
Efficacy and Synergy of Small Molecule Inhibitors Targeting FLT3-ITD+ Acute Myeloid Leukemia

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
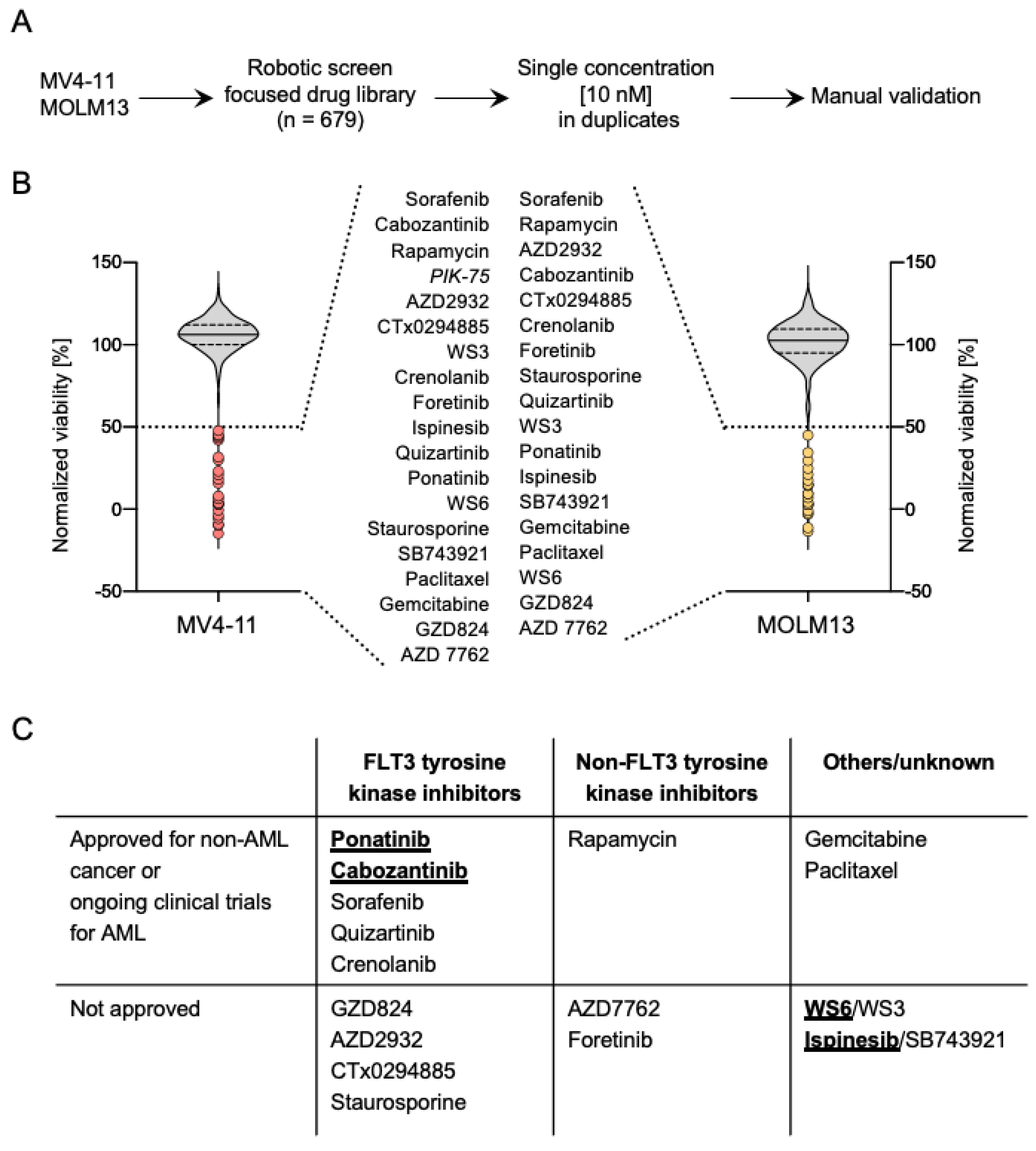
2.1. A High-Throughput Drug Screen Identifies Compounds That Exhibit a Strong Inhibitory Activity against FLT3-ITD+ AML
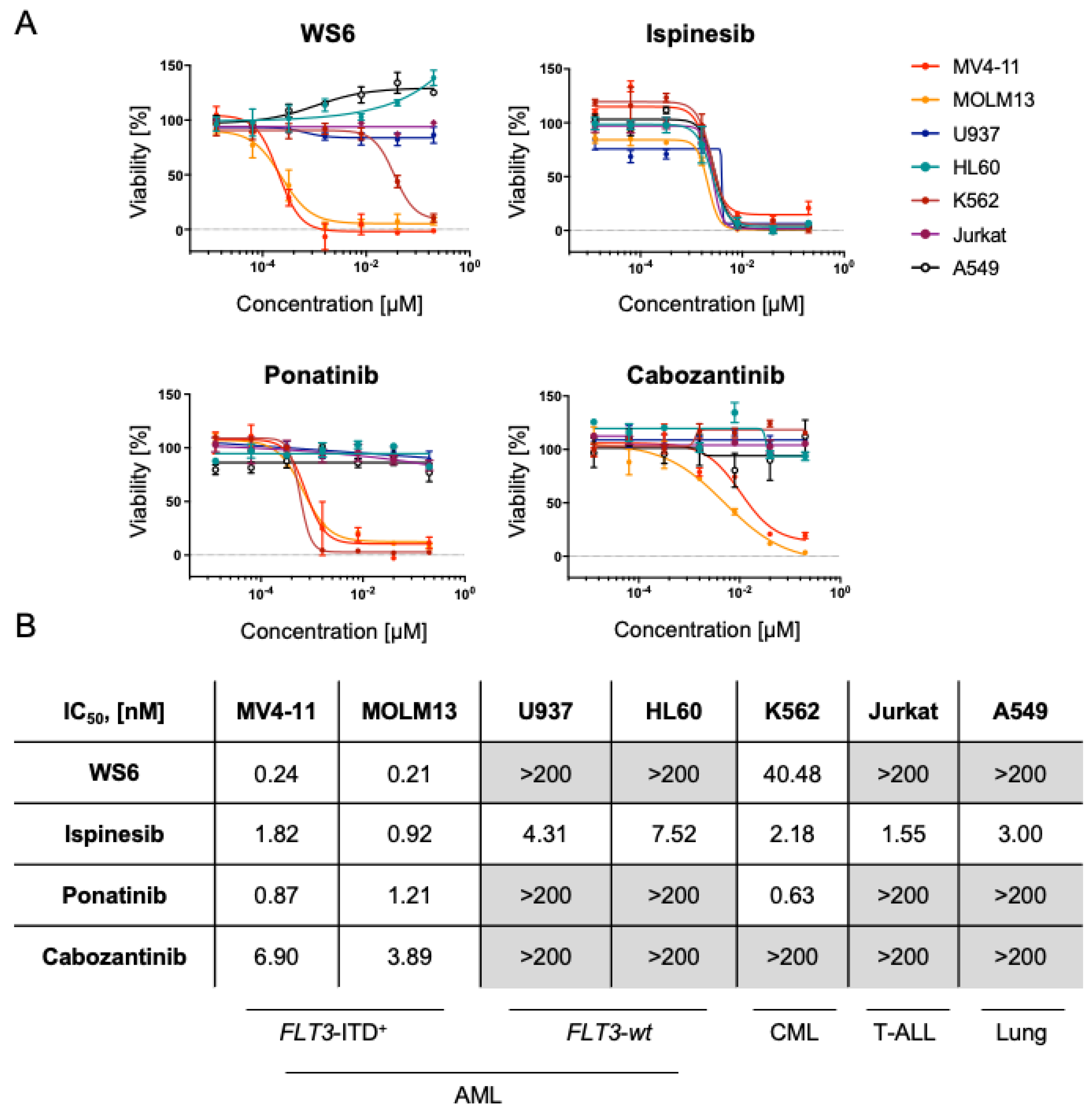
2.2. WS6, Ponatinib and Cabozantinib Are Selective for FLT3-ITD+ Compared to FLT3-wt AML
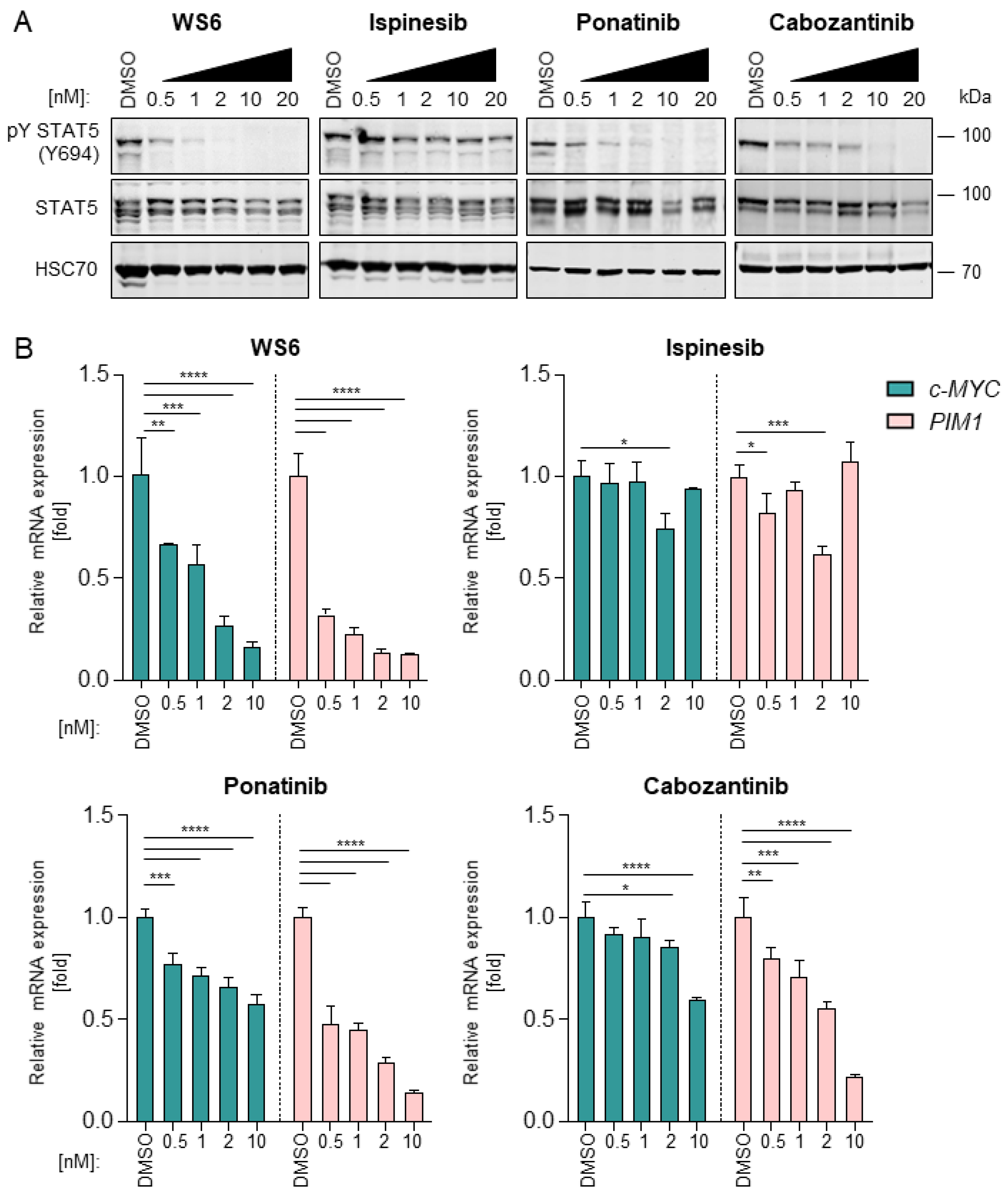
2.3. WS6, Ponatinib and Cabozantinib Inhibit the FLT3-STAT5 Axis
2.4. Ispinesib and WS6 Induce Apoptosis More Rapidly Than Ponatinib and Cabozantinib
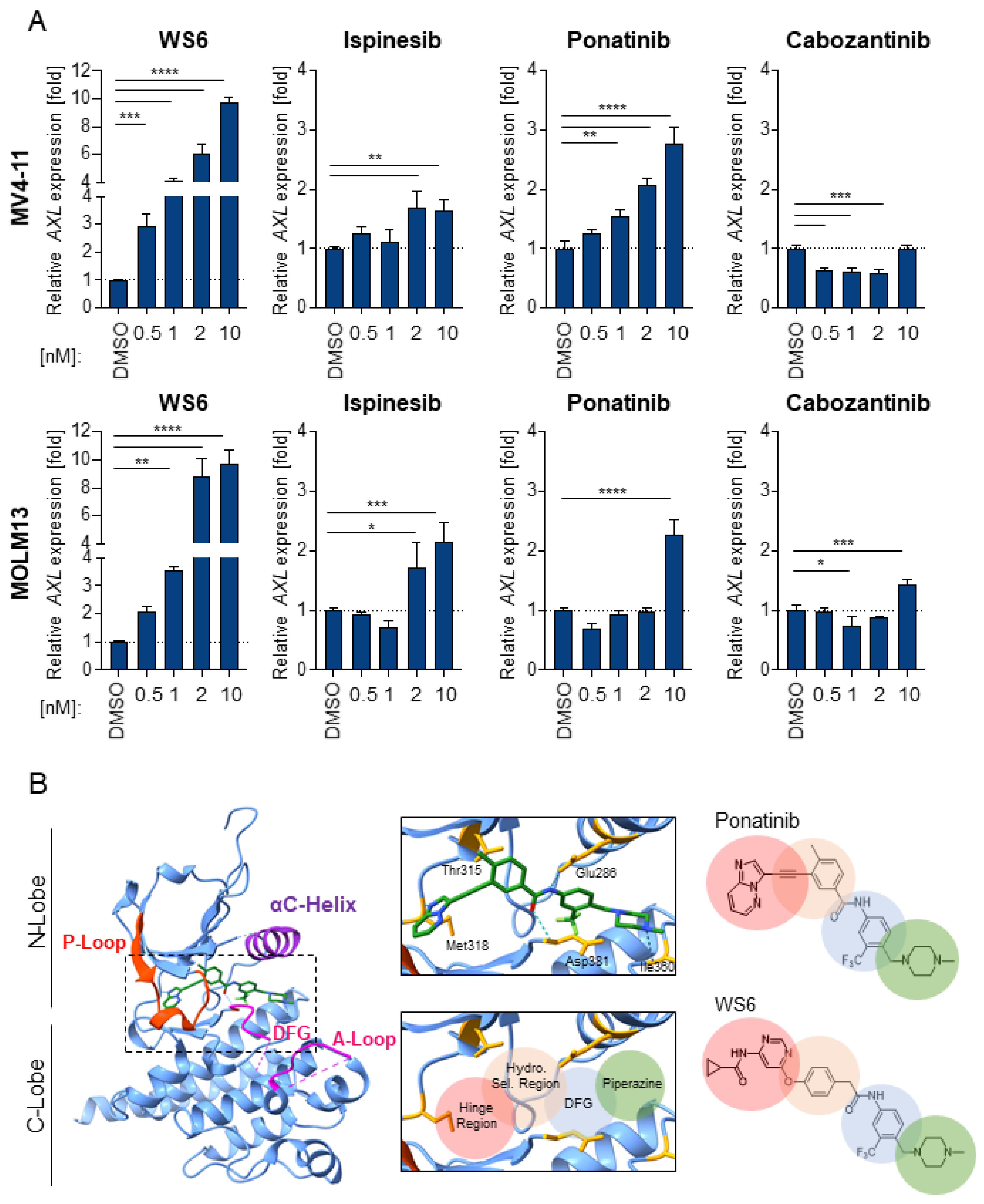
2.5. WS6 Action Shows Similarities to Ponatinib and Cabozantinib
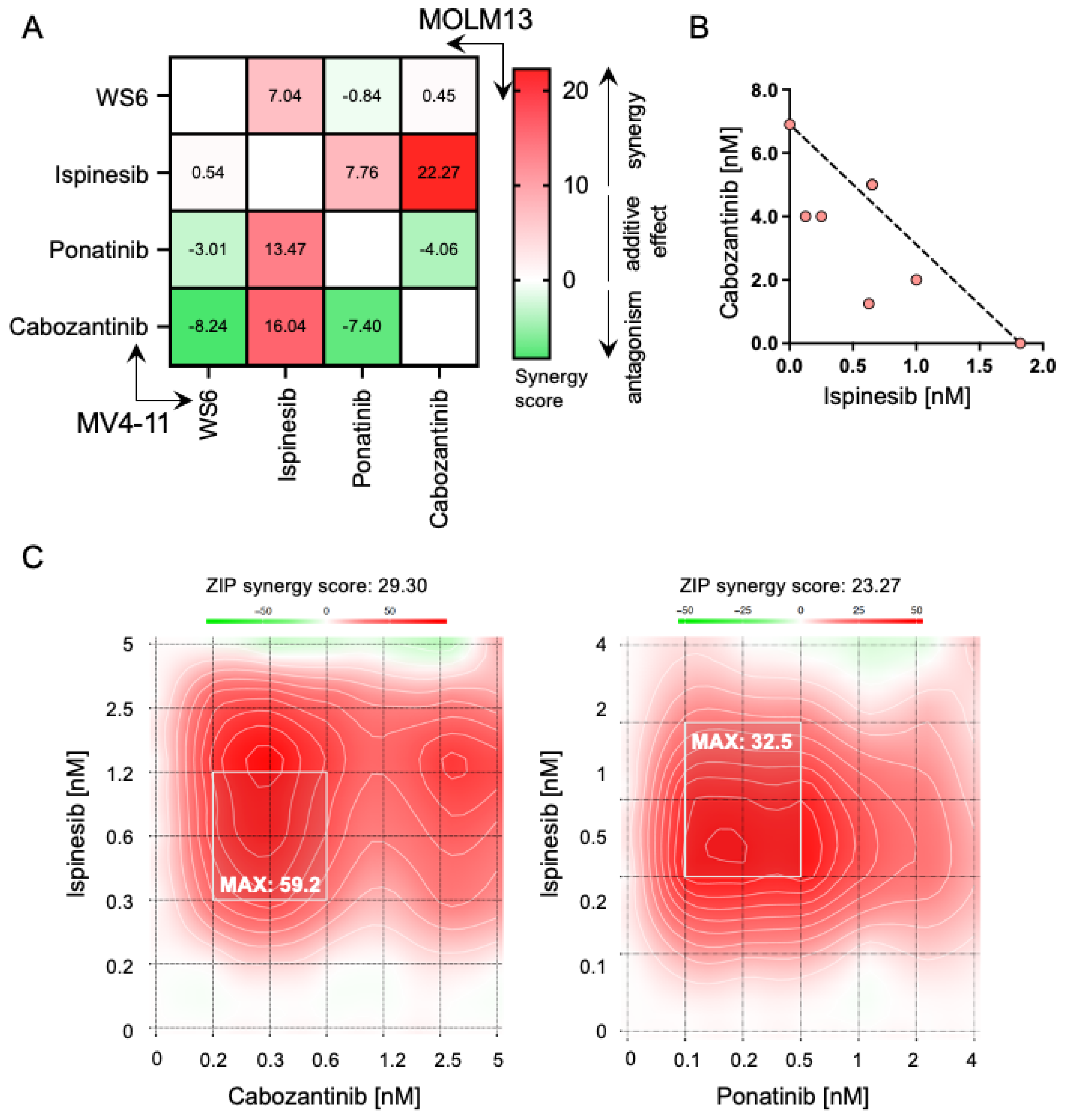
2.6. Ispinesib Synergizes with Cabozantinib and Ponatinib in Inhibiting AML Cells Growth
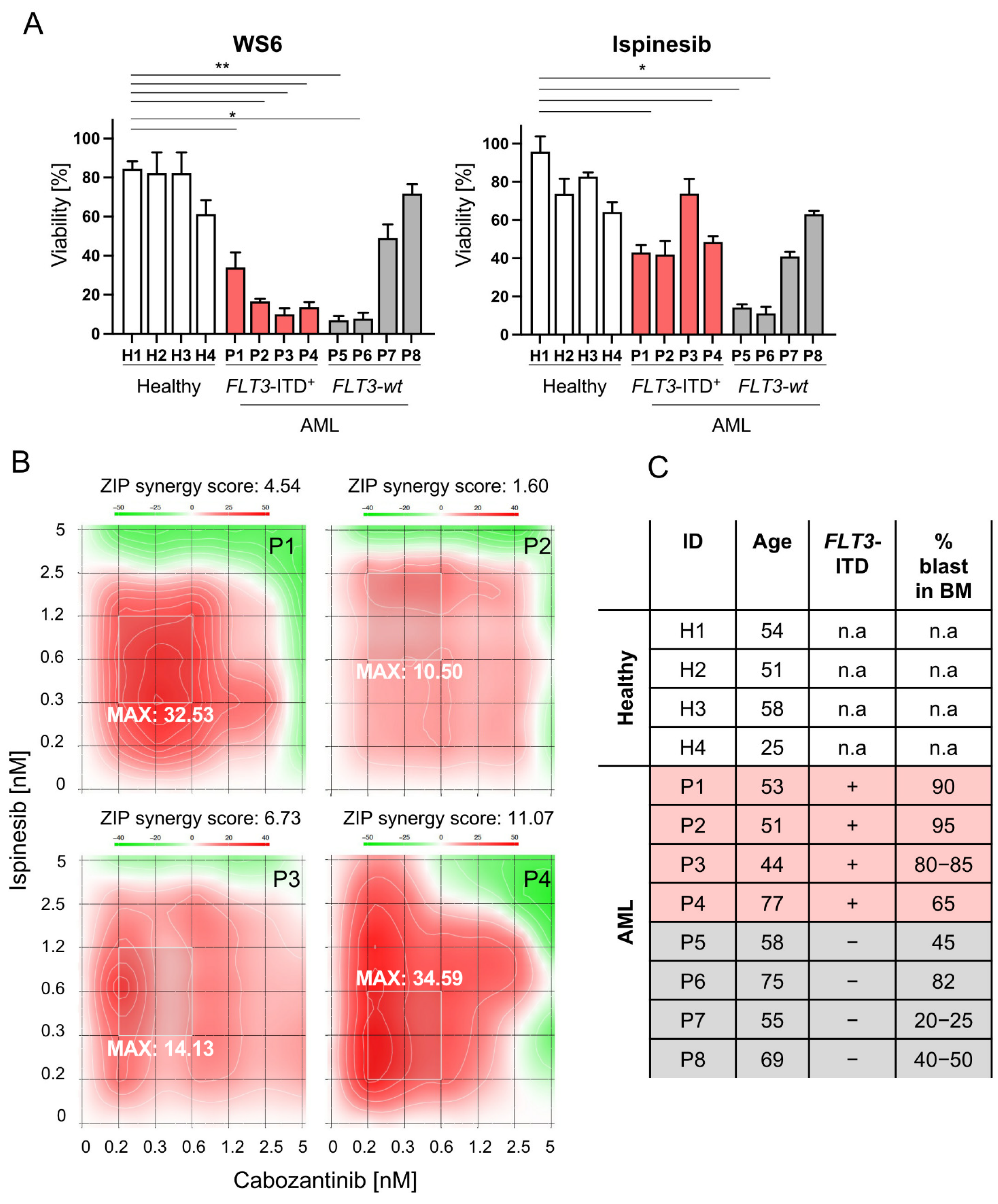
2.7. Selected Compounds as Well as Cabozantinib-Ispinesib Combination Are Effective in Leukemic Cells Derived from FLT3-ITD+ AML Patients
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Compounds
4.2. Patient Samples
4.3. High-Throughput Compound Screening
4.4. Cytotoxicity Assay
4.5. Synergy Screening
4.6. Immunoblotting
4.7. Gene Expression Analysis
4.8. CD34+ Annexin V/DAPI Staining
4.9. Annexin V/PI Staining
4.10. Caspase 3/7 Activity Assay for Apoptosis Detection
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, J.; Yu, Y.; Kaye, J.A.; Davis, K.L. Medicare Fee-for-Service Enrollees with Primary Acute Myeloid Leukemia: An Analysis of Treatment Patterns, Survival, and Healthcare Resource Utilization and Costs. Appl. Health Econ. Health Policy 2013, 11, 275–286. [Google Scholar] [CrossRef]
