
Molecular Activation of the Kv11.1 Channel Reprograms EMT in Colon Cancer by Inhibiting TGFβ Signaling via Activation of Calcineurin

 , ,
, ,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
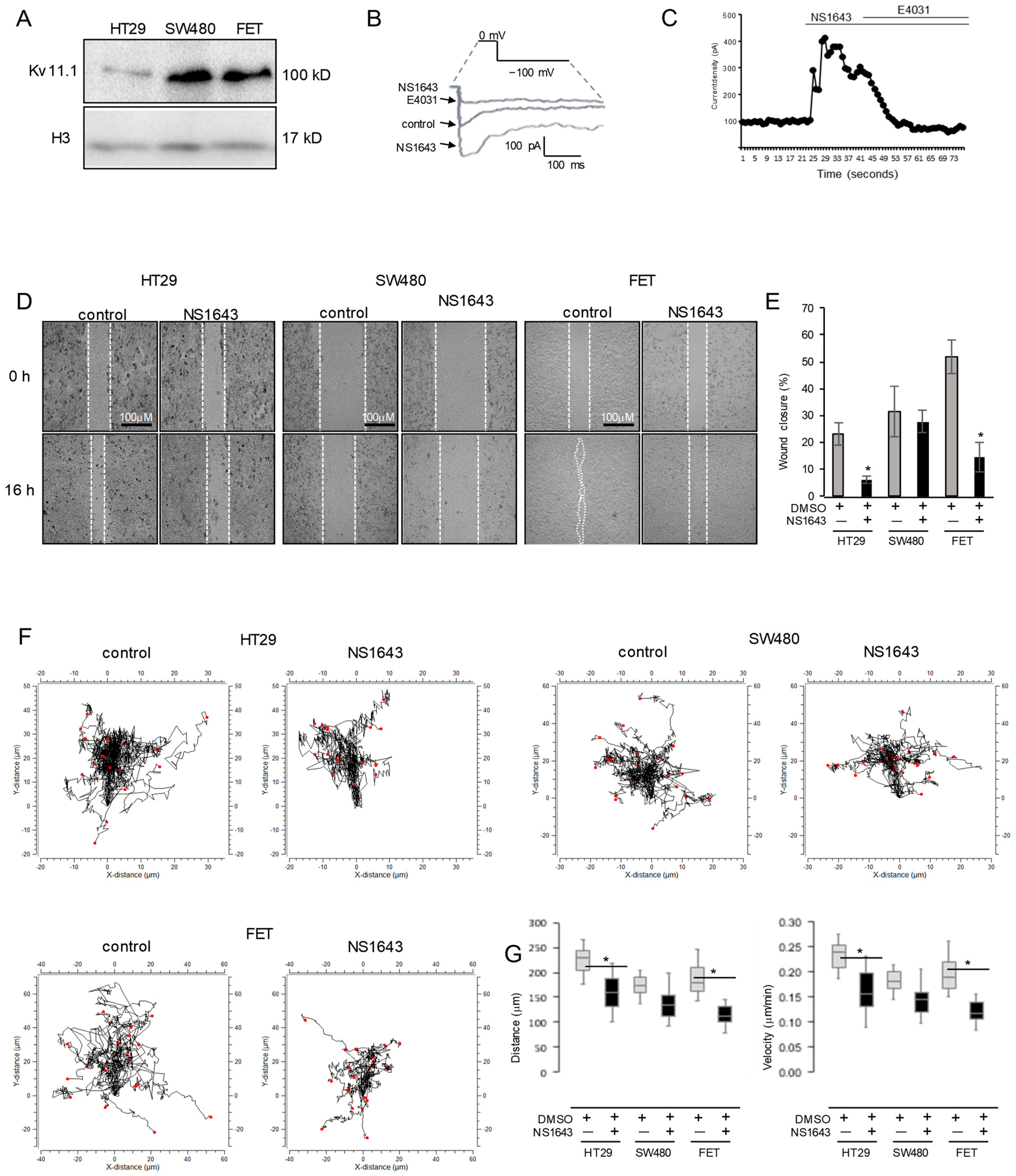
2.1. Cell Culture, Antibodies, and Reagents
2.2. Electrophysiology
2.3. RT-PCR Analysis and si-RNA
