expression are still unknown. That notwithstanding, evidence exists that compromised allelic exclusion leading to dual kappa/lambda expression might allow autoreactive cells to avoid clonal deletion, a mechanism described by the term receptor dilution [81].
2.6. Biological Implications: Cell of Origin
The identity and behaviour of tumour cells are largely controlled by the activity of transcriptional programs which reflect the programs active in, or available to, their cell(s)of origin, and/or their dysregulation by either cell-intrinsic genomic/epigenetic factors or cell-extrinsic interactions with the TME [84,85]. The biological and clinical significance of these variables has been well documented among mature B-cell tumours such as diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma (MCL), and SMZL [86,87,88].
The stem cell origin of the clonal BRAFV600E mutation in HCLc, together with the induction of a lethal haematopoietic disorder with features of HCLc in BRAFV600E mice [26], are consistent with the role of BRAFV600E as an early/initiating event in hairy cell leukemogenesis. However, there remains uncertainty about the nature of the mature B-cell population(s) expanded in HCLc and whether additional genetic and/or epigenetic alterations or a suitable TME are required to give rise to mature HCL cells. Dysregulation of the G1 phase of the cell cycle is a common finding in HCLc, but as yet, no subsequent genomic final transforming event has been discovered.
Data relevant to determining the COO in HCLc include the following:
- The presence of mutated IGHV genes, with evidence for antigen selection, in the majority of cases and preferential use of the IGHV4-34 gene in the minority of cases with low or no SHM;
This phenotype also delineates a subset of normal B cells present in blood and splenic red pulp but rarely in lymph nodes. These cells also lack expression of CD21 and the chemokine receptors CD185 (CXCR5) and CD184 (CXCR4), reflecting their distribution within lymphoid organs. They frequently express sIgG, consistent with the role of Tbet in regulating antibody class switching to IgG1 or IgG3. Cells with a CD11c+ Tbet+ phenotype are found within B-cell populations variously described as age-associated B cells, atypical B cells, and double-negative B cells. CD11c+, Tbet+ B cell numbers increase with age and are expanded in conditions associated with chronic antigenic stimulation such as infections with human immunodeficiency virus and malaria, and in autoimmune diseases such as SLE. However, the CD11c+ Tbet+ phenotype does not, by itself, identify a distinct B-cell population, nor a specific B-cell lineage, and can be found in activated naïve B cells and in memory B cells believed to be generated through follicular or extra-follicular maturation pathways [93,94,95,96,97,98,99].
Immunogenetic, transcriptomic, and epigenetic analysis of B cells based either on the expression of CD11chi, Tbet, or a double-negative phenotype (CD19+ IgD- CD27- CXCR5-) shows significant differences from canonical CD11-ve Tbet -memory B cells [96,100,101]. Interestingly, the CD11c+ cohort is associated with enrichment of IGHV4-34 gene usage [100]. Immunoglobulins encoded by the IGHV4-34 gene display autoreactivity to the I/i antigens present on erythrocytes by virtue of a germline motif within the VH FR1 and additionally show cross-reactivity with other self and microbial antigens. Naïve B cells expressing IGHV4-34 are often anergic, while this gene is largely excluded from switched memory and plasma cells. The persistence of this gene in a spleen-resident Tbet+ memory B-cell subset has been postulated to reflect either positive selection of B cells that may facilitate clearing of self-antigens or neutralisation of microbial antigens [102] or a defect in negative selection during GC transit [100].
It would be interesting to review the transcriptomic and methylation data in HCLc using normal splenic CD11c+ Tbet+ CD27- cells rather than CD27+ memory B cells as the comparator. However, currently, and as in CLL despite extensive studies [103], the COO of HCLc remains enigmatic.
2.7. Clinical Implications of Genetic Features
2.7.1. Diagnosis
The initial description of BRAFV600E in HCLc failed to find the same mutation in 195 cases of other mature B-cell tumours including CLL, follicular lymphoma, DLBCL, and other splenic lymphomas [9]. However, subsequent screening of larger cohorts of CLL and myeloma for both BRAFV600E and other BRAF hotspot mutations has identified a low incidence of predominantly subclonal V600E and non V600E BRAF mutations, usually associated with a poorer outcome [104,105,106,107,108].
Whilst a confident diagnosis of HCLc can be made without knowledge of the BRAF mutation status, the specificity of the BRAFV600E mutation for HCLc among splenic lymphomas is valuable when there is diagnostic uncertainty, and its presence underpins the use of targeted inhibitors. If it emerges that widely available diagnostic criteria are unable to distinguish BRAFV600E HCLc from BRAFWT HCLc, this would provide an additional rationale for BRAFV600E mutation screening. Allele-specific PCR performed on blood or marrow aspirate samples has superseded less sensitive molecular techniques such as Sanger sequencing, pyrosequencing, or melting curve analysis [109]. Digital, droplet PCR has comparable specificity and superior sensitivity to QT–PCR and is a potential method for MRD analysis [110]. Immunohistochemistry (IHC) using a BRAFV600E-specific antibody is an alternative method suitable for bone marrow trephine or other tissue sections, with comparable sensitivity and specificity to allele-specific PCR. Next-generation sequencing (NGS) also has high sensitivity but, currently, also has higher costs and longer turnaround time, compared with allele-specific tests [111].
2.7.2. Prognostic Significance of IGHV Gene Somatic Hypermutation Status
The clinical significance of IGHV gene SHM status in HCLc has been evaluated in two studies with discordant results. In a trial of single-agent cladribine in 58 previously untreated patients, all expressing annexin A1, failure to respond was observed in 5/6 patients with unmutated IGHV genes using a 98% cut-off value, only one of whom used IGHV4-34. Bulky splenomegaly, leucocytosis, and TP53 abnormalities were present in four, three, and two of the five cases, respectively [73].
In a cohort of 62 patients with HCLc and 20 with HCLv diagnosed according to the WHO 2008 criteria [112], IGHV4-34 was used in 6 (10%) of HCLc and 8 (40%) of HCLv cases, respectively, and was unmutated in all but 1 case, using a 98% cut-off value. A suboptimal response to first-line treatment with cladribine was seen in 4/6 IGHV4-34 HCLc positive cases, compared with 4/56 IGHV4-34 negative cases. A worse response was also seen in IGHV4-34 positive HCLv cases, suggesting that outcome was more closely related to IGHV4-34 status than to whether or not patients had HCL or HCLv. However, many of the HCLc cases were BRAFV600E negative [113].
2.7.3. BRAFV600E as a Therapeutic Target
The purine nucleoside analogues (PNAs), pentostatin, and cladribine remain the current treatment of choice for first-line therapy of HCLc. However, PNAs may cause short-term myelosuppression, with an increased risk of infection and an increased risk of secondary malignancies, and approximately 50% of patients eventually relapse. Single-agent vemurafenib or dabrafenib resulted in high overall response rates without minimal/measurable residual disease (MRD) negativity in relapsed/refractory HCL, but the median relapse-free survival in responders was less than 1 year [114,115]. In contrast, a phase II study of vemurafenib plus rituximab achieved a CR rate of 87%, of whom 65% were MRD negative and with relapse-free survival of 85% at a median follow-up of 34 months [116].
2.7.4. Genomic Abnormalities as Predictors of Drug Resistance
- To PNAs
Targeted mutational and copy number analysis showed no difference in the pattern of genomic abnormalities between treatment naïve cases and those refractory to a PNA [11]. Serial samples from two HCL-c cases tested both at diagnosis and relapse post-PNA therapy, revealed two additional subclonal mutations of BCOR (BCORE1430X) and XPO1 (XPO1E571K) in one case, while the second case remained genomically stable [42]. However, there is no clear evidence to suggest that genomic mutations confer resistance to PNAs in HCLc.
- To BRAF Inhibitors
Of 13 evaluable HCLc cases treated with vemurafenib, 6 showed persistence of ERK phosphorylation in bone marrow cells, suggesting that, in at least some patients, the growth of HCL cells remains dependent on MEK–ERK signalling, likely reactivated through mechanisms bypassing BRAF inhibition by vemurafenib. In support, targeted sequencing of 300 genes performed in one patient who was refractory to vemurafenib showed two separate activating subclonal KRAS mutations at relapse [116].
A further case with vemurafenib resistance had heterozygous deletions of BRAF, NF1, NF2, and TP53 and subclonal mutations in CREBBP and IRS1 in a pretreatment sample. NF1 and NF2 encode tumour suppressors that have been experimentally implicated in RAF inhibitor resistance in epithelial cancer cells [117] and downregulation of either or both Nf1 or Nf2 in Ba/F3 cells stably expressing BRAFV600E conferred vemurafenib resistance in vitro [11].
Seven distinct activating mutations in KRAS and two mutations in MAP2K1 were detected in the relapse sample of a patient resistant to a PNA and vemurafenib plus rituximab. Allele frequencies were consistent with the parallel, convergent evolution of multiple clones with KRAS mutations appearing before MAP2K1 mutations. Treatment with MEK inhibitor cobimetinib in combination with vemurafenib resulted in significant clinical and haematological improvement, associated with suppression of mutant allele frequencies for BRAF, KRAS, and MAP2K1 mutations and of ERK activity [118].
Elucidating the mechanisms of resistance to BRAF inhibitors in solid tumours, especially melanoma, is an area of intensive investigation. In addition to the selection of genomic mutations such as mutations of RAS or MAP2K1/MEK1 or of drug-tolerant persister cells, it is increasingly recognised that tumour cells may undergo non-genetic adaptive changes such as metabolic reprograming or reversion to a progenitor cell phenotype which result in drug resistance. It remains to be seen whether such adaptive changes will emerge in HCLc, a tumour with significantly less genomic complexity and instability [119,120,121,122,123].
