
Integration of Tumor Heterogeneity for Recurrence Prediction in Patients with Esophageal Squamous Cell Cancer

 ,
,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Sample Selection and Sequencing
2.2. Bioinformatic Analysis
2.3. Statistical Analysis
3. Results
3.1. Patient Characteristics
3.2. Overview of Genomic Alterations
3.3. Clinical Relevance of Genetic Alterations
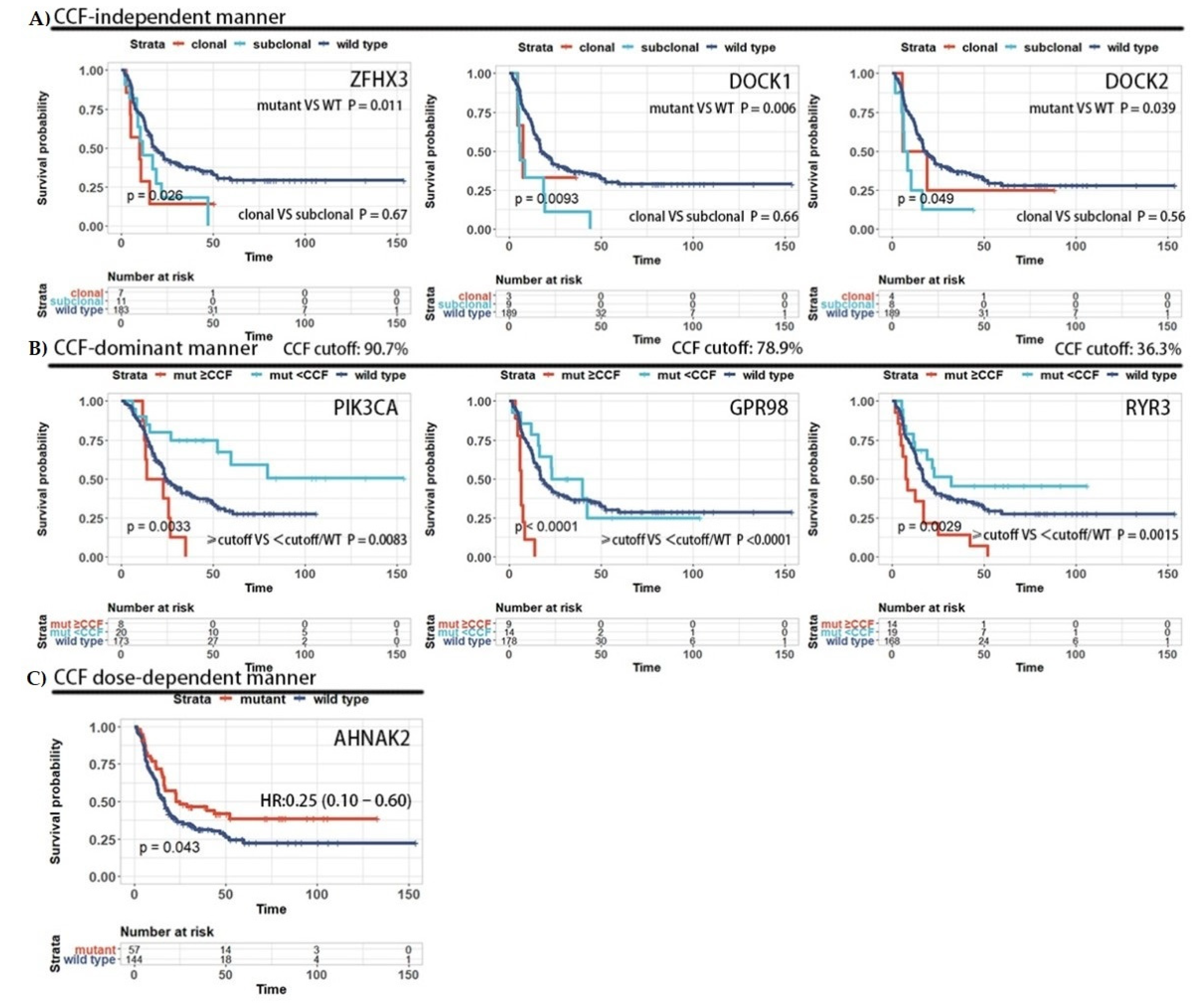
3.4. The Prognosis Effect of Mutations Is Related to Cancer Cell Fraction
3.5. Construction of a Recurrence Predictor Using Sequencing Data
3.6. Prognostic Value of the Eight-Gene Classifier
3.7. Specific Genotype of Long Survivors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, H.; Chen, Y.; Zhu, C.; Fang, W.; Yu, Z. Neoadjuvant Chemoradiotherapy Followed by SurgeryVersus Surgery Alone for Locally Advanced Squamous Cell Carcinoma of the Esophagus (NEOCRTEC5010): A Phase III Multicenter, Randomized, Open-Label Clinical Trial. J. Clin. Oncol. 2018, 36, 2796–2803. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, H.; Xi, R.; Cui, H.; Zhao, Y.; Xu, E.; Yan, T.; Lu, X.; Huang, F.; Kong, P.; et al. Whole-Genome sequencing of 508 patients identifies key molecular features associated with poor prognosis in esophageal squamous cell carcinoma. Cell. Res. 2020, 30, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zhao, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Shigaki, H.; Baba, Y.; Watanabe, M.; Murata, A.; Ishimoto, T.; Iwatsuki, M.; Iwagami, S.; Nosho, K.; Baba, H. PIK3CA mutation is associated with a favorable prognosis among patients with curatively resected esophageal squamous cell carcinoma. Clin. Cancer Res. 2013, 19, 2451–2459. [Google Scholar] [CrossRef] [Green Version]
- Yokota, T.; Serizawa, M.; Hosokawa, A.; Kusafuka, K.; Mori, K.; Sugiyama, T.; Tsubosa, Y.; Koh, Y. PIK3CA mutation is a favorable prognostic factor in esophageal cancer: Molecular profile by next-Generation sequencing using surgically resected formalin-Fixed, paraffin-Embedded tissue. BMC Cancer 2018, 18, 826. [Google Scholar] [CrossRef]
- Li, X.C.; Wang, M.Y.; Yang, M.; Dai, H.J.; Zhang, B.F.; Wang, W.; Chu, X.L.; Wang, X.; Zheng, H.; Niu, R.F.; et al. A mutational signature associated with alcohol consumption and prognostically significantly mutated driver genes in esophageal squamous cell carcinoma. Ann. Oncol. 2018, 29, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef]
- Rossi, D.; Khiabanian, H.; Spina, V.; Ciardullo, C.; Bruscaggin, A.; Famà, R.; Rasi, S.; Monti, S.; Deambrogi, C.; de Paoli, L.; et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 2014, 123, 2139–2147. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Chronic Myeloid Disorders Working Group of the International Cancer Genome, Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Nadeu, F.; Delgado, J.; Royo, C.; Baumann, T.; Stankovic, T.; Pinyol, M.; Jares, P.; Navarro, A.; Martin-Garcia, D.; Bea, S.; et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood 2016, 127, 2122–2130. [Google Scholar] [CrossRef]