- Gary Gilliland, D.; Griffin, J.D. The Roles of FLT3 in Hematopoiesis and Leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Pan, Y.; Guo, Y.; Zhao, W.; Ho, W.T.; Wang, J.; Xu, M.; Yang, F.C.; Zhao, Z.J. Tyrosine Kinase Inhibitors Targeting FLT3 in the Treatment of Acute Myeloid Leukemia. Stem Cell Investig. 2017, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, T.M.; Marini, B.L.; Bixby, D.L.; Perissinotti, A.J. Clinical Considerations for the Use of FLT3 Inhibitors in Acute Myeloid Leukemia. Crit. Rev. Oncol. Hematol. 2019, 141, 125–138. [Google Scholar] [CrossRef]
- Hannum, C.; Culpepper, J.; Campbell, D.; McClanahan, T.; Zurawski, S.; Kastelein, R.; Bazan, J.F.; Hudak, S.; Wagner, J.; Mattson, J.; et al. Ligand for FLT3/FLK2 Receptor Tyrosine Kinase Regulates Growth of Haematopoietic Stem Cells and Is Encoded by Variant RNAs. Nature 1994, 368, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An Overview on the Role of FLT3-Tyrosine Kinase Receptor in Acute Myeloid Leukemia: Biology and Treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M.; Schnetzke, U.; Spies-Weisshart, B.; Walther, M.; Fleischmann, M.; Hilgendorf, I.; Hochhaus, A.; Scholl, S. Impact of FLT3-ITD Diversity on Response to Induction Chemotherapy in Patients with Acute Myeloid Leukemia. Haematologica 2017, 102, e129. [Google Scholar] [CrossRef] [Green Version]
- Orlova, A.; Neubauer, H.A.; Moriggl, R. The Stromal Microenvironment Provides an Escape Route from FLT3 Inhibitors through the GAS6-AXL-STAT5 Axis. Haematologica 2019, 104, 1907–1909. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 Mutations in AML: Review of Current Knowledge and Evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with Acute Myeloid Leukemia and an Activating Mutation in FLT3 Respond to a Small-Molecule FLT3 Tyrosine Kinase Inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, G.; Kantarjian, H.; Ravandi, F.; Zhang, W.; Konopleva, M.; Wright, J.J.; Faderl, S.; Verstovsek, S.; Mathews, S.; Andreeff, M.; et