
High Metabolic Dependence on Oxidative Phosphorylation Drives Sensitivity to Metformin Treatment in MLL/AF9 Acute Myeloid Leukemia

 , ,
, , 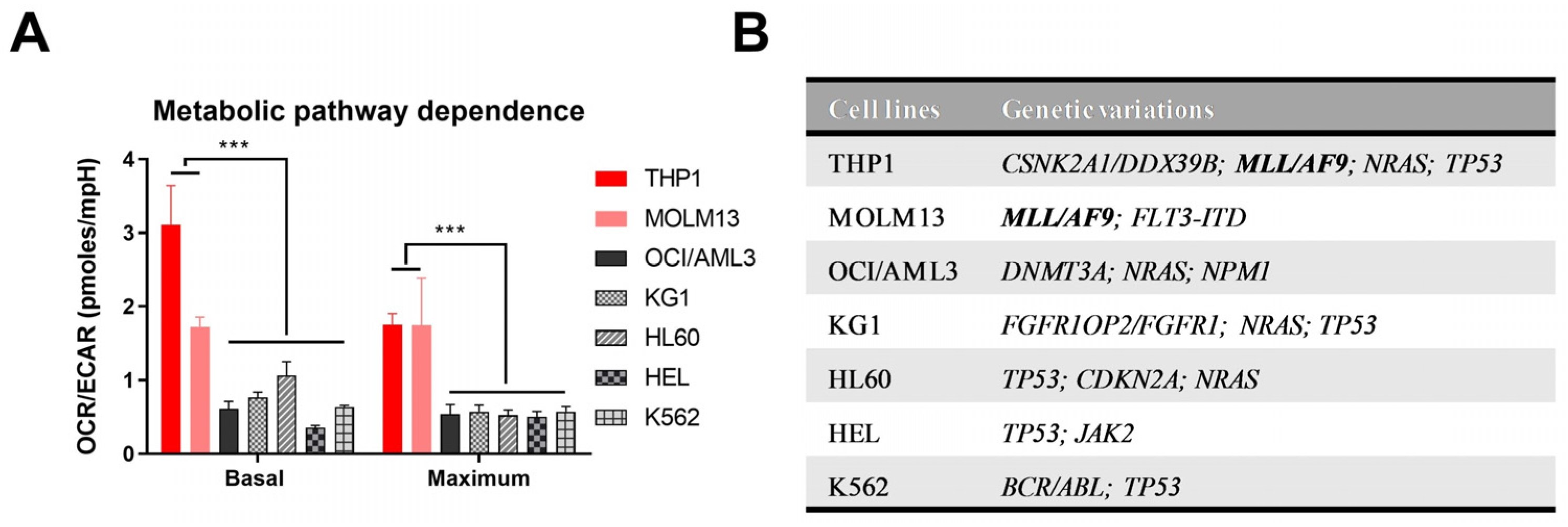{kind=link}
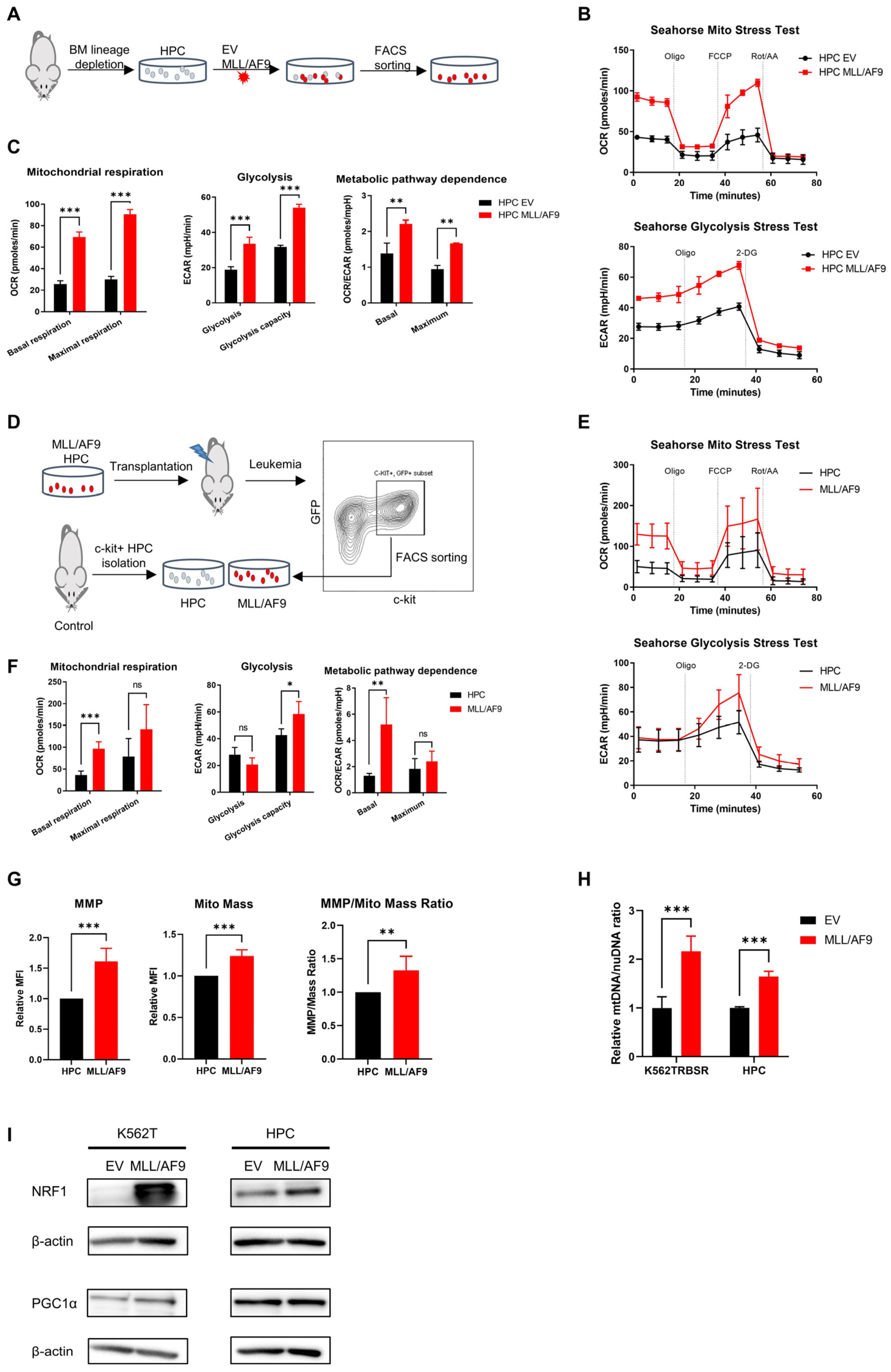{kind=link}
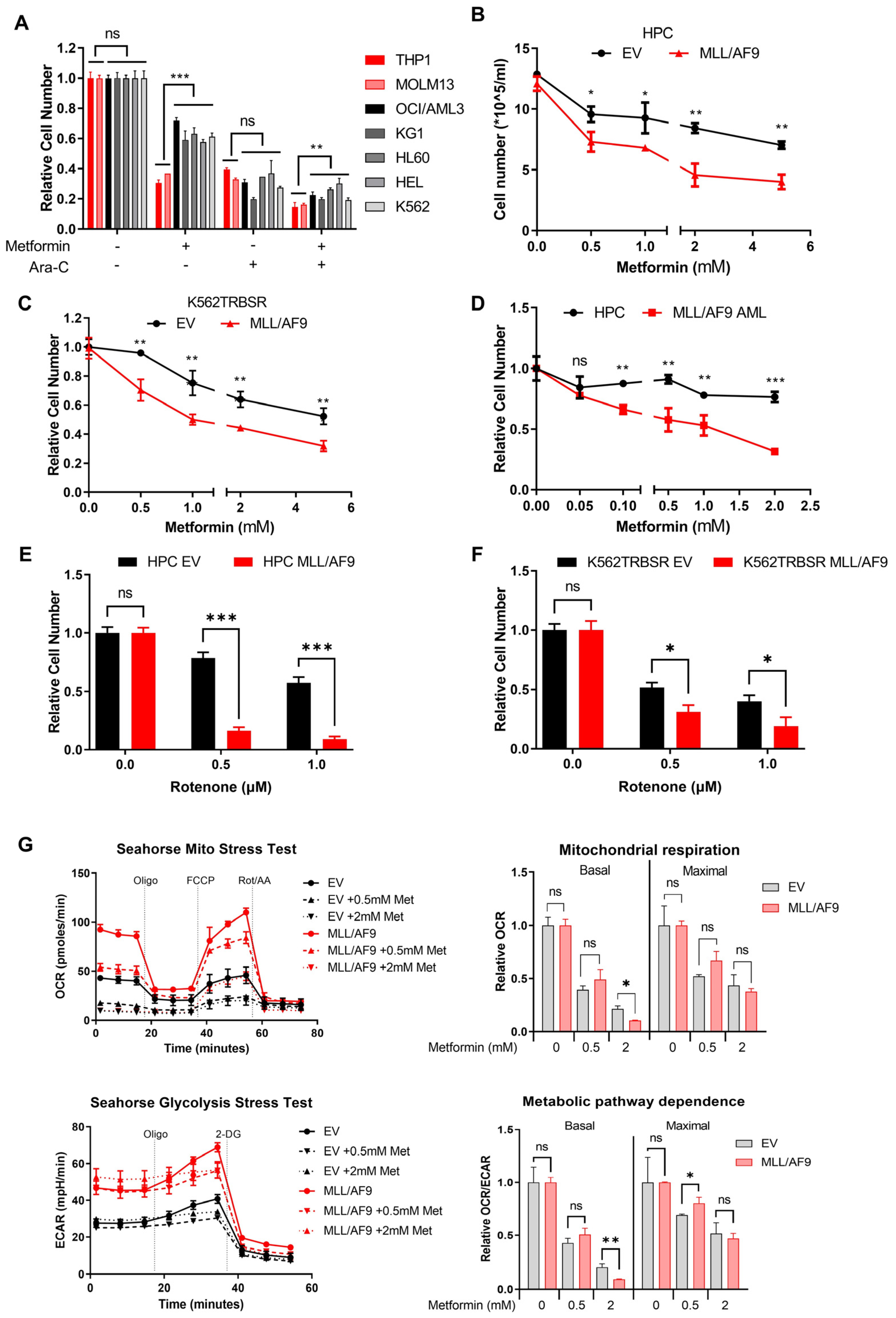{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Seahorse Extracellular Flux Analysis
2.3. Retrovirus Production and Transduction
2.4. MLL/AF9 Leukemia Mouse Models
2.5. Mitochondrial Membrane Potential (MMP), Mitochondrial Number Detection, and Cells Sorting
2.6. Metformin Treatment
2.7. Immunoblot Analysis
2.8. Mitochondrial DNA Quantification
2.9. Statistical Analysis
3. Results
3.1. Metabolic Pathway Dependence Analysis Shows Upregulated OXPHOS in MLL/AF9 AML Cells
3.2. MLL/AF9 Gene Expression Is Associated with Increased Mitochondrial Respiration
3.3. MLL/AF9 AML Cells Are More Sensitive to Metformin Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oran, B.; Weisdorf, D.J. Survival for older patients with acute myeloid leukemia: A population-based study. Haematologica 2012, 97, 1916–1924. [Google Scholar] [CrossRef]
- Yi, M.; Li, A.; Zhou, L.; Chu, Q.; Song, Y.; Wu, K.; Wu, K. The global burden and attributable risk factor analysis of acute myeloid leukemia in 195 countries and territories from 1990 to 2017: Estimates based on the global burden of disease study 2017. J. Hematol. Oncol. 2020, 13, 1–16. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef]
- Burnett, A.; Wetzler, M.; Löwenberg, B. Therapeutic Advances in Acute Myeloid Leukemia. J. Clin. Oncol. 2011, 29, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, R.; Paterno, G.; De Bellis, E.; Mercante, L.; Buzzatti, E.; Esposito, F.; Del Principe, M.I.; Maurillo, L.; Buccisano, F.; Venditti, A. Therapeutic Choice in Older Patients with Acute Myeloid Leukemia: A Matter of Fitness. Cancers 2020, 12, 120. [Google Scholar] [CrossRef] [Green Version]