- Pastore, A.; Jurinovic, V.; Kridel, R.; Hoster, E.; Staiger, A.M.; Szczepanowski, M.; Pott, C.; Kopp, N.; Murakami, M.; Horn, H.; et al. Integration of gene mutations in risk prognostication for patients receiving first-Line immunochemotherapy for follicular lymphoma: A retrospective analysis of a prospective clinical trial and validation in a population-Based registry. Lancet Oncol. 2015, 16, 1111–1122. [Google Scholar] [CrossRef]
- Wang, H.L.; Liu, P.F.; Yue, J.; Jiang, W.H.; Cui, Y.L.; Ren, H.; Wang, H.; Zhuang, Y.; Liu, Y.; Jiang, D.; et al. Somatic gene mutation signatures predict cancer type and prognosis in multiple cancers with pan-Cancer 1000 gene panel. Cancer Lett. 2020, 470, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. pROC: An open-Source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; van Loo, P.; Wedge, D.C.; Alexandrov, L.B.; Greenman, C.D.; Lau, K.W.; Raine, K.; Jones, D.; Marshall, J.; Ramakrishna, M.; et al. Breast Cancer Working Group of the International Cancer Genome, The life history of 21 breast cancers. Cell 2012, 149, 994–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dentro, S.C.; Wedge, D.C.; van Loo, P. Principles of Reconstructing the Subclonal Architecture of Cancers. Cold Spring Harb. Perspect Med. 2017, 7, a026625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shan, L.; Zhang, S.; Ying, J.; Xue, L.; Yuan, Y.; Xie, Y.; Lu, N. PIK3CA gene mutations and overexpression: Implications for prognostic biomarker and therapeutic target in Chinese esophageal squamous cell carcinoma. PLoS ONE 2014, 9, e103021. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Lin, L.H.; Yu, S.Y.; Kao, S.Y.; Chang, K.W.; Cheng, H.W.; Liu, C.J. FAT1 somatic mutations in head and neck carcinoma are associated with tumor progression and survival. Carcinogenesis 2018, 39, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.J.; Miranda, M.A.; O’Hern, M.J.; McElroy, J.P.; Coombes, K.R.; Bundschuh, R.; Cohn, D.E.; Mutch, D.G.; Goodfellow, P.J. Patterns of CTCF and ZFHX3 Mutation and Associated Outcomes in Endometrial Cancer. J. Natl. Cancer Inst. 2015, 107, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Chen, H.; Zhou, D.; Zhang, J.; Chen, Y.; Liu, Q.; Ai, D.; Zhu, H.; Chu, L.; Ren, W.; et al. Comparative genomic analysis of esophageal squamous cell carcinoma between Asian and Caucasian patient populations. Nat. Commun. 2017, 8, 1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Li, R. Variable Selection via Nonconcave Penalized Likelihood and its Oracle Properties. J. Am. Stat. Assoc. 2001, 96, 1348–1360. [Google Scholar] [CrossRef]