al. Phase I Study of Sorafenib in Patients with Refractory or Relapsed Acute Leukemias. Haematologica 2011, 96, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.; Perl, A.E.; Döhner, H.; Kantarjian, H.; Martinelli, G.; Kovacsovics, T.; Rousselot, P.; Steffen, B.; Dombret, H.; Estey, E.; et al. Quizartinib, an FLT3 Inhibitor, as Monotherapy in Patients with Relapsed or Refractory Acute Myeloid Leukaemia: An Open-Label, Multicentre, Single-Arm, Phase 2 Trial. Lancet Oncol. 2018, 19, 889–903. [Google Scholar] [CrossRef]
- Cortes, J.E.; Tallman, M.S.; Schiller, G.J.; Trone, D.; Gammon, G.; Goldberg, S.L.; Perl, A.E.; Marie, J.-P.; Martinelli, G.; Kantarjian, H.M.; et al. Phase 2b Study of 2 Dosing Regimens of Quizartinib Monotherapy in FLT3-ITD-Mutated, Relapsed or Refractory AML. Blood 2018, 132, 598–607. [Google Scholar] [CrossRef]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective Inhibition of FLT3 by Gilteritinib in Relapsed or Refractory Acute Myeloid Leukaemia: A Multicentre, First-in-Human, Open-Label, Phase 1-2 Study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Smith, C.C.; Wang, Q.; Chin, C.-S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD Mutations in FLT3 as a Therapeutic Target in Human Acute Myeloid Leukaemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef] [Green Version]
- Von Bubnoff, N.; Engh, R.A.; Aberg, E.; Sänger, J.; Peschel, C.; Duyster, J. FMS-like Tyrosine Kinase 3-Internal Tandem Duplication Tyrosine Kinase Inhibitors Display a Nonoverlapping Profile of Resistance Mutations in Vitro. Cancer Res. 2009, 69, 3032–3041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.-T.; Levis, M.; Small, D. Prolonged Exposure to FLT3 Inhibitors Leads to Resistance via Activation of Parallel Signaling Pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumas, P.