2.4. Wound-Healing Assay
2.5. Video Microscopy and Single Cell Tracking
2.6. Flow Cytometry Analysis
2.7. Cell Cycle Assay
2.8. Cell Spheroid Formation and Drug Treatment on Agarose-Microwells
2.9. Cell Apoptosis Assay
2.10. Statistical Analysis
3. Results
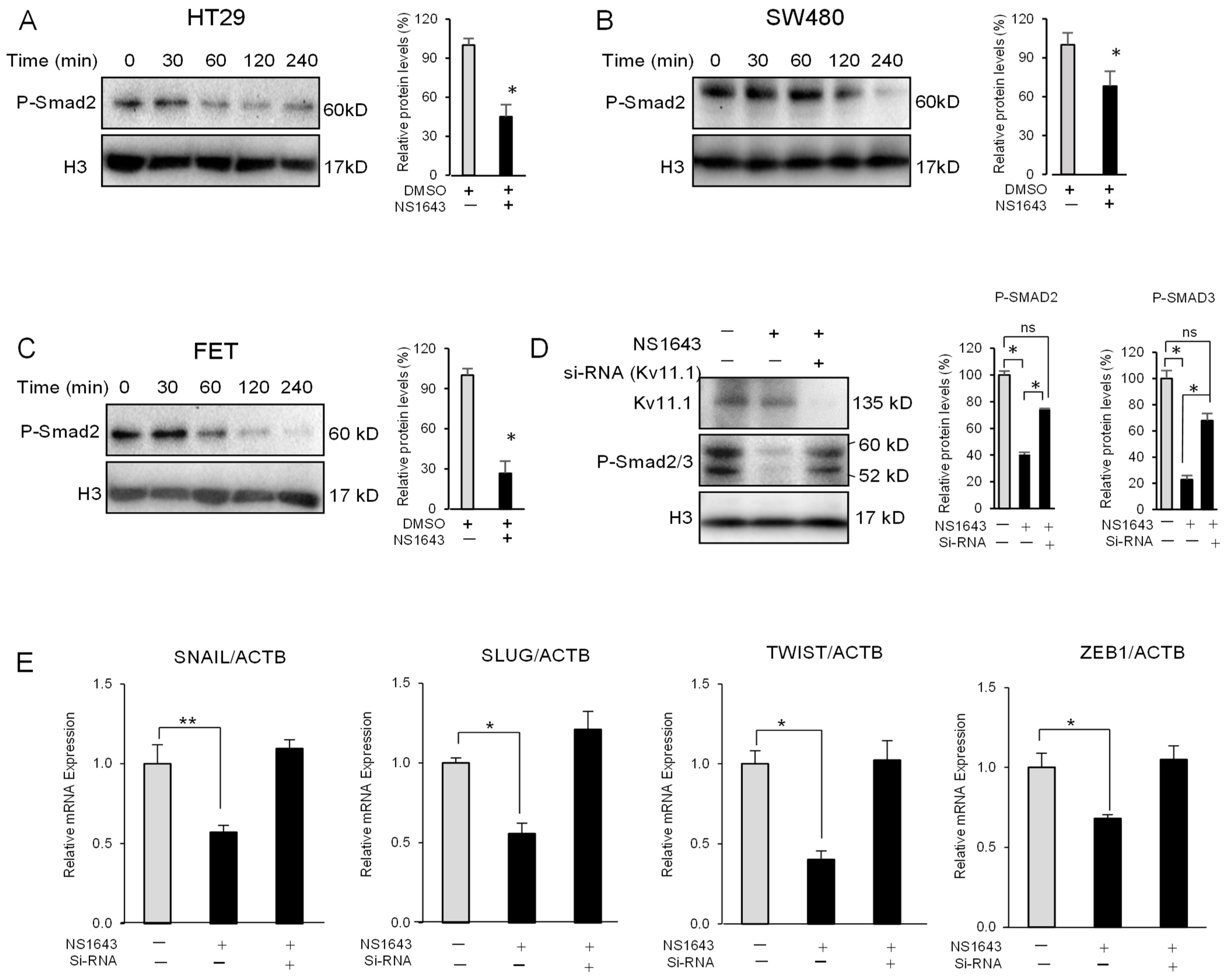
3.1. Activation of the Kv11.1 Channel Inhibits Colon Cancer Cells Motility