3. Hairy Cell Variant
3.1. Cytogenetic and Copy Number Abnormalities
CBA showed an abnormal karyotype in 12/17 (71%) of cases, of which 5 (29%) were complex, defined as three or more chromosomal abnormalities. Recurrent aberrations included 17p abnormalities and del(18q) each in three cases. FISH analysis showed TP53 deletion or monosomy 17 in 5 of 12 (42%) cases and ATM deletion or monosomy 11 was detected in 2 of 9 (22.2%) cases. One case showed del(7q) by both conventional karyotype and FISH analysis [124]. Using single-nucleotide polymorphism (SNP) arrays in 15 previously untreated cases, CNAs were identified in 14 (93%) cases, with a mean of 7.9 abnormalities per case. Although the data are limited, combined CBA and SNP results suggest a greater degree of genomic complexity in HCLv than HCLc. Gains on chromosome 5 were identified in 5 cases and deletions of 17p and 7q in five and three cases, respectively [47]. Copy number analysis of regions covered in a targeted sequencing study identified 7q deletions and also recurrent 3p deletions which included a critical tumour suppressor locus encoding VHL, SETD2, BAP1, and PBRM1 [11].
3.1.1. Recurring Mutations
Information on the genomic landscape of HCLv is also based on limited data—namely, WGS in 7 cases published in abstract from only [125], WES in 7 cases [43] targeted sequencing using a cancer gene panel in 12 cases [11,42], and targeted sequencing of MAP2K1 in 25 cases [42,124,126] and of TP53 in 30 cases [127]. No case had the BRAF-V600E mutation. Recurring mutations have been found in TP53, among cases with a 17p deletion [47], in MAP2K1, U2AF1, and KDM6A, as discussed below, and in ARID1A and CREBBP, while single mutations were identified in CCND3, in genes involved in transcriptional regulation (CEBPA, DDX3X, and PBRM1) and chromatin remodelling (KMT2C and KDM5C).
- MAP2K1
Waterfall first identified MAP2K1 mutations in HCLv in 10/24 (41%) cases. The mutations mapped predominantly to the regions encoding the negative regulatory region and catalytic core (Figure 5) and are functionally active, increasing the basal enzymatic activity of MEK, encoded by MAP2K1 [128]. The only non-missense mutation was a 48 bp in-frame deletion (amino acids 42 through 57) that almost entirely removed the autoinhibitory helix A13. Subsequent studies have confirmed the finding of recurring MAP2K1 mutations but at a varying and predominantly lower incidence, with 2/4, 3/8, 2/11, and 1/14 cases and an overall incidence of 8/37 (22%) [11,42,124,126].
- U2AF1
U2AF1 encodes the U2AF1 protein which heterodimerises with U2AF2, to form a key component of the splicesome. Among haematological malignancies, U2AF1 and U2AF2 mutations are largely restricted to myeloid neoplasms, especially high-risk myelodysplastic syndromes and acute myeloid leukaemia in which U2AF1 mutants may alter the differential splicing of many genes that affect various biological pathways, including DNA damage response (ATR and FANCA) and epigenetic regulation (H2AFY, ASXL1, BCOR, and DNMT3B). Subclonal hotspot p.Ser34Phe U2AF1 mutations were identified in 2/7 cases of HCLv [43] and in one case which underwent a high-grade transformation in a lymph node 7 years after presentation, the mutation was found in both pre- and post-transformation samples [129]. The biological significance of U2AF1 mutations in HCLv is unknown, but they are potential targets for splicing inhibitors.
- KDM6A
KDM6A encodes a lysine demethylase protein that removes di- and tri-methyl groups from lysine 27 of Histone 3 (H3K27). Potentially deleterious mutations resulting in the loss of the highly conserved C-terminal region of KDM6A, essential for its demethylase activity, were identified in 2/4 cases, one of which was unresponsive to first- and second-line therapies [42]. Their biological and clinical significance in HCLv is unknown but the loss of KDM6A activity may sensitise tumour cells to demethylating agents such as EZH2 inhibitors [130].
3.1.2. Immunogenetic Features
Immunogenetic profiling in HCLv shows distinct differences in the incidence of somatic hypermutation and IGHV gene usage, compared with HCLc. A study of 41 patients revealed that 22% had truly unmutated IGHV genes, with 100% germline identity, and 5% were borderline mutated, with 97–99.9% germline identity. The most commonly used gene was IGHV4-34, in 17% of cases, of which 67% were truly unmutated [74]. Similar findings were reported in 26 patients, with 5/28 (18%) of rearrangements truly unmutated and 10/28 (36%) borderline mutated. IGHV4-34 was used in 10 (36%) cases, and all were unmutated, using a 98% cut-off [75].
3.2. Clinical Implications
3.2.1. Prognostic Significance of TP53 Aberrations
A significant association was found between 17p deletion and shorter overall survival [127]. In a recently reported phase II study of CDA plus rituximab in 20 patients, the overall CR rate was 95%, and 80% achieved bone marrow MRD negativity at 6 months. The five patients with a TP53 mutation had a significantly shorter PFS and OS than those with wild-type TP53 [131].
3.2.2. Targeted Therapy
In contrast to HCLc, current treatments for HCLv are suboptimal, with chemoimmunotherapy remaining the preferred initial therapy [6,132]. A patient with IGHV4-34 expressing HCLv who had relapsed with skin nodules following multiple previous treatments, including chemoimmunotherapy and allogeneic transplantation, was found to have a somatic MAP2K1 p.K57N mutation, with a VAF of 43.26% in skin and 20.08% in blood. He received the MEK inhibitor trametinib and achieved a partial response [133]. Based on partial or complete remissions following compassionate use of MEK inhibitors in patients with either HCLc with wild-type BRAF or HCLv, use of the MEK inhibitor binimetinib is currently being explored in a phase II trial of both patient groups who have relapsed or refractory disease, have received at least one course of a purine analogue, and who require further treatment. Trial inclusion criteria do not include a requirement to show evidence for dysregulation of the MAPK pathway such as a MAP2K1 mutation or increased pERK expression [134].
4. Splenic Diffuse Red Pulp Lymphoma
4.1. Cytogenetic and Copy Number Abnormalities
The largest studies in SDRPL employing CBA identified cytogenetic abnormalities in 35–57% of cases, of which 13% had a complex karyotype. The most frequent abnormality was 7q deletion in 18–25%, while recurring trisomies of chromosomes 3, 12, or 18 were also seen [7,64,135]. In contrast, copy number analysis in 16 cases using array-comparative genomic hybridisation identified aberrations in 69% of samples, including recurrent losses of 10q23, 14q31–q32, and 17p13 in three, and 9p21 in two cases. Deletion of 7q31.3–q32.3 was present in only one case, and trisomy 3 or 18 were not detected [136].
4.2. Genomic Mutations
As with HCLc, the data are limited and based on WES in 33 cases, targeted sequencing using a panel of 109 genes relevant to lymphomagenesis in 42 cases, and targeted sequencing of CCND3 in 34 cases and of BRAF, MAP2K1, MYD88, NOTCH1, NOTCH2, SF3B1 and TP53 in 23–36 cases. The most frequent recurring abnormalities involved CCND3 and BCOR, as described below, while single- or low-frequency recurrent mutations were found in genes encoding proteins involved in cell cycle regulation, epigenetic regulation, the RAS–MAPK pathway, NF-κB and NOTCH signalling, cytoskeleton, and cell–matrix interactions [64,135,136]. No BRAFV600E mutations were found, but a single BRAF mutation (p.G469A) was identified in a case with a non-HCLc (CD103+, CD25-, CD123-) immunophenotype, expression of an unmutated IGHV4-34-encoded BcR IG and a MAP2K1 mutation, highlighting the overlap of genomic and immunogenetic features in some cases diagnosed as HCLv or SDRPL.
4.2.1. CCND3 Mutations
CCND3 located at 12p13 encodes cyclin D3, required in normal B cells for the proliferative expansion of pre-B cells and of B cells within the dark zone of germinal centres [137,138]. In addition, cyclin D3 has several non-canonical functions which include the activation or repression of transcription either directly or via the recruitment of chromatin modifiers to gene promotors [139].
CCND3 mutations have been identified in 20–24% cases of SDRPL and almost invariably comprise missense variants in the negative regulatory proline, glutamic acid, serine, and threonine (PEST) domain, involving the amino acids T283, P284, and I290 which are part of a phosphorylation motif that regulates cyclin D3 phosphorylation and stability [64,140].
Cyclin D3 was shown to be overexpressed in >50% of tumour cells from splenectomy samples in all cases with a CCND3 mutation and also in 19/24 cases without a CCND3 mutation. CCND3/IGH translocations resulting in cyclin D3 overexpression have previously been documented in other B-cell lymphomas, but no CCND3 translocations were detected using a CCND3 Break Apart FISH Probe Kit [140]. Currently, neither the functional consequences of cyclin D3 overexpression in SDRPL nor the explanation for cyclin D3 overexpression in CCND3 WT cases is understood. However, a subsequent study in cyclin D1 negative MCL expressing cyclin D3 revealed cryptic insertion of the IGK/L enhancer upstream of the CCND3 gene that was undetectable by standard FISH probes and was associated with CCND3 overexpression [141].
4.2.2. BCOR Abnormalities
The BCL6 co-repressor (BCOR) gene, located at Xp11.4., encodes the widely expressed BCOR protein whose function is highly tissue specific. In germinal centres (GC), BCOR interacts with polycomb repressive complex 1 (PCR1) and BCL6, facilitating the transient repression of genes associated with DNA damage response, cell cycle checkpoint control, GC exit, and plasma cell differentiation [142].
BCOR mutations, found in 16% of SDRPL cases, are characterised by splicing site (1/6), nonsense (2/6), and frameshift (3/6) alterations across the coding sequence, consistent with loss of function, as seen in other lymphoid and myeloid malignancies. Overall, 4/6 mutations exhibited a high variant allele frequency. Additionally, loss of the BCOR locus due either to a microdeletion or loss of a whole X chromosome was found in four SDRPL female patients, with no BCOR mutation within the remaining allele, resulting in an overall incidence of BCOR abnormalities in 11/42 cases.
4.3. Immunogenetic Features
The great majority (79–89%) of cases have been found to carry hypermutated IGHV genes (<100% identity), with overrepresentation of the IGHV3-23 and IGHV4-34 genes. Of 10 cases using IGHV4-34, 6 were borderline unmutated, and 4 were truly unmutated, comparable to the findings in HCLv. IGHV1-2 usage was confined to a single case [64,135]. Broadly similar findings were reported in another study of 13 patients [136].