-Y.; Naudin, C.; Martin-Lannerée, S.; Izac, B.; Casetti, L.; Mansier, O.; Rousseau, B.; Artus, A.; Dufossée, M.; Giese, A.; et al. Hematopoietic Niche Drives FLT3-ITD Acute Myeloid Leukemia Resistance to Quizartinib via STAT5- and Hypoxia- Dependent up-Regulation of AXL. Haematologica 2019, 104, 2017–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.-K.; Mundy-Bosse, B.; Whitman, S.P.; Zhang, X.; Warner, S.L.; Bearss, D.J.; Blum, W.; Marcucci, G.; Caligiuri, M.A. Receptor Tyrosine Kinase Axl Is Required for Resistance of Leukemic Cells to FLT3-Targeted Therapy in Acute Myeloid Leukemia. Leukemia 2015, 29, 2382–2389. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Cortes, J.; Ravandi, F.; Patel, K.P.; Burger, J.A.; Konopleva, M.; Kantarjian, H. Secondary Mutations as Mediators of Resistance to Targeted Therapy in Leukemia. Blood 2015, 125, 3236–3245. [Google Scholar] [CrossRef]
- Chen, S.-H.; Lahav, G. Two Is Better than One; toward a Rational Design of Combinatorial Therapy. Curr. Opin. Struct. Biol. 2016, 41, 145–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. Ponatinib: A Novel Multi-Tyrosine Kinase Inhibitor against Human Malignancies. OncoTargets Ther. 2019, 12, 635–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grüllich, C. Cabozantinib: Multi-Kinase Inhibitor of MET, AXL, RET, and VEGFR2. Recent Results Cancer Res. 2018, 211, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional Genomic Landscape of Acute Myeloid Leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Purcell, J.W.; Davis, J.; Reddy, M.; Martin, S.; Samayoa, K.; Vo, H.; Thomsen, K.; Bean, P.; Kuo, W.L.; Ziyad, S.; et al. Activity of the Kinesin Spindle Protein Inhibitor Ispinesib (SB-715992) in Models of Breast Cancer. Clin. Cancer Res. 2010, 16, 566–576. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Tremblay, M.S.; Deshmukh, V.A.; Wang, W.; Filippi, C.M.; Harb, G.; Zhang, Y.; Kamireddy, A.; Baaten, J.E.; Jin, Q.; et al. Small-Molecule Inducer of β Cell Proliferation Identified by High-Throughput Screening. J. Am. Chem. Soc. 2013, 135, 1669–1672. [Google Scholar] [CrossRef] [PubMed]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 Mutations in Acute Myeloid Leukemia: Therapeutic Paradigm beyond Inhibitor Development. Cancer Sci. 2020, 111, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Wang, A.; Liao, H.; Chen, C.; Hou, H.; Hu, C.; Tien, H.; Ou, D.; Lin, L. Cabozantinib Is Selectively Cytotoxic in Acute Myeloid Leukemia Cells with FLT3 -Internal Tandem Duplication (FLT3 -ITD). Cancer Lett. 2016, 376, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Caldera, M.; Müller, F.; Kaltenbrunner, I.; Licciardello, M.P.; Lardeau, C.-H.; Kubicek, S.; Menche, J. Mapping the Perturbome Network of Cellular Perturbations. Nat. Commun. 2019, 10, 5140. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated Data and New Features for Efficient Prediction of Protein Targets of Small Molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong-Ly, K.C.; Peterson, J.R. The Human Kinome and Kinase Inhibition. Curr. Protoc. Pharmacol. 2013, 60, 2.9.1–2.9.14. [Google Scholar] [CrossRef] [Green Version]
- Tanneeru, K.; Guruprasad, L. Ponatinib Is a Pan-BCR-ABL Kinase Inhibitor: MD Simulations and SIE Study. PLoS ONE 2013, 8, e78556. [Google Scholar] [CrossRef]
- Zhou, T.; Commodore, L.; Huang, W.-S.; Wang, Y.; Thomas, M.; Keats, J.; Xu, Q.; Rivera, V.M.; Shakespeare, W.C.; Clackson, T.; et al. Structural Mechanism of the Pan-BCR-ABL Inhibitor Ponatinib (AP24534): Lessons for Overcoming Kinase Inhibitor Resistance. Chem. Biol. Drug Des. 2011, 77, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ghiaur, G.; Levis, M. Mechanisms of Resistance to FLT3 Inhibitors and the Role of the Bone Marrow Microenvironment. Hematol./Oncol. Clin. N. Am. 2017, 31, 681–692. [Google Scholar] [CrossRef]