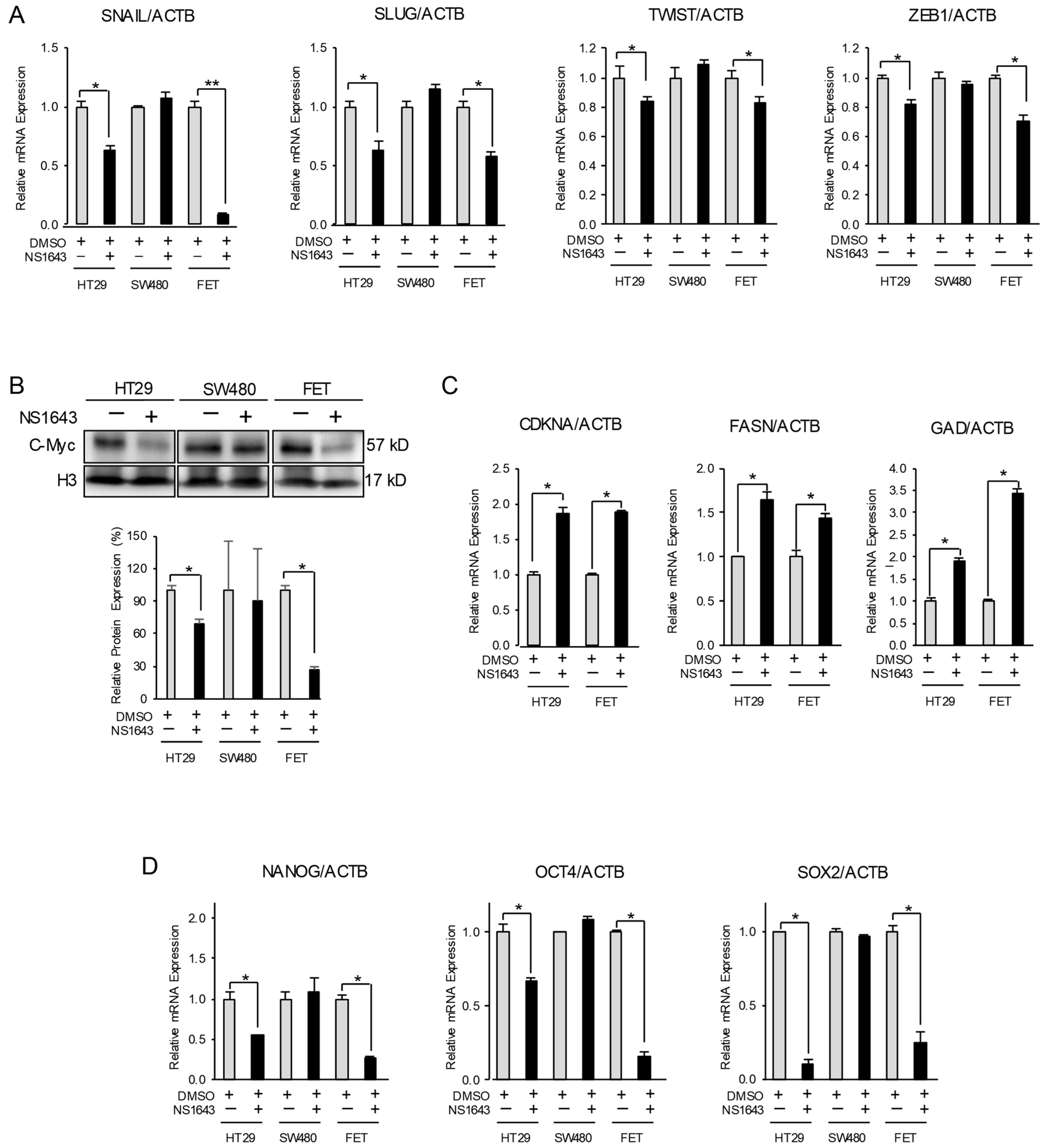
3.2. Kv11.1 Activity Inhibits Mesenchymal and Stemness Phenotype in Colon Cancer Cells
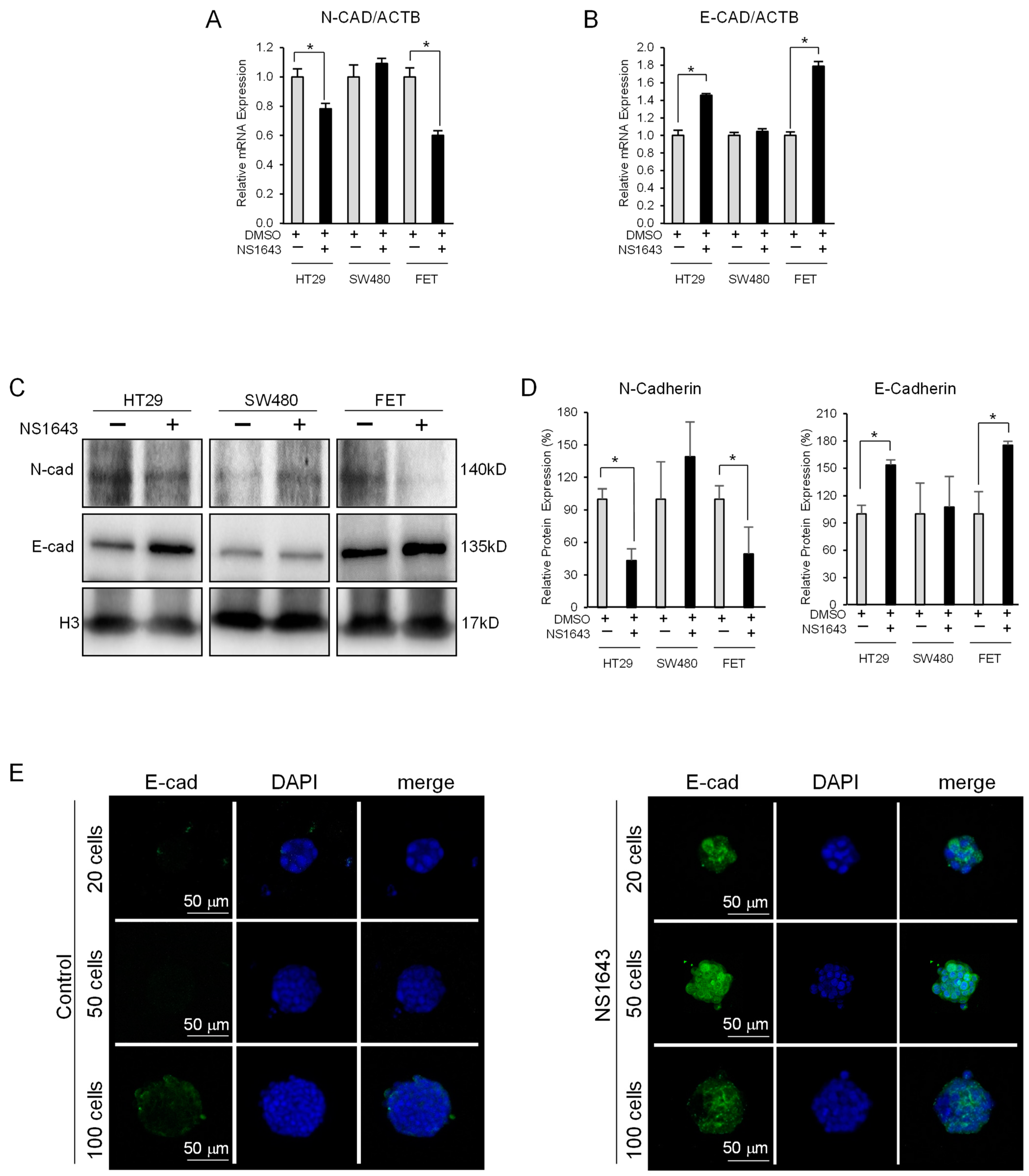