5. Conclusions and Future Studies
A major focus of this review was the key role that the discovery of the almost ubiquitous clonal BRAFV600E mutation has played in understanding the biology of HCLc and its importance both in differential diagnosis and as a therapeutic target. However, there remain many unanswered questions regarding the diagnosis and biology of both HCLc and, particularly, HCLv and SDRPL. Of greater clinical importance is an unmet need for potentially curative non-chemotherapeutic regimens for HCLc and more effective treatments for HCLv which additional genetic data may help to resolve.
While the finding of a BRAFV600E mutation in HCLc unequivocally identifies a disorder with largely uniform laboratory and clinical features, methylome and clinical course, the pathogenesis of less frequent features such as skeletal involvement, found in 3% of cases [144], and a propensity to autoimmune disease [145] remain unexplained. Additionally, there is still much to learn about the incidence, biology, and optimal management of cases with a typical HCLc phenotype that lacks the BRAFV600E mutation or another mechanism for BRAF upregulation.
It is also unlikely to be coincidental that BRAF WT HCLc cases display enrichment for IGHV4-34 gene usage, frequently accompanied by activating MAP2K1 mutations, and that these two features are also found in a subset of cases with HCLv, raising questions about the inter-relationship between these two patient groups. If IGHV4-34-positive, MAP2K1-mutated cases of HCLc and HCLv do exhibit the typical phenotypes of HCLc and HCLv, respectively, what might account for the differences between the two phenotypes?
There is also uncertainty about the relationship between HCLv and SDRPL, given their many overlapping features and the current absence of disease-defining genetic abnormalities. The absence of reports of the rare cases of SDRPL with progressive disease acquiring typical features of HCLv such as TP53 abnormalities or prominent nucleoli would suggest they are not simply different stages of a single disease.
These uncertainties are, in large part, a consequence of the rarity of these disorders, the lack of cell lines and animal models, and the difficulty in obtaining tumour cells, especially in HCLc, where the circulating tumour cell count is usually low, bone marrow aspiration is unsuccessful, and splenectomy rarely performed [146,147]. This is reflected in the lack of genomic data on HCLc and especially HCLv and SDRPL, compared with that available in the more common B-cell tumours, such that the published genomic landscapes are unlikely to reflect the full range or true incidence of CNAs and mutations present in all three disorders.
New biological insights are likely to require studies in larger multi-institution patient cohorts, together with the application of newer technologies such as WGS, and transcriptomic and epigenetic analyses, both at the bulk and single-cell levels, comparing data from tumour cells with that from normal splenic B-cell subsets.
It is conceivable that these studies, in conjunction with a more detailed analysis of the TME, may lead to the identification of new disease subsets within or spanning the current diagnoses of BRAF WT HCLc, HCLv, and SDRPL, offer new insights into their cells of origin, and give rise to a more genetically based classification, offering more precise diagnostic and prognostic features and targeted therapies.
Author Contributions
Conceptualisation, D.O. and K.S.; data curation, A.M.; writing—original draft preparation, D.O.; writing—review and editing, D.O., K.S., A.M. and J.S. All authors have read and agreed to the published version of the manuscript.
Funding
A.M. is funded by the University of Southampton Centre for Cancer Immunology Talent Fund; J.S. is supported by the Cancer Research UK (CRUK) ECRIN-M3 program (grant C42023/A29370); the CRUK B-cell malignancy program (C2750/A23669); and CRUK Southampton Centre core funding (C36811/A29101).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouroncle, B.A.; Wiseman, B.K.; Doan, C.A. Leukemic reticuloendotheliosis. Blood 1958, 13, 609–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, M.; Dearden, C. Hairy Cell Leukaemia. Curr. Oncol. Rep. 2020, 22, 42. [Google Scholar] [CrossRef] [PubMed]
- Maitre, E.; Cornet, E.; Troussard, X. Hairy cell leukemia: 2020 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2019, 94, 1413–1422. [Google Scholar] [CrossRef]
- Cawley, J.C.; Burns, G.F.; Hayhoe, F.G. A chronic lymphoproliferative disorder with distinctive features: A distinct variant of hairy-cell leukaemia. Leuk. Res. 1980, 4, 547–559. [Google Scholar] [CrossRef]
- Matutes, E. Diagnostic and therapeutic challenges in hairy cell leukemia-variant: Where are we in 2021? Expert Rev. Hematol. 2021, 14, 355–363. [Google Scholar] [CrossRef]
- Traverse-Glehen, A.; Baseggio, L.; Bauchu, E.C.; Morel, D.; Gazzo, S.; Ffrench, M.; Verney, A.; Rolland, D.; Thieblemont, C.; Magaud, J.P.; et al. Splenic red pulp lymphoma with numerous basophilic villous lymphocytes: A distinct clinicopathologic and molecular entity? Blood 2008, 111, 2253–2260. [Google Scholar] [CrossRef] [Green Version]
- Kanellis, G.; Mollejo, M.; Montes-Moreno, S.; Rodriguez-Pinilla, S.M.; Cigudosa, J.C.; Algara, P.; Montalban, C.; Matutes, E.; Wotherspoon, A.; Piris, M.A. Splenic diffuse red pulp small B-cell lymphoma: Revision of a series of cases reveals characteristic clinico-pathological features. Haematologica 2010, 95, 1122–1129. [Google Scholar] [CrossRef] [Green Version]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [Green Version]
- Tiacci, E.; Schiavoni, G.; Martelli, M.P.; Boveri, E.; Pacini, R.; Tabarrini, A.; Zibellini, S.; Santi, A.; Pettirossi, V.; Fortini, E.; et al. Constant activation of the RAF-MEK-ERK pathway as a diagnostic and therapeutic target in hairy cell leukemia. Haematologica 2013, 98, 635–639. [Google Scholar] [CrossRef]
- Durham, B.H.; Getta, B.; Dietrich, S.; Taylor, J.; Won, H.; Bogenberger, J.M.; Scott, S.; Kim, E.; Chung, Y.R.; Chung, S.S.; et al. Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood 2017, 130, 1644–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Mooi, W.J.; Peeper, D.S. BRAF(E600) in benign and malignant human tumours. Oncogene 2008, 27, 877–895. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.E.; Merad, M.; McClain, K.L. Langerhans-Cell Histiocytosis. N. Engl. J. Med. 2018, 379, 856–868. [Google Scholar] [CrossRef]
- Gulati, N.; Allen, C.E. Langerhans cell histiocytosis: Version 2021. Hematol. Oncol. 2021, 39 (Suppl. S1), 15–23. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.L.; Arber, D.A.; Pittaluga, S.; Martinez, A.; Burke, J.S.; Raffeld, M.; Camos, M.; Warnke, R.; Jaffe, E.S. Clonally related follicular lymphomas and histiocytic/dendritic cell sarcomas: Evidence for transdifferentiation of the follicular lymphoma clone. Blood 2008, 111, 5433–5439. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.T.; Lakshmanan, A.; Lehman, A.; Harrington, B.K.; Lucas, F.M.; Tran, M.; Sass, E.J.; Long, M.; Flechtner, A.D.; Jaynes, F.; et al. BRAF(V600E) accelerates disease progression and enhances immune suppression in a mouse model of B-cell leukemia. Blood Adv. 2017, 1, 2147–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, C.; Lack, J.; Skarshaug, S.; Pham, T.A.; Abdullaev, Z.; Xi, L.; Pack, S.; Pittaluga, S.; Jaffe, E.S.; Raffeld, M. The mutational landscape of histiocytic sarcoma associated with lymphoid malignancy. Mod. Pathol. 2021, 34, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Kang, B.; Petkovich, D.A.; Bhandari, Y.R.; In, J.; Stein-O’Brien, G.; Kong, X.; Xie, W.; Zachos, N.; Maegawa, S.; et al. Aging-like Spontaneous Epigenetic Silencing Facilitates Wnt Activation, Stemness, and Braf(V600E)-Induced Tumorigenesis. Cancer Cell 2019, 35, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Baggiolini, A.; Callahan, S.J.; Montal, E.; Weiss, J.M.; Trieu, T.; Tagore, M.M.; Tischfield, S.E.; Walsh, R.M.; Suresh, S.; Fan, Y.; et al. Developmental chromatin programs determine oncogenic competence in melanoma. Science 2021, 373, eabc1048. [Google Scholar] [CrossRef] [PubMed]
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiewicz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Lutz, C.; Buss, E.C.; Nowak, D.; et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017, 19, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triana, S.; Vonficht, D.; Jopp-Saile, L.; Raffel, S.; Lutz, R.; Leonce, D.; Antes, M.; Hernandez-Malmierca, P.; Ordonez-Rueda, D.; Ramasz, B.; et al. Single-cell proteo-genomic reference maps of the hematopoietic system enable the purification and massive profiling of precisely defined cell states. Nat. Immunol. 2021, 22, 1577–1589. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Kim, E.; Park, J.H.; Chung, Y.R.; Lito, P.; Teruya-Feldstein, J.; Hu, W.; Beguelin, W.; Monette, S.; Duy, C.; et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci. Transl. Med. 2014, 6, 238ra271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loghavi, S.; Khoury, J.D. Langerhans cell histiocytosis in a patient with hairy cell leukemia: A tale of divergence. Blood 2017, 129, 1563. [Google Scholar] [CrossRef]