- Ianevski, A.; He, L.; Aittokallio, T.; Tang, J. SynergyFinder: A Web Application for Analyzing Drug Combination Dose-Response Matrix Data. Bioinformatics 2017, 33, 2413–2415. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual Analytics of Multi-Drug Combination Synergies. Nucleic Acids Res. 2020, 48, 488–493. [Google Scholar] [CrossRef]
- Zhang, Y.; Linn, D.; Liu, Z.; Melamed, J.; Tavora, F.; Young, C.Y.; Burger, A.M.; Hamburger, A.W. EBP1, an ErbB3-Binding Protein, Is Decreased in Prostate Cancer and Implicated in Hormone Resistance. Mol. Cancer Ther. 2008, 7, 3176–3186. [Google Scholar] [CrossRef] [Green Version]
- Squatrito, M.; Mancino, M.; Donzelli, M.; Areces, L.B.; Draetta, G.F. EBP1 Is a Nucleolar Growth-Regulating Protein That Is Part of Pre-Ribosomal Ribonucleoprotein Complexes. Oncogene 2004, 23, 4454–4465. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.W.; Bélanger, K.; Rao, S.C.; Petrella, T.M.; Tozer, R.G.; Wood, L.; Savage, K.J.; Eisenhauer, E.A.; Synold, T.W.; Wainman, N.; et al. A Phase II Study of Ispinesib (SB-715992) in Patients with Metastatic or Recurrent Malignant Melanoma: A National Cancer Institute of Canada Clinical Trials Group Trial. Investig. New Drugs 2008, 26, 249–255. [Google Scholar] [CrossRef]
- Lee, L.; Hizukuri, Y.; Severson, P.; Powell, B.; Zhang, C.; Ma, Y.; Narahara, M.; Sumi, H.; Hernandez, D.; Rajkhowa, T.; et al. A Novel Combination Regimen of BET and FLT3 Inhibition for FLT3-ITD Acute Myeloid Leukemia. Haematologica 2021, 106, 1022–1033. [Google Scholar] [CrossRef]