- Castro, I.; Sampaio-Marques, B.; Ludovico, P. Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells 2019, 8, 967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Tcheng, M.; Roma, A.; Ahmed, N.; Smith, R.W.; Jayanth, P.; Minden, M.D.; Schimmer, A.D.; Hess, D.A.; Hope, K.; Rea, K.A.; et al. Very long chain fatty acid metabolism is required in acute myeloid leukemia. Blood 2021, 137, 3518–3532. [Google Scholar] [CrossRef]
- Stergiou, I.E.; Kapsogeorgou, E.K. Autophagy and Metabolism in Normal and Malignant Hematopoiesis. Int. J. Mol. Sci. 2021, 22, 8540. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Grønningsæter, I.S.; Reikvam, H.; Aasebø, E.; Bartaula-Brevik, S.; Tvedt, T.H.; Bruserud, Ø.; Hatfield, K.J. Targeting Cellular Metabolism in Acute Myeloid Leukemia and the Role of Patient Heterogeneity. Cells 2020, 9, 1155. [Google Scholar] [CrossRef]
- Ngo, V.N.; Davis, R.E.; Lamy, L.; Yu, X.; Zhao, H.; Lenz, G.; Lam, L.T.; Dave, S.; Yang, L.; Powell, J.; et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 2006, 441, 106–110. [Google Scholar] [CrossRef]
- Hönes, J.M.; Thivakaran, A.; Botezatu, L.; Patnana, P.; da Conceição Castro, S.V.; Al-Matary, Y.S.; Schütte, J.; Fischer, K.B.I.I.; Vassen, L.; Görgens, A.; et al. Enforced GFI1 expression impedes human and murine leukemic cell growth. Sci. Rep. 2017, 7, 15720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Chapple, R.H.; Lin, A.; Kitano, A.; Nakada, D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell 2015, 17, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naviaux, R.K.; Costanzi, E.; Haas, M.; Verma, I.M. The pCL vector system: Rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 1996, 70, 5701–5705. [Google Scholar] [CrossRef] [Green Version]