- Bigenwald, C.; Le Berichel, J.; Wilk, C.M.; Chakraborty, R.; Chen, S.T.; Tabachnikova, A.; Mancusi, R.; Abhyankar, H.; Casanova-Acebes, M.; Laface, I.; et al. BRAF(V600E)-induced senescence drives Langerhans cell histiocytosis pathophysiology. Nat. Med. 2021, 27, 851–861. [Google Scholar] [CrossRef]
- Biavasco, R.; Lettera, E.; Giannetti, K.; Gilioli, D.; Beretta, S.; Conti, A.; Scala, S.; Cesana, D.; Gallina, P.; Norelli, M.; et al. Oncogene-induced senescence in hematopoietic progenitors features myeloid restricted hematopoiesis, chronic inflammation and histiocytosis. Nat. Commun. 2021, 12, 4559. [Google Scholar] [CrossRef]
- Sivina, M.; Burger, J.A. The importance of the tissue microenvironment in hairy cell leukemia. Best Pract. Res. Clin. Haematol. 2015, 28, 208–216. [Google Scholar] [CrossRef]
- Bohn, J.P.; Salcher, S.; Pircher, A.; Untergasser, G.; Wolf, D. The Biology of Classic Hairy Cell Leukemia. Int. J. Mol. Sci. 2021, 22, 7780. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Liso, A.; Tiacci, E.; Benedetti, R.; Pulsoni, A.; Foa, R.; Di Raimondo, F.; Ambrosetti, A.; Califano, A.; Klein, U.; et al. Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J. Exp. Med. 2004, 199, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4519–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettirossi, V.; Santi, A.; Imperi, E.; Russo, G.; Pucciarini, A.; Bigerna, B.; Schiavoni, G.; Fortini, E.; Spanhol-Rosseto, A.; Sportoletti, P.; et al. BRAF inhibitors reverse the unique molecular signature and phenotype of hairy cell leukemia and exert potent antileukemic activity. Blood 2015, 125, 1207–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, E.M.; Bench, A.J.; van’t Veer, M.B.; Wright, P.; Bloxham, D.M.; Follows, G.A.; Scott, M.A. High resolution melting analysis for detection of BRAF exon 15 mutations in hairy cell leukaemia and other lymphoid malignancies. Br. J. Haematol. 2011, 155, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Arcaini, L.; Zibellini, S.; Boveri, E.; Riboni, R.; Rattotti, S.; Varettoni, M.; Guerrera, M.L.; Lucioni, M.; Tenore, A.; Merli, M.; et al. The BRAF V600E mutation in hairy cell leukemia and other mature B-cell neoplasms. Blood 2012, 119, 188–191. [Google Scholar] [CrossRef] [Green Version]
- Blombery, P.A.; Wong, S.Q.; Hewitt, C.A.; Dobrovic, A.; Maxwell, E.L.; Juneja, S.; Grigoriadis, G.; Westerman, D.A. Detection of BRAF mutations in patients with hairy cell leukemia and related lymphoproliferative disorders. Haematologica 2012, 97, 780–783. [Google Scholar] [CrossRef] [Green Version]
- Xi, L.; Arons, E.; Navarro, W.; Calvo, K.R.; Stetler-Stevenson, M.; Raffeld, M.; Kreitman, R.J. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012, 119, 3330–3332. [Google Scholar] [CrossRef] [Green Version]
- Rogers, K.A.; Andritsos, L.A.; Wei, L.; McLaughlin, E.M.; Ruppert, A.S.; Anghelina, M.; Blachly, J.S.; Call, T.; Chihara, D.; Dauki, A.; et al. Phase 2 study of ibrutinib in classic and variant hairy cell leukemia. Blood 2021, 137, 3473–3483. [Google Scholar] [CrossRef]
- Tschernitz, S.; Flossbach, L.; Bonengel, M.; Roth, S.; Rosenwald, A.; Geissinger, E. Alternative BRAF mutations in BRAF V600E-negative hairy cell leukaemias. Br. J. Haematol. 2014, 165, 529–533. [Google Scholar] [CrossRef]
- Thompson, E.R.; Lim, K.J.C.; Kuzich, J.A.; McBean, M.; Westerman, D.; Tam, C.S.; Blombery, P. Detection of an IGH-BRAF fusion in a patient with BRAF Val600Glu negative hairy cell leukemia. Leuk. Lymphoma 2020, 61, 2024–2026. [Google Scholar] [CrossRef] [PubMed]
- Maitre, E.; Bertrand, P.; Maingonnat, C.; Viailly, P.J.; Wiber, M.; Naguib, D.; Salaun, V.; Cornet, E.; Damaj, G.; Sola, B.; et al. New generation sequencing of targeted genes in the classical and the variant form of hairy cell leukemia highlights mutations in epigenetic regulation genes. Oncotarget 2018, 9, 28866–28876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterfall, J.J.; Arons, E.; Walker, R.L.; Pineda, M.; Roth, L.; Killian, J.K.; Abaan, O.D.; Davis, S.R.; Kreitman, R.J.; Meltzer, P.S. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat. Genet. 2014, 46, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Haglund, U.; Juliusson, G.; Stellan, B.; Gahrton, G. Hairy cell leukemia is characterized by clonal chromosome abnormalities clustered to specific regions. Blood 1994, 83, 2637–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierlamm, J.; Stefanova, M.; Wlodarska, I.; Michaux, L.; Hinz, K.; Penas, E.M.; Maes, B.; Hagemeijer, A.; De Wolf-Peeters, C.; Hossfeld, D.K. Chromosomal gains and losses are uncommon in hairy cell leukemia: A study based on comparative genomic hybridization and interphase fluorescence in situ hybridization. Cancer Genet. Cytogenet. 2001, 128, 164–167. [Google Scholar] [CrossRef]
- Andersen, C.L.; Gruszka-Westwood, A.; Ostergaard, M.; Koch, J.; Jacobsen, E.; Kjeldsen, E.; Nielsen, B. A narrow deletion of 7q is common to HCL, and SMZL, but not CLL. Eur. J. Haematol. 2004, 72, 390–402. [Google Scholar] [CrossRef]
- Hockley, S.L.; Morgan, G.J.; Leone, P.E.; Walker, B.A.; Morilla, A.; Else, M.; Wotherspoon, A.; Dearden, C.; Catovsky, D.; Gonzalez, D.; et al. High-resolution genomic profiling in hairy cell leukemia-variant compared with typical hairy cell leukemia. Leukemia 2011, 25, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.; Kwee, I.; Young, K.H.; Zucca, E.; Gaidano, G.; Forconi, F.; Bertoni, F. Genome-wide high resolution DNA profiling of hairy cell leukaemia. Br. J. Haematol. 2013, 162, 566–569. [Google Scholar] [CrossRef]
- Dietrich, S.; Hullein, J.; Lee, S.C.; Hutter, B.; Gonzalez, D.; Jayne, S.; Dyer, M.J.; Oles, M.; Else, M.; Liu, X.; et al. Recurrent CDKN1B (p27) mutations in hairy cell leukemia. Blood 2015, 126, 1005–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston-Bell, N.J.; Tapper, W.; Gibson, J.; Bryant, D.; Moreno, Y.; John, M.; Ennis, S.; Kluin-Nelemans, H.C.; Collins, A.R.; Sahota, S.S. Exome Sequencing in Classic Hairy Cell Leukaemia Reveals Widespread Variation in Acquired Somatic Mutations between Individual Tumours Apart from the Signature BRAF V(600)E Lesion. PLoS ONE 2016, 11, e0149162. [Google Scholar] [CrossRef] [Green Version]
- Bencivenga, D.; Caldarelli, I.; Stampone, E.; Mancini, F.P.; Balestrieri, M.L.; Della Ragione, F.; Borriello, A. p27(Kip1) and human cancers: A reappraisal of a still enigmatic protein. Cancer Lett. 2017, 403, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.M.; Braun, M.; Strmiska, V.; Sicinski, P. Targeting cell-cycle machinery in cancer. Cancer Cell 2021, 39, 759–778. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Chiarle, R.; Lestani, M.; Menestrina, F.; Montagna, L.; Ambrosetti, A.; Prolla, G.; Pizzolo, G.; Doglioni, C.; Piva, R.; et al. Low expression of p27 and low proliferation index do not correlate in hairy cell leukaemia. Br. J. Haematol. 2000, 111, 263–271. [Google Scholar] [CrossRef]
- Bhatt, K.V.; Spofford, L.S.; Aram, G.; McMullen, M.; Pumiglia, K.; Aplin, A.E. Adhesion control of cyclin D1 and p27Kip1 levels is deregulated in melanoma cells through BRAF-MEK-ERK signaling. Oncogene 2005, 24, 3459–3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, K.V.; Hu, R.; Spofford, L.S.; Aplin, A.E. Mutant B-RAF signaling and cyclin D1 regulate Cks1/S-phase kinase-associated protein 2-mediated degradation of p27Kip1 in human melanoma cells. Oncogene 2007, 26, 1056–1066. [Google Scholar] [CrossRef] [Green Version]
- le Sage, C.; Nagel, R.; Egan, D.A.; Schrier, M.; Mesman, E.; Mangiola, A.; Anile, C.; Maira, G.; Mercatelli, N.; Ciafre, S.A.; et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007, 26, 3699–3708. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, Y.; Brahmachary, M.; Tiacci, E.; Dalla-Favera, R.; Falini, B.; Basso, K. A microRNA signature specific for hairy cell leukemia and associated with modulation of the MAPK-JNK pathways. Leukemia 2012, 26, 2564–2567. [Google Scholar] [CrossRef] [Green Version]
- Hoek, K.L.; Gordy, L.E.; Collins, P.L.; Parekh, V.V.; Aune, T.M.; Joyce, S.; Thomas, J.W.; Van Kaer, L.; Sebzda, E. Follicular B cell trafficking within the spleen actively restricts humoral immune responses. Immunity 2010, 33, 254–265. [Google Scholar] [CrossRef] [Green Version]
- Hart, G.T.; Wang, X.; Hogquist, K.A.; Jameson, S.C. Kruppel-like factor 2 (KLF2) regulates B-cell reactivity, subset differentiation, and trafficking molecule expression. Proc. Natl. Acad. Sci. USA 2011, 108, 716–721. [Google Scholar] [CrossRef] [Green Version]