- Chang, E.; Ganguly, S.; Rajkhowa, T.; Gocke, C.D.; Levis, M.; Konig, H. The Combination of FLT3 and DNA Methyltransferase Inhibition Is Synergistically Cytotoxic to FLT3/ITD Acute Myeloid Leukemia Cells. Leukemia 2016, 30, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.A.; Berger-Becvar, A.; de Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic Inhibition of STAT5 in Acute Myeloid Leukemia. Leukemia 2018, 32, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Orlova, A.; Wagner, C.; de Araujo, E.D.; Bajusz, D.; Neubauer, H.A.; Herling, M.; Gunning, P.T.; Keserű, G.M.; Moriggl, R. Direct Targeting Options for STAT3 and STAT5 in Cancer. Cancers 2019, 11, 1930. [Google Scholar] [CrossRef] [Green Version]
- Scholl, S.; Fleischmann, M.; Schnetzke, U.; Heidel, F.H. Molecular Mechanisms of Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia: Ongoing Challenges and Future Treatments. Cells 2020, 9, 2493. [Google Scholar] [CrossRef]
- Smith, C.C.; Viny, A.D.; Massi, E.; Kandoth, C.; Socci, N.D.; Rapaport, F.; Najm, M.; Medina-Martinez, J.S.; Papaemmanuil, E.; Tarver, T.C.; et al. Recurrent Mutations in Cyclin D3 Confer Clinical Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 4003–4011. [Google Scholar] [CrossRef]
- Fernandez, S.; Desplat, V.; Villacreces, A.; Guitart, A.V.; Milpied, N.; Pigneux, A.; Vigon, I.; Pasquet, J.-M.; Dumas, P.-Y. Targeting Tyrosine Kinases in Acute Myeloid Leukemia: Why, Who and How? Int. J. Mol. Sci. 2019, 20, 3429. [Google Scholar] [CrossRef] [Green Version]
- Rummelt, C.; Gorantla, S.P.; Meggendorfer, M.; Charlet, A.; Endres, C.; Döhner, K.; Heidel, F.H.; Fischer, T.; Haferlach, T.; Duyster, J.; et al. Activating JAK-Mutations Confer Resistance to FLT3 Kinase Inhibitors in FLT3-ITD Positive AML in Vitro and in Vivo. Leukemia 2021, 35, 2017–2029. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Protein Name | Target Class |
| MAP kinase signal-integrating kinase 2 | MKNK2 | Kinase |
| MAP kinase-interacting serine/threonine-protein kinase MNK1 | MKNK1 | Kinase |