- Frontzek, F.; Staiger, A.M.; Zapukhlyak, M.; Xu, W.; Bonzheim, I.; Borgmann, V.; Sander, P.; Baptista, M.J.; Heming, J.-N.; Berning, P.; et al. Molecular and functional profiling identifies therapeutically targetable vulnerabilities in plasmablastic lymphoma. Nat. Commun. 2021, 12, 5183. [Google Scholar] [CrossRef]
- Thivakaran, A.; Botezatu, L.; Hönes, J.M.; Schütte, J.; Vassen, L.; Al-Matary, Y.S.; Patnana, P.; Zeller, A.; Heuser, M.; Thol, F.; et al. Gfi1b: A key player in the genesis and maintenance of acute myeloid leukemia and myelodysplastic syndrome. Haematologica 2018, 103, 614–625. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, M.J.; Luchsinger, L.L.; Corrigan, D.J.; Williams, L.J.; Snoeck, H.-W. Dye-Independent Methods Reveal Elevated Mitochondrial Mass in Hematopoietic Stem Cells. Cell Stem Cell 2017, 21, 725–729.e4. [Google Scholar] [CrossRef] [Green Version]
- Botezatu, L.; Michel, L.C.; Helness, A.; Vadnais, C.; Makishima, H.; Hönes, J.M.; Robert, F.; Vassen, L.; Thivakaran, A.; Al-Matary, Y.; et al. Epigenetic therapy as a novel approach for GFI136N-associated murine/human AML. Exp. Hematol. 2016, 44, 713–726.e14. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Seneviratne, A.K.; Xu, M.; Henao, J.J.A.; Fajardo, V.A.; Hao, Z.; Voisin, V.; Xu, G.W.; Hurren, R.; Kim, S.; MacLean, N.; et al. The Mitochondrial Transacylase, Tafazzin, Regulates AML Stemness by Modulating Intracellular Levels of Phospholipids. Cell Stem Cell 2019, 24, 621–636.e16. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Maldonado, E.; Rojas, D.A.; Urbina, F.; Solari, A. The Use of Antioxidants as Potential Co-Adjuvants to Treat Chronic Chagas Disease. Antioxidants 2021, 10, 1022. [Google Scholar] [CrossRef] [PubMed]