- Campos-Martin, Y.; Martinez, N.; Martinez-Lopez, A.; Cereceda, L.; Casado, F.; Algara, P.; Oscier, D.; Menarguez, F.J.; Garcia, J.F.; Piris, M.A.; et al. Clinical and diagnostic relevance of NOTCH2-and KLF2-mutations in splenic marginal zone lymphoma. Haematologica 2017, 102, e310–e312. [Google Scholar] [CrossRef] [Green Version]
- Oquendo, C.J.; Parker, H.; Oscier, D.; Ennis, S.; Gibson, J.; Strefford, J.C. The (epi)genomic landscape of splenic marginal zone lymphoma, biological implications, clinical utility, and future questions. J. Transl. Genet. Genom. 2021, 5, 89–111. [Google Scholar] [CrossRef]
- Piva, R.; Deaglio, S.; Famà, R.; Buonincontri, R.; Scarfò, I.; Bruscaggin, A.; Mereu, E.; Serra, S.; Spina, V.; Brusa, D.; et al. The Krüppel-like factor 2 transcription factor gene is recurrently mutated in splenic marginal zone lymphoma. Leukemia 2015, 29, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Clipson, A.; Wang, M.; de Leval, L.; Ashton-Key, M.; Wotherspoon, A.; Vassiliou, G.; Bolli, N.; Grove, C.; Moody, S.; Escudero-Ibarz, L.; et al. KLF2 mutation is the most frequent somatic change in splenic marginal zone lymphoma and identifies a subset with distinct genotype. Leukemia 2015, 29, 1177–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jallades, L.; Baseggio, L.; Sujobert, P.; Huet, S.; Chabane, K.; Callet-Bauchu, E.; Verney, A.; Hayette, S.; Desvignes, J.P.; Salgado, D.; et al. Exome sequencing identifies recurrent BCOR alterations and the absence of KLF2, TNFAIP3 and MYD88 mutations in splenic diffuse red pulp small B-cell lymphoma. Haematologica 2017, 102, 1758–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jay, J.J.; Brouwer, C. Lollipops in the Clinic: Information Dense Mutation Plots for Precision Medicine. PLoS ONE 2016, 11, e0160519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemov, A.; Pathak, A.; Jones, S.J.; Dewan, R.; Merberg, J.; Karra, S.; Kim, J.; Arons, E.; Ravichandran, S.; Luke, B.T.; et al. In search of genetic factors predisposing to familial hairy cell leukemia (HCL): Exome-sequencing of four multiplex HCL pedigrees. Leukemia 2020, 34, 1934–1938. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Allis, C.D.; Wang, G.G. The language of chromatin modification in human cancers. Nat. Rev. Cancer 2021, 21, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Fagan, R.J.; Dingwall, A.K. COMPASS Ascending: Emerging clues regarding the roles of MLL3/KMT2C and MLL2/KMT2D proteins in cancer. Cancer Lett. 2019, 458, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Arribas, A.J.; Rinaldi, A.; Chiodin, G.; Kwee, I.; Mensah, A.A.; Cascione, L.; Rossi, D.; Kanduri, M.; Rosenquist, R.; Zucca, E.; et al. Genome-wide promoter methylation of hairy cell leukemia. Blood Adv. 2019, 3, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Gemenetzi, K.; Agathangelidis, A.; Zaragoza-Infante, L.; Sofou, E.; Papaioannou, M.; Chatzidimitriou, A.; Stamatopoulos, K. B Cell Receptor Immunogenetics in B Cell Lymphomas: Immunoglobulin Genes as Key to Ontogeny and Clinical Decision Making. Front. Oncol. 2020, 10, 67. [Google Scholar] [CrossRef] [Green Version]
- Brezinschek, H.P.; Foster, S.J.; Brezinschek, R.I.; Dorner, T.; Domiati-Saad, R.; Lipsky, P.E. Analysis of the human VH gene repertoire. Differential effects of selection and somatic hypermutation on human peripheral CD5(+)/IgM+ and CD5(−)/IgM+ B cells. J. Clin. Investig. 1997, 99, 2488–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forconi, F.; Sozzi, E.; Rossi, D.; Sahota, S.S.; Amato, T.; Raspadori, D.; Trentin, L.; Leoncini, L.; Gaidano, G.; Lauria, F. Selective influences in the expressed immunoglobulin heavy and light chain gene repertoire in hairy cell leukemia. Haematologica 2008, 93, 697–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forconi, F.; Sozzi, E.; Cencini, E.; Zaja, F.; Intermesoli, T.; Stelitano, C.; Rigacci, L.; Gherlinzoni, F.; Cantaffa, R.; Baraldi, A.; et al. Hairy cell leukemias with unmutated IGHV genes define the minor subset refractory to single-agent cladribine and with more aggressive behavior. Blood 2009, 114, 4696–4702. [Google Scholar] [CrossRef] [PubMed]
- Hockley, S.L.; Giannouli, S.; Morilla, A.; Wotherspoon, A.; Morgan, G.J.; Matutes, E.; Gonzalez, D. Insight into the molecular pathogenesis of hairy cell leukaemia, hairy cell leukaemia variant and splenic marginal zone lymphoma, provided by the analysis of their IGH rearrangements and somatic hypermutation patterns. Br. J. Haematol. 2010, 148, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Arons, E.; Roth, L.; Sapolsky, J.; Suntum, T.; Stetler-Stevenson, M.; Kreitman, R.J. Evidence of canonical somatic hypermutation in hairy cell leukemia. Blood 2011, 117, 4844–4851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forconi, F.; Sahota, S.S.; Raspadori, D.; Ippoliti, M.; Babbage, G.; Lauria, F.; Stevenson, F.K. Hairy cell leukemia: At the crossroad of somatic mutation and isotype switch. Blood 2004, 104, 3312–3317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Zan, H.; Pone, E.J.; Mai, T.; Casali, P. Immunoglobulin class-switch DNA recombination: Induction, targeting and beyond. Nat. Rev. Immunol. 2012, 12, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, X.; Ba, Z.; Liang, Z.; Dring, E.W.; Hu, H.; Lou, J.; Kyritsis, N.; Zurita, J.; Shamim, M.S.; et al. The fundamental role of chromatin loop extrusion in physiological V(D)J recombination. Nature 2019, 573, 600–604. [Google Scholar] [CrossRef]
- Giachino, C.; Padovan, E.; Lanzavecchia, A. κ+λ+ dual receptor B cells are present in the human peripheral repertoire. J. Exp. Med. 1995, 181, 1245–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, L.D.; Zhao, Y.; Lutalo, P.M.; D’Cruz, D.P.; Cason, J.; Silva, J.S.; Dunn-Walters, D.K.; Nayar, S.; Cope, A.P.; Spencer, J. Immunoglobulin light chain allelic inclusion in systemic lupus erythematosus. Eur. J. Immunol. 2015, 45, 2409–2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, J.J.; Rezanka, L.J.; Lustig, A.; Fischer, R.T.; Yoder, J.; Marshall, S.; Longo, D.L. Autoreactive B cells escape clonal deletion by expressing multiple antigen receptors. J. Immunol. 2000, 164, 4111–4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, H.; Weigert, M. Autoreactive B cells in the marginal zone that express dual receptors. J. Exp. Med. 2002, 195, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Fournier, E.M.; Velez, M.G.; Leahy, K.; Swanson, C.L.; Rubtsov, A.V.; Torres, R.M.; Pelanda, R. Dual-reactive B cells are autoreactive and highly enriched in the plasmablast and memory B cell subsets of autoimmune mice. J. Exp. Med. 2012, 209, 1797–1812. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Holmes, A.B.; Corinaldesi, C.; Shen, Q.; Kumar, R.; Compagno, N.; Wang, Z.; Nitzan, M.; Grunstein, E.; Pasqualucci, L.; Dalla-Favera, R.; et al. Single-cell analysis of germinal-center B cells informs on lymphoma cell of origin and outcome. J. Exp. Med. 2020, 217, e20200483. [Google Scholar] [CrossRef]
- Nadeu, F.; Martin-Garcia, D.; Clot, G.; Diaz-Navarro, A.; Duran-Ferrer, M.; Navarro, A.; Vilarrasa-Blasi, R.; Kulis, M.; Royo, R.; Gutierrez-Abril, J.; et al. Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes. Blood 2020, 136, 1419–1432. [Google Scholar] [CrossRef]
- Bonfiglio, F.; Bruscaggin, A.; Guidetti, F.; Terzi di Bergamo, L.; Faderl, M.R.; Spina, V.; Condoluci, A.; Bonomini, L.; Forestieri, G.; Koch, R.; et al. Genetic and Phenotypic Attributes of Splenic Marginal Zone Lymphoma. Blood 2021. [Google Scholar] [CrossRef]
- Dorfman, D.M.; Hwang, E.S.; Shahsafaei, A.; Glimcher, L.H. T-bet, a T-cell-associated transcription factor, is expressed in a subset of B-cell lymphoproliferative disorders. Am. J. Clin. Pathol. 2004, 122, 292–297. [Google Scholar] [CrossRef]
- Toth-Liptak, J.; Piukovics, K.; Borbenyi, Z.; Demeter, J.; Bagdi, E.; Krenacs, L. A comprehensive immunophenotypic marker analysis of hairy cell leukemia in paraffin-embedded bone marrow trephine biopsies—A tissue microarray study. Pathol. Oncol. Res. 2015, 21, 203–211. [Google Scholar] [CrossRef]
- Johrens, K.; Moos, V.; Schneider, T.; Anagnostopoulos, I. T-box-expressed-in-T-cells (T-bet) expression by the tumor cells of hairy-cell leukemia correlates with interferon-gamma production. Leuk. Lymphoma 2009, 50, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Gao, Q.; Chan, A.; Lewis, N.; Sigler, A.; Pichardo, J.; Xiao, W.; Roshal, M.; Dogan, A. Hairy cell leukemia expresses programmed death-1. Blood Cancer J. 2020, 10, 115. [Google Scholar] [CrossRef] [PubMed]
- Cancro, M.P. Age-Associated B Cells. Annu. Rev. Immunol. 2020, 38, 315–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsner, R.A.; Shlomchik, M.J. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 2020, 53, 1136–1150. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.R.; Tsai, A.G.; Oliveria, J.P.; Hartmann, F.J.; Kimmey, S.C.; Calderon, A.A.; Borges, L.; Glass, M.C.; Wagar, L.E.; Davis, M.M.; et al. An Integrated Multi-omic Single-Cell Atlas of Human B Cell Identity. Immunity 2020, 53, 217–232.e215. [Google Scholar] [CrossRef]