- Meng, X.; Chen, X.; Lu, P.; Ma, W.; Yue, D.; Song, L.; Fan, Q. MicroRNA-202 inhibits tumor progression by targeting LAMA1 in esophageal squamous cell carcinoma. Biochem. Biophys Res Commun. 2016, 473, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Michaud, D.S.; Ruan, M.; Koestler, D.C.; Pei, D.; Marsit, C.J.; de Vivo, I.; Kelsey, K.T. Epigenome-Wide Association Study Using Prediagnostic Bloods Identifies New Genomic Regions Associated With Pancreatic Cancer Risk. JNCI Cancer Spectr. 2020, 4, pkaa041. [Google Scholar] [CrossRef]
- Yang, B.; Wu, A.; Hu, Y.; Tao, C.; Wang, J.M.; Lu, Y.; Xing, R. Mucin 17 inhibits the progression of human gastric cancer by limiting inflammatory responses through a MYH9-p53-RhoA regulatory feedback loop. J. Exp. Clin. Cancer Res. 2019, 38, 283. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Sheldon, H.; Martincorena, I.; van Loo, P.; Gundem, G.; Wedge, D.C.; Ramakrishna, M.; Cooke, S.L.; Pillay, N.; et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat. Genet. 2014, 46, 376–379. [Google Scholar] [CrossRef]
- Wang, M.; Li, X.; Zhang, J.; Yang, Q.; Chen, W.; Jin, W.; Huang, Y.R.; Yang, R.; Gao, W.Q. AHNAK2 is a Novel Prognostic Marker and Oncogenic Protein for Clear Cell Renal Cell Carcinoma. Theranostics 2017, 7, 1100–1113. [Google Scholar] [CrossRef]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012, 485, 502–506. [Google Scholar] [CrossRef]
- He, S.; Lin, J.; Yu, S.; Sun, S. Upregulation of PREX2 promotes the proliferation and migration of hepatocellular carcinoma cells via PTEN-AKT signaling. Oncol. Lett. 2016, 11, 2223–2228. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, D.; He, S.; Harada, K.; Pizzi, M.P.; Lu, Y.; Guan, P.; Chen, L.; Wang, R.; Zhang, S.; Sewastjanow-Silva, M.A.; et al. Integrated genomic profiling and modelling for risk stratification in patients with advanced oesophagogastric adenocarcinoma. Gut 2021, 70, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Yoon, C.; Lee, J.H.; Chang, K.K.; Lin, J.X.; Kim, Y.H.; Kook, M.C.; Aksoy, B.A.; Park, D.J.; Ashktorab, H.; et al. KMT2C Mutations in Diffuse-Type Gastric Adenocarcinoma Promote Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2018, 24, 6556–6569. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Training Set (N = 143) | Validation Set (N = 58) | Full Set (N = 201) | |
|---|---|---|---|---|
| N (%) | N (%) | p | N (%) | |
| Sex | 0.002 | |||
| Female/Male | 17/126 (11.9/88.1) | 18/40 (31.0/69) | 35/166 (17.4/82.6) | |
| 0.206 | ||||
| <60/≥60 | 70/73 (49.0/51.0) | 22/36 (37.9/62.1) | 92/109 (45.8/54.2) | |
| 0.222 | ||||
| <4 cm/≥4 cm | 67/76 (46.9/53.1) | 21/37 (36.2/63.8) | 88/113 (43.8/56.2) | |
| Smoking status | 0.063 | |||
| Yes/No | 98/45 (68.5/31.5) | 31/27 (53.5/46.5) | 129/72 (64.2/35.8) | |
| Alcoholism | 0.520 | |||
| Yes/No | 73/70 (51.0/49.0) | 26/32 (44.8/55.2) | 99/102 (49.3/50.7) | |