- Luna-Yolba, R.; Marmoiton, J.; Gigo, V.; Marechal, X.; Boet, E.; Sahal, A.; Alet, N.; Abramovich, I.; Gottlieb, E.; Visentin, V.; et al. Disrupting Mitochondrial Electron Transfer Chain Complex I Decreases Immune Checkpoints in Murine and Human Acute Myeloid Leukemic Cells. Cancers 2021, 13, 3499. [Google Scholar] [CrossRef]
- Bindels, E.M.J.; Havermans, M.; Lugthart, S.; Erpelinck, C.; Wocjtowicz, E.; Krivtsov, A.V.; Rombouts, E.; Armstrong, S.A.; Taskesen, E.; Haanstra, J.R.; et al. EVI1 is critical for the pathogenesis of a subset of MLL-AF9–rearranged AMLs. Blood 2012, 119, 5838–5849. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Sawa, D.; Kinoshita, M.; Yamada, A.; Kamimura, S.; Suekane, A.; Ogoh, H.; Matsuo, H.; Adachi, S.; Taga, T.; et al. EVI1 triggers metabolic reprogramming associated with leukemogenesis and increases sensitivity to L-asparaginase. Haematologica 2020, 105, 2118–2129. [Google Scholar] [CrossRef]
- Wong, N.-H.M.; So, C.W.E. Novel therapeutic strategies for MLL-rearranged leukemias. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194584. [Google Scholar] [CrossRef]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Alibhai, S.M.H.; Leach, M.; Minden, M.D.; Brandwein, J. Outcomes and quality of care in acute myeloid leukemia over 40 years. Cancer 2009, 115, 2903–2911. [Google Scholar] [CrossRef]
- Stuani, L.; Riols, F.; Millard, P.; Sabatier, M.; Batut, A.; Saland, E.; Viars, F.; Tonini, L.; Zaghdoudi, S.; Linares, L.K.; et al. Stable Isotope Labeling Highlights Enhanced Fatty Acid and Lipid Metabolism in Human Acute Myeloid Leukemia. Int. J. Mol. Sci. 2018, 19, 3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roca-Portoles, A.; Rodriguez-Blanco, G.; Sumpton, D.; Cloix, C.; Mullin, M.; Mackay, G.M.; O’Neill, K.; Lemgruber, L.; Luo, X.; Tait, S.W.G. Venetoclax causes metabolic reprogramming independent of BCL-2 inhibition. Cell Death