| Tyrosine-protein kinase SRC | SRC | Kinase |
| Serine/threonine-protein kinase B-RAF | BRAF | Kinase |
| Tyrosine-protein kinase ABL | ABL1 | Kinase |
| Tyrosine-protein kinase LCK | LCK | Kinase |
| Vascular endothelial growth factor receptor 2 | VEGFR-2/KDR | Kinase |
| Tyrosine-protein kinase TIE-2 | TEK | Kinase |
| Tyrosine-protein kinase Lyn | LYN | Kinase |
| Serine/threonine-protein kinase RAF | RAF1 | Kinase |
| Gene | Sequence | Fragment Size |
|---|---|---|
| c-MYC | fwd: TTTCGGGTAGTGGAAAACCA | 90 bp |
| rev: CACCGAGTCGTAGTCGAGGT | ||
| PIM-1 | fwd: CTCAAGCTCATCGACTTCGG | 105 bp |
| rev: ATGGTAGCGGATCCACTCTG | ||
| AXL | fwd: GTTTGGAGCTGTGATGGAAGGC | 120 bp |
| rev: CGCTTCACTCAGGAAATCCTCC | ||
| GAPDH | fwd: AAGGTGAAGGTCGGAGTCAA | 108 bp |
| rev: AATGAAGGGGTCATTGATGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bregante, J.; Schönbichler, A.; Pölöske, D.; Degenfeld-Schonburg, L.; Monzó Contreras, G.; Hadzijusufovic, E.; de Araujo, E.D.; Valent, P.; Moriggl, R.; Orlova, A. Efficacy and Synergy of Small Molecule Inhibitors Targeting FLT3-ITD+ Acute Myeloid Leukemia. Cancers 2021, 13, 6181. https://doi.org/10.3390/cancers13246181
Bregante J, Schönbichler A, Pölöske D, Degenfeld-Schonburg L, Monzó Contreras G, Hadzijusufovic E, de Araujo ED, Valent P, Moriggl R, Orlova A. Efficacy and Synergy of Small Molecule Inhibitors Targeting FLT3-ITD+ Acute Myeloid Leukemia. Cancers. 2021; 13(24):6181. https://doi.org/10.3390/cancers13246181
Chicago/Turabian StyleBregante, Javier, Anna Schönbichler, Daniel Pölöske, Lina Degenfeld-Schonburg, Garazi Monzó Contreras, Emir Hadzijusufovic, Elvin D. de Araujo, Peter Valent, Richard Moriggl, and Anna Orlova. 2021. "Efficacy and Synergy of Small Molecule Inhibitors Targeting FLT3-ITD+ Acute Myeloid Leukemia" Cancers 13, no. 24: 6181. https://doi.org/10.3390/cancers13246181