3.3. Kv11.1 Activator Stimulates Expression of the Epithelial Marker E-Cadherin in Colon Cancer Cells
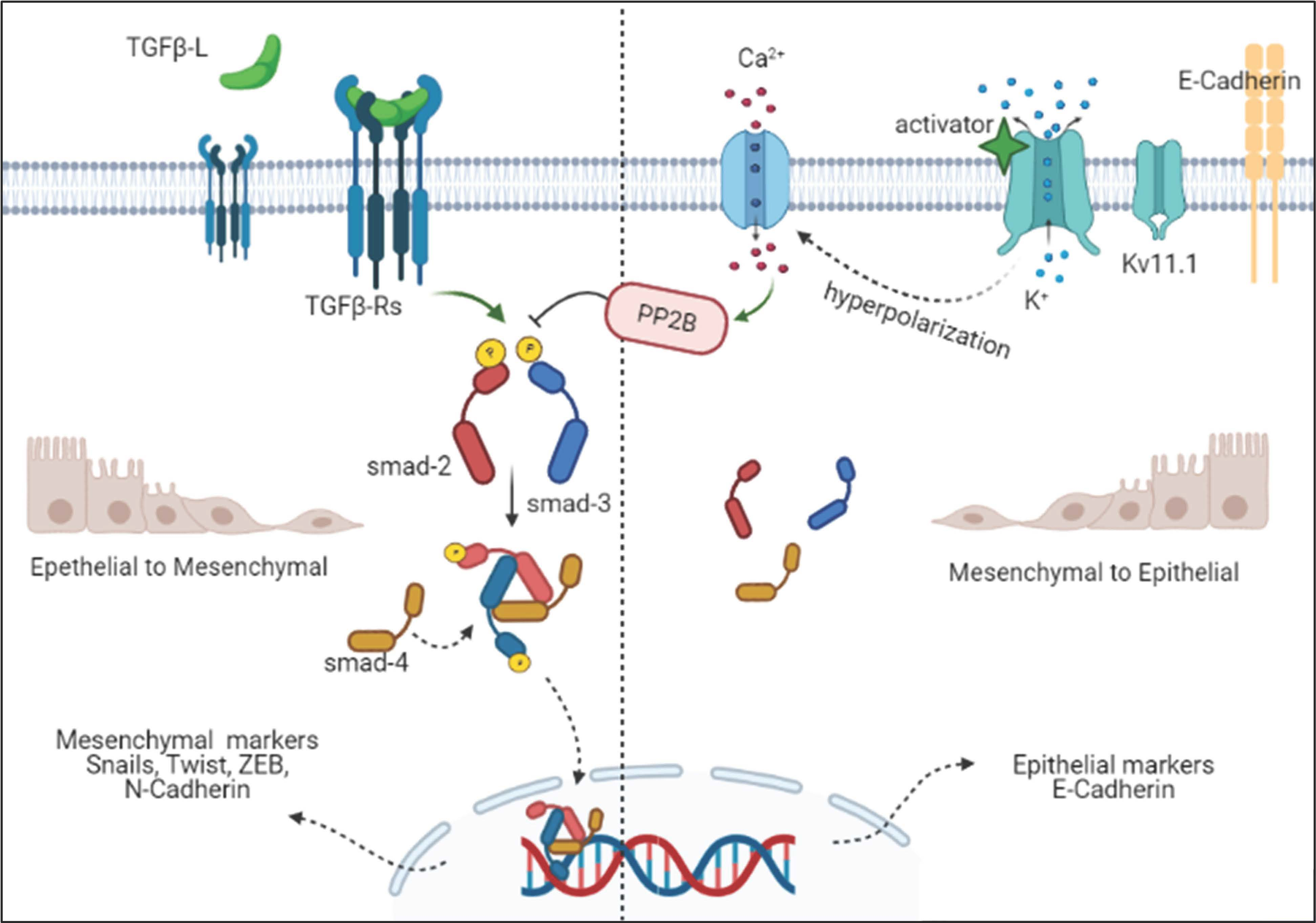