Dis. 2020, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Kalpage, H.A.; Wang, D.; Edwards, H.; Hüttemann, M.; Ma, J.; Su, Y.; Carter, J.; Li, X.; Polin, L.; et al. Cotargeting of Mitochondrial Complex I and Bcl-2 Shows Antileukemic Activity against Acute Myeloid Leukemia Cells Reliant on Oxidative Phosphorylation. Cancers 2020, 12, 2400. [Google Scholar] [CrossRef]
- Lee, J.; Yesilkanal, A.E.; Wynne, J.P.; Frankenberger, C.; Liu, J.; Yan, J.; Elbaz, M.; Rabe, D.C.; Rustandy, F.D.; Tiwari, P.; et al. Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature 2019, 568, 254–258. [Google Scholar] [CrossRef]
- De Beauchamp, L.; Himonas, E.; Helgason, G.V. Mitochondrial metabolism as a potential therapeutic target in myeloid leukaemia. Leukemia 2021, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Patnana, P.K.; Xie, X.; Frank, D.; Nimmagadda, S.C.; Rosemann, A.; Liebmann, M.; Klotz, L.; Opalka, B.; Khandanpour, C. High Metabolic Dependence on Oxidative Phosphorylation Drives Sensitivity to Metformin Treatment in MLL/AF9 Acute Myeloid Leukemia. Cancers 2022, 14, 486. https://doi.org/10.3390/cancers14030486
Liu L, Patnana PK, Xie X, Frank D, Nimmagadda SC, Rosemann A, Liebmann M, Klotz L, Opalka B, Khandanpour C. High Metabolic Dependence on Oxidative Phosphorylation Drives Sensitivity to Metformin Treatment in MLL/AF9 Acute Myeloid Leukemia. Cancers. 2022; 14(3):486. https://doi.org/10.3390/cancers14030486
Chicago/Turabian StyleLiu, Longlong, Pradeep Kumar Patnana, Xiaoqing Xie, Daria Frank, Subbaiah Chary Nimmagadda, Annegret Rosemann, Marie Liebmann, Luisa Klotz, Bertram Opalka, and Cyrus Khandanpour. 2022. "High Metabolic Dependence on Oxidative Phosphorylation Drives Sensitivity to Metformin Treatment in MLL/AF9 Acute Myeloid Leukemia" Cancers 14, no. 3: 486. https://doi.org/10.3390/cancers14030486