| Differentiation | 0.343 | |||
| Well/Moderate/Poor | 17/71/55 (11.9/49.7/38.5) | 8/34/16 (13.8/58.6/27.6) | 25/105/71 (12.4/52.2/35.3) | |
| Surgical approach | 0.609 | |||
| Left thoracotomy | 49(34.3) | 17(29.3) | 66(32.8) | |
| Right thoracotomy | 94(65.7) | 41(70.7) | 135(67.2) | |
| Lesion location | 0.151 | |||
| Upper/Middle/Lower | 8/54/51 (5.6/58.7/35.7) | 8/31/19 (13.8/53.4/32.8) | 16/115/70 (7.9/57.2/34.9) | |
| 0.848 | ||||
| 23/120(16.1/83.9) | 8/50(13.7/86.3) | 31/170 (15.4/84.6) | ||
| 1.000 | ||||
| 70/73 (49.0/51.0) | 29/29 (50.0/50.0) | 99/102(49.3/20.7) | ||
| 0.696 | ||||
| ≥21/<21 | 108/35 (75.5/24.5) | 46/12 (79.3/20.7) | 154/47 (76.4/23.4) | |
| Adjuvant therapy | 1.00 | |||
| Yes/No | 43/100 (30.1/69.9) | 17/41 (29.3/70.7) | 60/141 (29.8/69.2) | |
| Variables | Entire Cohort | Training Cohort | Validation Cohort | |||
|---|---|---|---|---|---|---|
| HR (95%CI) | p | HR (95%CI) | p | HR (95%CI) | p | |
| Sex (male vs. female) | 0.45 (0.26–0.77) | 0.004 | 1.18 (0.61–2.31) | 0.625 | 0.13 (0.05–0.38) | 0.0001 |
| (≥60 vs. <60) | 0.75 (0.52–1.09) | 0.130 | 0.67 (0.44–1.03) | 0.071 | 0.45 (0.20–0.99) | 0.046 |
| Surgical approach (right vs. left thoracotomy) | 0.72 (0.50–1.05) | 0.102 | 0.52 (0.33–0.82) | 0.005 | 1.17 (0.54–2.56) | 0.688 |
| Alb (≥40 vs. <40) | 0.46 (0.11–2.01) | 0.304 | 1.86 (0.24–14.18) | 0.548 | 0.30 (0.033–2.56) | 0.294 |
(≥21 vs. <21) | 1.19 (0.83–1.71) | 0.351 | 0.99 (0.64–1.53) | 0.952 | 1.51 (0.74–3.08) | 0.262 |
) | 1.03 (0.60–1.77) | 0.902 | 1.31 (0.69–2.50) | 0.408 | 0.43 (0.15–1.28) | 0.128 |
) | 2.37 (1.63–3.44) | <0.001 | 2.71 (1.73–4.25) | <0.001 | 2.29 (1.07–4.92) | 0.033 |
| Genetic model (low risk) | - | - | - | - | - | - |
| Intermediate risk | 2.74 (1.59–4.72) | <0.001 | 2.65 (1.40–5.03) | 0.003 | 3.52 (1.23–10.10) | 0.019 |
| High risk | 6.50 (3.60–11.75) | <0.001 | 6.74 (3.34–13.58) | <0.001 | 8.19 (2.484–27.01) | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mai, Z.; Liu, Q.; Wang, X.; Xie, J.; Yuan, J.; Zhong, J.; Fang, S.; Xie, X.; Yang, H.; Wen, J.; et al. Integration of Tumor Heterogeneity for Recurrence Prediction in Patients with Esophageal Squamous Cell Cancer. Cancers 2021, 13, 6084. https://doi.org/10.3390/cancers13236084
Mai Z, Liu Q, Wang X, Xie J, Yuan J, Zhong J, Fang S, Xie X, Yang H, Wen J, et al. Integration of Tumor Heterogeneity for Recurrence Prediction in Patients with Esophageal Squamous Cell Cancer. Cancers. 2021; 13(23):6084. https://doi.org/10.3390/cancers13236084
Chicago/Turabian StyleMai, Zihang, Qianwen Liu, Xinye Wang, Jiaxin Xie, Jianye Yuan, Jian Zhong, Shuogui Fang, Xiuying Xie, Hong Yang, Jing Wen, and et al. 2021. "Integration of Tumor Heterogeneity for Recurrence Prediction in Patients with Esophageal Squamous Cell Cancer" Cancers 13, no. 23: 6084. https://doi.org/10.3390/cancers13236084