- Johnson, J.L.; Rosenthal, R.L.; Knox, J.J.; Myles, A.; Naradikian, M.S.; Madej, J.; Kostiv, M.; Rosenfeld, A.M.; Meng, W.; Christensen, S.R.; et al. The Transcription Factor T-bet Resolves Memory B Cell Subsets with Distinct Tissue Distributions and Antibody Specificities in Mice and Humans. Immunity 2020, 52, 842–855.e846. [Google Scholar] [CrossRef]
- Sutton, H.J.; Aye, R.; Idris, A.H.; Vistein, R.; Nduati, E.; Kai, O.; Mwacharo, J.; Li, X.; Gao, X.; Andrews, T.D.; et al. Atypical B cells are part of an alternative lineage of B cells that participates in responses to vaccination and infection in humans. Cell Rep. 2021, 34, 108684. [Google Scholar] [CrossRef]
- Sanz, I.; Wei, C.; Jenks, S.A.; Cashman, K.S.; Tipton, C.; Woodruff, M.C.; Hom, J.; Lee, F.E. Challenges and Opportunities for Consistent Classification of Human B Cell and Plasma Cell Populations. Front. Immunol. 2019, 10, 2458. [Google Scholar] [CrossRef] [Green Version]
- Stewart, A.; Ng, J.C.; Wallis, G.; Tsioligka, V.; Fraternali, F.; Dunn-Walters, D.K. Single-Cell Transcriptomic Analyses Define Distinct Peripheral B Cell Subsets and Discrete Development Pathways. Front. Immunol. 2021, 12, 602539. [Google Scholar] [CrossRef]
- Maul, R.W.; Catalina, M.D.; Kumar, V.; Bachali, P.; Grammer, A.C.; Wang, S.; Yang, W.; Hasni, S.; Ettinger, R.; Lipsky, P.E.; et al. Transcriptome and IgH Repertoire Analyses Show That CD11c(hi) B Cells Are a Distinct Population With Similarity to B Cells Arising in Autoimmunity and Infection. Front. Immunol. 2021, 12, 649458. [Google Scholar] [CrossRef]
- Scharer, C.D.; Blalock, E.L.; Mi, T.; Barwick, B.G.; Jenks, S.A.; Deguchi, T.; Cashman, K.S.; Neary, B.E.; Patterson, D.G.; Hicks, S.L.; et al. Epigenetic programming underpins B cell dysfunction in human SLE. Nat. Immunol. 2019, 20, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Pugh-Bernard, A.E.; Silverman, G.J.; Cappione, A.J.; Villano, M.E.; Ryan, D.H.; Insel, R.A.; Sanz, I. Regulation of inherently autoreactive VH4-34 B cells in the maintenance of human B cell tolerance. J. Clin. Investig. 2001, 108, 1061–1070. [Google Scholar] [CrossRef]
- Ng, A.; Chiorazzi, N. Potential Relevance of B-cell Maturation Pathways in Defining the Cell(s) of Origin for Chronic Lymphocytic Leukemia. Hematol. Oncol. Clin. N. Am. 2021, 35, 665–685. [Google Scholar] [CrossRef]
- Gimenez, N.; Martinez-Trillos, A.; Montraveta, A.; Lopez-Guerra, M.; Rosich, L.; Nadeu, F.; Valero, J.G.; Aymerich, M.; Magnano, L.; Rozman, M.; et al. Mutations in the RAS-BRAF-MAPK-ERK pathway define a specific subgroup of patients with adverse clinical features and provide new therapeutic options in chronic lymphocytic leukemia. Haematologica 2019, 104, 576–586. [Google Scholar] [CrossRef]
- Vendramini, E.; Bomben, R.; Pozzo, F.; Benedetti, D.; Bittolo, T.; Rossi, F.M.; Dal Bo, M.; Rabe, K.G.; Pozzato, G.; Zaja, F.; et al. KRAS, NRAS, and BRAF mutations are highly enriched in trisomy 12 chronic lymphocytic leukemia and are associated with shorter treatment-free survival. Leukemia 2019, 33, 2111–2115. [Google Scholar] [CrossRef] [Green Version]
- Blakemore, S.J.; Clifford, R.; Parker, H.; Antoniou, P.; Stec-Dziedzic, E.; Larrayoz, M.; Davis, Z.; Kadalyayil, L.; Colins, A.; Robbe, P.; et al. Clinical significance of TP53, BIRC3, ATM and MAPK-ERK genes in chronic lymphocytic leukaemia: Data from the randomised UK LRF CLL4 trial. Leukemia 2020, 34, 1760–1774. [Google Scholar] [CrossRef]
- Andrulis, M.; Lehners, N.; Capper, D.; Penzel, R.; Heining, C.; Huellein, J.; Zenz, T.; von Deimling, A.; Schirmacher, P.; Ho, A.D.; et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov. 2013, 3, 862–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustad, E.H.; Dai, H.Y.; Hov, H.; Coward, E.; Beisvag, V.; Myklebost, O.; Hovig, E.; Nakken, S.; Vodak, D.; Meza-Zepeda, L.A.; et al. BRAF V600E mutation in early-stage multiple myeloma: Good response to broad acting drugs and no relation to prognosis. Blood Cancer J. 2015, 5, e299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiacci, E.; Schiavoni, G.; Forconi, F.; Santi, A.; Trentin, L.; Ambrosetti, A.; Cecchini, D.; Sozzi, E.; Francia di Celle, P.; Di Bello, C.; et al. Simple genetic diagnosis of hairy cell leukemia by sensitive detection of the BRAF-V600E mutation. Blood 2012, 119, 192–195. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, F.; Paolicchi, M.; Ghio, F.; Ciabatti, E.; Grassi, S.; Salehzadeh, S.; Ercolano, G.; Metelli, M.R.; Del Re, M.; Iovino, L.; et al. The Droplet Digital PCR: A New Valid Molecular Approach for the Assessment of B-RAF V600E Mutation in Hairy Cell Leukemia. Front. Pharmacol. 2016, 7, 363. [Google Scholar] [CrossRef] [Green Version]
- Cardus, B.; Colling, R.; Hamblin, A.; Soilleux, E. Comparison of methodologies for the detection of BRAF mutations in bone marrow trephine specimens. J. Clin. Pathol. 2019, 72, 406–411. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Vardiman, J.W., Eds.; WHO Press: Geneva, Switzerland, 2008. [Google Scholar]
- Arons, E.; Suntum, T.; Stetler-Stevenson, M.; Kreitman, R.J. VH4-34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy. Blood 2009, 114, 4687–4695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiacci, E.; Park, J.H.; De Carolis, L.; Chung, S.S.; Broccoli, A.; Scott, S.; Zaja, F.; Devlin, S.; Pulsoni, A.; Chung, Y.R.; et al. Targeting Mutant BRAF in Relapsed or Refractory Hairy-Cell Leukemia. N. Engl. J. Med. 2015, 373, 1733–1747. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; De Carolis, L.; Simonetti, E.; Merluzzi, M.; Bennati, A.; Perriello, V.M.; Pucciarini, A.; Santi, A.; Venanzi, A.; Pettirossi, V.; et al. Safety and efficacy of the BRAF inhibitor dabrafenib in relapsed or refractory hairy cell leukemia: A pilot phase-2 clinical trial. Leukemia 2021, 35, 3314–3318. [Google Scholar] [CrossRef]
- Tiacci, E.; De Carolis, L.; Simonetti, E.; Capponi, M.; Ambrosetti, A.; Lucia, E.; Antolino, A.; Pulsoni, A.; Ferrari, S.; Zinzani, P.L.; et al. Vemurafenib plus Rituximab in Refractory or Relapsed Hairy-Cell Leukemia. N. Engl. J. Med. 2021, 384, 1810–1823. [Google Scholar] [CrossRef]
- Whittaker, S.R.; Theurillat, J.P.; Van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L.A. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013, 3, 350–362. [Google Scholar] [CrossRef] [Green Version]
- Caeser, R.; Collord, G.; Yao, W.Q.; Chen, Z.; Vassiliou, G.S.; Beer, P.A.; Du, M.Q.; Scott, M.A.; Follows, G.A.; Hodson, D.J. Targeting MEK in vemurafenib-resistant hairy cell leukemia. Leukemia 2019, 33, 541–545. [Google Scholar] [CrossRef]
- Marine, J.C.; Dawson, S.J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef]
- Ciriello, G.; Magnani, L. The many faces of cancer evolution. iScience 2021, 24, 102403. [Google Scholar] [CrossRef]
- Emert, B.L.; Cote, C.J.; Torre, E.A.; Dardani, I.P.; Jiang, C.L.; Jain, N.; Shaffer, S.M.; Raj, A. Variability within rare cell states enables multiple paths toward drug resistance. Nat. Biotechnol. 2021, 39, 865–876. [Google Scholar] [CrossRef]
- Sadras, T.; Chan, L.N.; Xiao, G.; Muschen, M. Metabolic Gatekeepers of Pathological B Cell Activation. Annu. Rev. Pathol. 2021, 16, 323–349. [Google Scholar] [CrossRef] [PubMed]
- Waldschmidt, J.M.; Kloeber, J.A.; Anand, P.; Frede, J.; Kokkalis, A.; Dimitrova, V.; Potdar, S.; Nair, M.S.; Vijaykumar, T.; Im, N.G.; et al. Single-Cell Profiling Reveals Metabolic Reprogramming as a Resistance Mechanism in BRAF-Mutated Multiple Myeloma. Clin. Cancer Res. 2021, 27, 6432–6444. [Google Scholar] [CrossRef] [PubMed]
- Angelova, E.A.; Medeiros, L.J.; Wang, W.; Muzzafar, T.; Lu, X.; Khoury, J.D.; Ravandi, F.; Patel, K.P.; Hu, Z.; Kanagal-Shamanna, R. Clinicopathologic and molecular features in hairy cell leukemia-variant: Single institutional experience. Mod. Pathol. 2018, 31, 1717–1732. [Google Scholar] [CrossRef]
- Haferlach, T.; Nadarajah, N.; Meggendorfer, M.; Haferlach, C.; Kern, W. Whole Genome Sequencing in Hairy Cell Leukemia-Variant (HCL-v) and Splenic Marginal Zone Lymphoma (SMZL). Blood 2017, 130, 4003. [Google Scholar] [CrossRef]
- Mason, E.F.; Brown, R.D.; Szeto, D.P.; Gibson, C.J.; Jia, Y.; Garcia, E.P.; Jacobson, C.A.; Dal Cin, P.; Kuo, F.C.; Pinkus, G.S.; et al. Detection of activating MAP2K1 mutations in atypical hairy cell leukemia and hairy cell leukemia variant. Leuk. Lymphoma 2017, 58, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Hockley, S.L.; Else, M.; Morilla, A.; Wotherspoon, A.; Dearden, C.; Catovsky, D.; Gonzalez, D.; Matutes, E. The prognostic impact of clinical and molecular features in hairy cell leukaemia variant and splenic marginal zone lymphoma. Br. J. Haematol. 2012, 158, 347–354. [Google Scholar] [CrossRef]
- Hanrahan, A.J.; Sylvester, B.E.; Chang, M.T.; Elzein, A.; Gao, J.; Han, W.; Liu, Y.; Xu, D.; Gao, S.P.; Gorelick, A.N.; et al. Leveraging Systematic Functional Analysis to Benchmark an In Silico Framework Distinguishes Driver from Passenger MEK Mutants in Cancer. Cancer Res. 2020, 80, 4233–4243. [Google Scholar] [CrossRef]
- Zanelli, M.; Ragazzi, M.; Valli, R.; Piattoni, S.; Alvarez De Celis, M.I.; Farnetti, E.; Orcioni, G.F.; Longo, R.; Ascani, S.; Falini, B.; et al. Transformation of IGHV4-34+ hairy cell leukaemia-variant with U2AF1 mutation into a clonally-related high grade B-cell lymphoma responding to immunochemotherapy. Br. J. Haematol. 2016, 173, 491–495. [Google Scholar] [CrossRef]
- Ler, L.D.; Ghosh, S.; Chai, X.; Thike, A.A.; Heng, H.L.; Siew, E.Y.; Dey, S.; Koh, L.K.; Lim, J.Q.; Lim, W.K.; et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci. Transl. Med. 2017, 9, eaai8312. [Google Scholar] [CrossRef]
- Chihara, D.; Arons, E.; Stetler-Stevenson, M.; Yuan, C.; Wang, H.W.; Zhou, H.; Raffeld, M.; Xi, L.; Steinberg, S.M.; Feurtado, J.; et al. Long term follow-up of a phase II study of cladribine with concurrent rituximab with hairy cell leukemia variant. Blood Adv. 2021, 5, 4807–4816. [Google Scholar] [CrossRef]
- Liu, Q.; Harris, N.; Epperla, N.; Andritsos, L.A. Current and Emerging Therapeutic Options for Hairy Cell Leukemia Variant. OncoTargets Ther. 2021, 14, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Andritsos, L.A.; Grieselhuber, N.R.; Anghelina, M.; Rogers, K.A.; Roychowdhury, S.; Reeser, J.W.; Timmers, C.D.; Freud, A.G.; Blachly, J.S.; Lucas, D.M.; et al. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk. Lymphoma 2018, 59, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- Binimetinib for People with Relapsed/Refractory BRAF Wild Type Hairy Cell Leukemia and Variant. Available online: https://clinicaltrials.gov/ct2/show/NCT04322383 (accessed on 20 December 2021).
- Traverse-Glehen, A.; Verney, A.; Gazzo, S.; Jallades, L.; Chabane, K.; Hayette, S.; Coiffier, B.; Callet-Bauchu, E.; Ffrench, M.; Felman, P.; et al. Splenic diffuse red pulp lymphoma has a distinct pattern of somatic mutations amongst B-cell malignancies. Leuk. Lymphoma 2017, 58, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Martinez, D.; Navarro, A.; Martinez-Trillos, A.; Molina-Urra, R.; Gonzalez-Farre, B.; Salaverria, I.; Nadeu, F.; Enjuanes, A.; Clot, G.; Costa, D.; et al. NOTCH1, TP53, and MAP2K1 Mutations in Splenic Diffuse Red Pulp Small B-cell Lymphoma Are Associated With Progressive Disease. Am. J. Surg. Pathol. 2016, 40, 192–201. [Google Scholar] [CrossRef]
- Ramezani-Rad, P.; Chen, C.; Zhu, Z.; Rickert, R.C. Cyclin D3 Governs Clonal Expansion of Dark Zone Germinal Center B Cells. Cell Rep. 2020, 33, 108403. [Google Scholar] [CrossRef] [PubMed]
- Pae, J.; Ersching, J.; Castro, T.B.R.; Schips, M.; Mesin, L.; Allon, S.J.; Ordovas-Montanes, J.; Mlynarczyk, C.; Melnick, A.; Efeyan, A.; et al. Cyclin D3 drives inertial cell cycling in dark zone germinal center B cells. J. Exp. Med. 2021, 218, e20201699. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Malumbres, M.; Sicinski, P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat. Rev. Mol. Cell Biol. 2016, 17, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Curiel-Olmo, S.; Mondejar, R.; Almaraz, C.; Mollejo, M.; Cereceda, L.; Mares, R.; Derdak, S.; Campos-Martin, Y.; Batlle, A.; Gonzalez de Villambrosia, S.; et al. Splenic diffuse red pulp small B-cell lymphoma displays increased expression of cyclin D3 and recurrent CCND3 mutations. Blood 2017, 129, 1042–1045. [Google Scholar] [CrossRef] [Green Version]
- Martin-Garcia, D.; Navarro, A.; Valdes-Mas, R.; Clot, G.; Gutierrez-Abril, J.; Prieto, M.; Ribera-Cortada, I.; Woroniecka, R.; Rymkiewicz, G.; Bens, S.; et al. CCND2 and CCND3 hijack immunoglobulin light-chain enhancers in cyclin D1(-) mantle cell lymphoma. Blood 2019, 133, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Mlynarczyk, C.; Fontan, L.; Melnick, A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol. Rev. 2019, 288, 214–239. [Google Scholar] [CrossRef] [Green Version]
- Sportoletti, P.; Sorcini, D.; Falini, B. BCOR gene alterations in hematologic diseases. Blood 2021, 138, 2455–2468. [Google Scholar] [CrossRef] [PubMed]
- Robak, P.; Jesionek-Kupnicka, D.; Kupnicki, P.; Polliack, A.; Robak, T. Bone lesions in hairy cell leukemia: Diagnosis and treatment. Eur. J. Haematol. 2020, 105, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Dasanu, C.A.; Van den Bergh, M.; Pepito, D.; Alvarez Argote, J. Autoimmune disorders in patients with hairy cell leukemia: Are they more common than previously thought? Curr. Med. Res. Opin. 2015, 31, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Weston-Bell, N.J.; Hendriks, D.; Sugiyarto, G.; Bos, N.A.; Kluin-Nelemans, H.C.; Forconi, F.; Sahota, S.S. Hairy cell leukemia cell lines expressing annexin A1 and displaying B-cell receptor signals characteristic of primary tumor cells lack the signature BRAF mutation to reveal unrepresentative origins. Leukemia 2013, 27, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Martelli, M.P.; Tiacci, E. BRAF V600E mutation in hairy cell leukemia: From bench to bedside. Blood 2016, 128, 1918–1927. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The RAS–RAF–MEK–ERK signal transduction cascade is one of four mitogen-activated protein kinase (MAPK) cascades which are activated in response to extracellular signals. RAS activation occurs within biomolecular condensates at the inner part of the cell membrane and recruits members of the RAF kinase family (A-RAF, B-RAF, and C-RAF/RAF-1) to the plasma membrane for activation. Active RAF kinases phosphorylate downstream mitogen-activated protein kinase/extracellular signal-regulated kinase ERK kinase (MEK). A transient tetramer, consisting of two RAF-MEK dimers, is formed to facilitate MEK activation by RAF. Active MEK then dually phosphorylates its only downstream targets, extracellular signal-related kinases 1 and 2 (ERK1/2). In contrast, ERK1/2 has extremely broad substrate specificity and is capable of activating both nuclear and cytosolic targets, many of which are transcription factors essential for the regulation of cell proliferation, survival, growth, metabolism, migration, and differentiation. In addition, ERKs also phosphorylate RAFs themselves at specific inhibitory amino acid residues, which releases RAF from RAS and extinguishes the signal via a negative feedback mechanism [12,13]. Created with Biorender.com (accessed on 28 December 2021).
Figure 1.
The RAS–RAF–MEK–ERK signal transduction cascade is one of four mitogen-activated protein kinase (MAPK) cascades which are activated in response to extracellular signals. RAS activation occurs within biomolecular condensates at the inner part of the cell membrane and recruits members of the RAF kinase family (A-RAF, B-RAF, and C-RAF/RAF-1) to the plasma membrane for activation. Active RAF kinases phosphorylate downstream mitogen-activated protein kinase/extracellular signal-regulated kinase ERK kinase (MEK). A transient tetramer, consisting of two RAF-MEK dimers, is formed to facilitate MEK activation by RAF. Active MEK then dually phosphorylates its only downstream targets, extracellular signal-related kinases 1 and 2 (ERK1/2). In contrast, ERK1/2 has extremely broad substrate specificity and is capable of activating both nuclear and cytosolic targets, many of which are transcription factors essential for the regulation of cell proliferation, survival, growth, metabolism, migration, and differentiation. In addition, ERKs also phosphorylate RAFs themselves at specific inhibitory amino acid residues, which releases RAF from RAS and extinguishes the signal via a negative feedback mechanism [12,13]. Created with Biorender.com (accessed on 28 December 2021).

Figure 2.
The BRAF protein includes three highly conserved regions—CR1 which functions as an auto-inhibitor of the BRAF kinase domain and contains a RAS-GTP binding domain (RBD), CR2 which acts as a flexible hinge between CR1 and CR3, and CR3, the kinase domain, which comprises multiple subregions including the P loop, the dimerisation interface (DIF), the DFG motif, and the activation segment. In the wild-type protein, inactive RAF exists in an auto-inhibited state. Under activating conditions, RAS-GTP binds to the RBD, disrupting auto-inhibition. BRAF is then phosphorylated at T599 and S602 within the DFG motif and activation segments, destabilising interactions with the P loop and allowing the activation segment to flip into its active conformation. The majority of BRAF mutants are located within either the P loop or the activation segment and adjacent DFG motif. The BRAF-V600E mutation occurs in the kinase activation segment, thereby inducing a change to the active conformation independently from upstream RAS activation. This results in constitutive kinase activity and aberrant signalling through the RAF–MEK–ERK pathway [14,15]. Created with Biorender.com (accessed on 28 December 2021).
Figure 2.
The BRAF protein includes three highly conserved regions—CR1 which functions as an auto-inhibitor of the BRAF kinase domain and contains a RAS-GTP binding domain (RBD), CR2 which acts as a flexible hinge between CR1 and CR3, and CR3, the kinase domain, which comprises multiple subregions including the P loop, the dimerisation interface (DIF), the DFG motif, and the activation segment. In the wild-type protein, inactive RAF exists in an auto-inhibited state. Under activating conditions, RAS-GTP binds to the RBD, disrupting auto-inhibition. BRAF is then phosphorylated at T599 and S602 within the DFG motif and activation segments, destabilising interactions with the P loop and allowing the activation segment to flip into its active conformation. The majority of BRAF mutants are located within either the P loop or the activation segment and adjacent DFG motif. The BRAF-V600E mutation occurs in the kinase activation segment, thereby inducing a change to the active conformation independently from upstream RAS activation. This results in constitutive kinase activity and aberrant signalling through the RAF–MEK–ERK pathway [14,15]. Created with Biorender.com (accessed on 28 December 2021).

Figure 3.
Mitogenic signals received during the G1 phase of the cell cycle, partially mediated through RAS-induced ERK signalling, upregulate the D-type cyclins D1, D2, D3, encoded by CCND1, CCND2, and CCND3, respectively. These bind and activate their catalytic partners, CDK4 or CDK6, whose activity is, in turn, negatively regulated by the INK4 family of inhibitors which include p16INK4A and p15INKB encoded by CDKN2A and CDKN2B, respectively. The formation of stable cyclin D-CDK4/6 complexes also requires the KIP/CIP proteins p21CIP1, p27KIP1, and p57KIP2, encoded by CDKN1A, CDKN1B, and CDKN1C, respectively, which serve as assembly factors for cyclin D-CDK4/6 but also act as inhibitors of Cdk2–cyclin E complexes required for transition into S phase [52]. Created with Biorender.com (accessed on 28 December 2021).
Figure 3.
Mitogenic signals received during the G1 phase of the cell cycle, partially mediated through RAS-induced ERK signalling, upregulate the D-type cyclins D1, D2, D3, encoded by CCND1, CCND2, and CCND3, respectively. These bind and activate their catalytic partners, CDK4 or CDK6, whose activity is, in turn, negatively regulated by the INK4 family of inhibitors which include p16INK4A and p15INKB encoded by CDKN2A and CDKN2B, respectively. The formation of stable cyclin D-CDK4/6 complexes also requires the KIP/CIP proteins p21CIP1, p27KIP1, and p57KIP2, encoded by CDKN1A, CDKN1B, and CDKN1C, respectively, which serve as assembly factors for cyclin D-CDK4/6 but also act as inhibitors of Cdk2–cyclin E complexes required for transition into S phase [52]. Created with Biorender.com (accessed on 28 December 2021).

Figure 4.
Distribution of KLF2 mutations in HCLc and SMZL. Mutations were compiled from [42,62,64]. Diagram produced using Lollipops [65].

Figure 5.
Distribution of MAP2K1 mutations in BRAFWT HCLc, HCLv, and SDRPL. Mutations compiled from [11,42,43,62]. Diagram produced using Lollipops [65].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
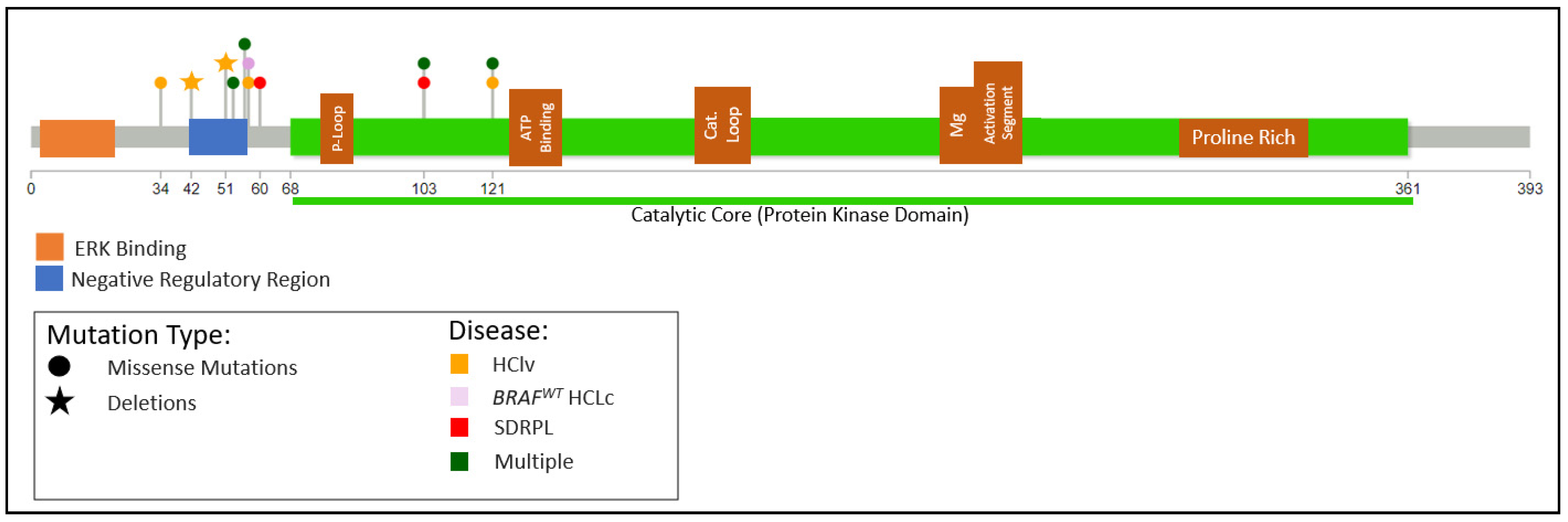{kind=link}
Table 1.
Key Differences between HCLc, HCLv, and SDRPL.
| HCLc | HCLv | SDRPL | ||
|---|---|---|---|---|
| Demographics | Incidence | 0.4/100,000 | 0.03/100,000 | ? |
| Median age | 55–63 | 70 | 70 | |
| M:F ratio | 3–4: 1 | 1–2: 1 | 1.6–2.4: 1 | |
| Haematology | Monocytopenia | Yes | No | No |
| Nucleolus | Inconspicuous | Single prominent | Inconspicuous | |
| Immunophenotype | Surface IgH | Usually, multiple isotypes | Usually IgG +/− other isotypes | M+/−D, M+G, G |
| CD11c | Strong | Strong | Moderate | |
| CD25 | Strong | Negative | Negative (weak in 3%) | |
| CD103 | Strong | Moderate | Negative (weak in 33%) | |
| CD123 | Strong | Negative | Negative (weak in 15%) | |
| CD27 | Negative | ? | Negative (positive in 20%) | |
| CD200 | Strong | Weak or negative | Weak | |
| Immunohistochemistry | Annexin A1 | Positive | Negative | Negative |
| Cyclin D1 | Positive | Negative | Negative | |
| Outcome | Need for treatment | Yes | Yes | Approx. 50% |
Table 2.
Most frequent features of HCLc, HCLv, and SDRPL.
| HCLc | HCLv | SDRPL | ||||
|---|---|---|---|---|---|---|
| HCLc BRAF WT | HCLc BRAF Mutated or Upregulated | |||||
| Recurring CNAs * | 7q Loss | Y | Y | Y | ||
| 8q Loss (MAPK15) | Y | N | N | |||
| 17p Loss | Rare | Y | Rare | |||
| X or Xp Loss (BCOR) | N | N | Y | |||
| 5q Gain | Y | Y | N | |||
| Genomic Mutations | MAPK Pathway | BRAFV600E | 0% | >99% | 0% | 0% |
| MAP2K1 | 22% | 0% | 22–41% | 7–13% | ||
| Cell Cycle | CDKN1B | 16% | ||||
| CCND3 | 0% | 13% | 21–24% | |||
| Epigenetic Regulators ** | KMT2C | 15% | 25% | |||
| KDM6A | 0% | 50% | ||||
| CREBBP | 5% | 12–25% | ||||
| ARID1A | 4% | 4% | 9% | |||
| Transcriptional Repressors | BCOR | 0% | 0% | 16% | ||
| NFKβ Pathway | KLF2 | 13% | 0% | 2% | ||
| Spliceosome | U2AF1 | 0% | 2/7 | 0% | ||
| TP53 | 2% | |||||
| IGHV Genes | Homology | 100% | <5% | 18–22% | 11% | |
| 98–100% | 17–20% | 27–53% | 14% | |||
| <98% | 80% | 47–73% | 86% | |||
| Gene Usage | IGHV3-23 | 7–10% | 14% | |||
| IGHV3-30 | 7–10% | 6% | ||||
| IGHV4-34 | 50% | 7–10% | 17–36% | 22% | ||
* % incidence of CNAs is not provided due to wide variations among studies. ** % incidence of mutations in epigenetic regulators is based on small samples.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Oscier, D.; Stamatopoulos, K.; Mirandari, A.; Strefford, J. The Genomics of Hairy Cell Leukaemia and Splenic Diffuse Red Pulp Lymphoma. Cancers 2022, 14, 697. https://doi.org/10.3390/cancers14030697
AMA Style
Oscier D, Stamatopoulos K, Mirandari A, Strefford J. The Genomics of Hairy Cell Leukaemia and Splenic Diffuse Red Pulp Lymphoma. Cancers. 2022; 14(3):697. https://doi.org/10.3390/cancers14030697
Chicago/Turabian StyleOscier, David, Kostas Stamatopoulos, Amatta Mirandari, and Jonathan Strefford. 2022. "The Genomics of Hairy Cell Leukaemia and Splenic Diffuse Red Pulp Lymphoma" Cancers 14, no. 3: 697. https://doi.org/10.3390/cancers14030697
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.