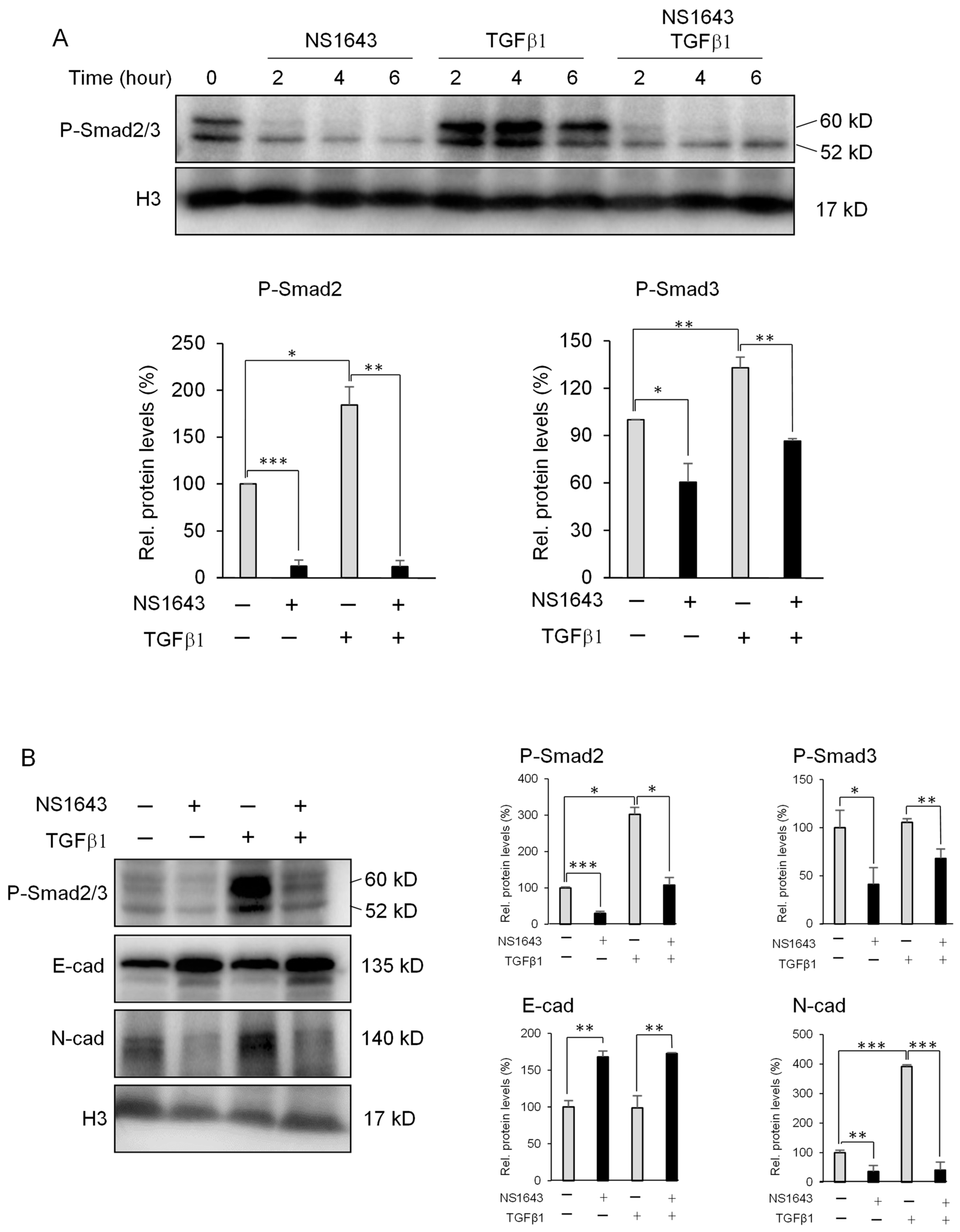
3.4. Kv11.1 Activity Inhibits TGFβ Signaling
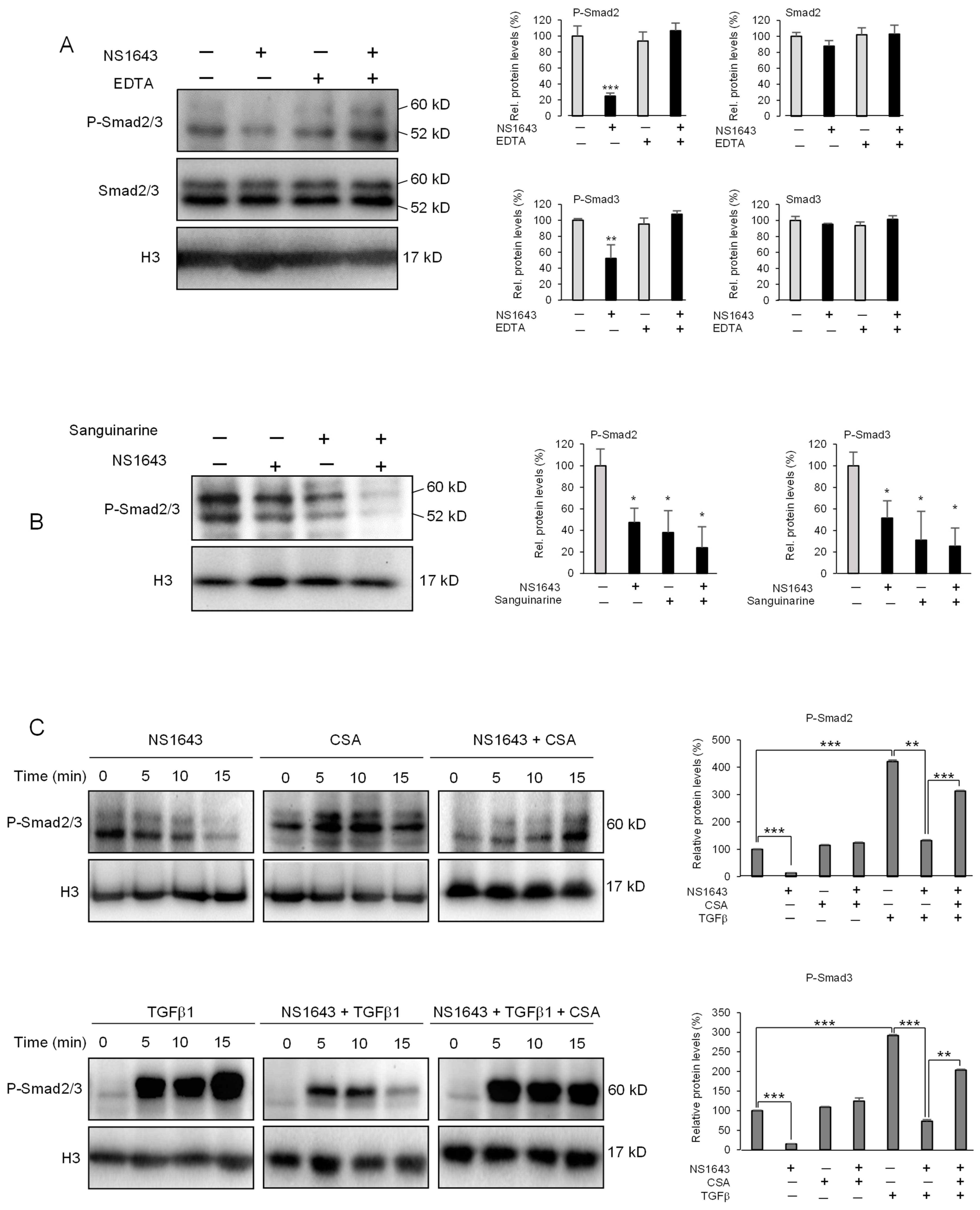
3.5. Kv11.1 Activation Inhibits TGFβ Signaling Via Calcineurin
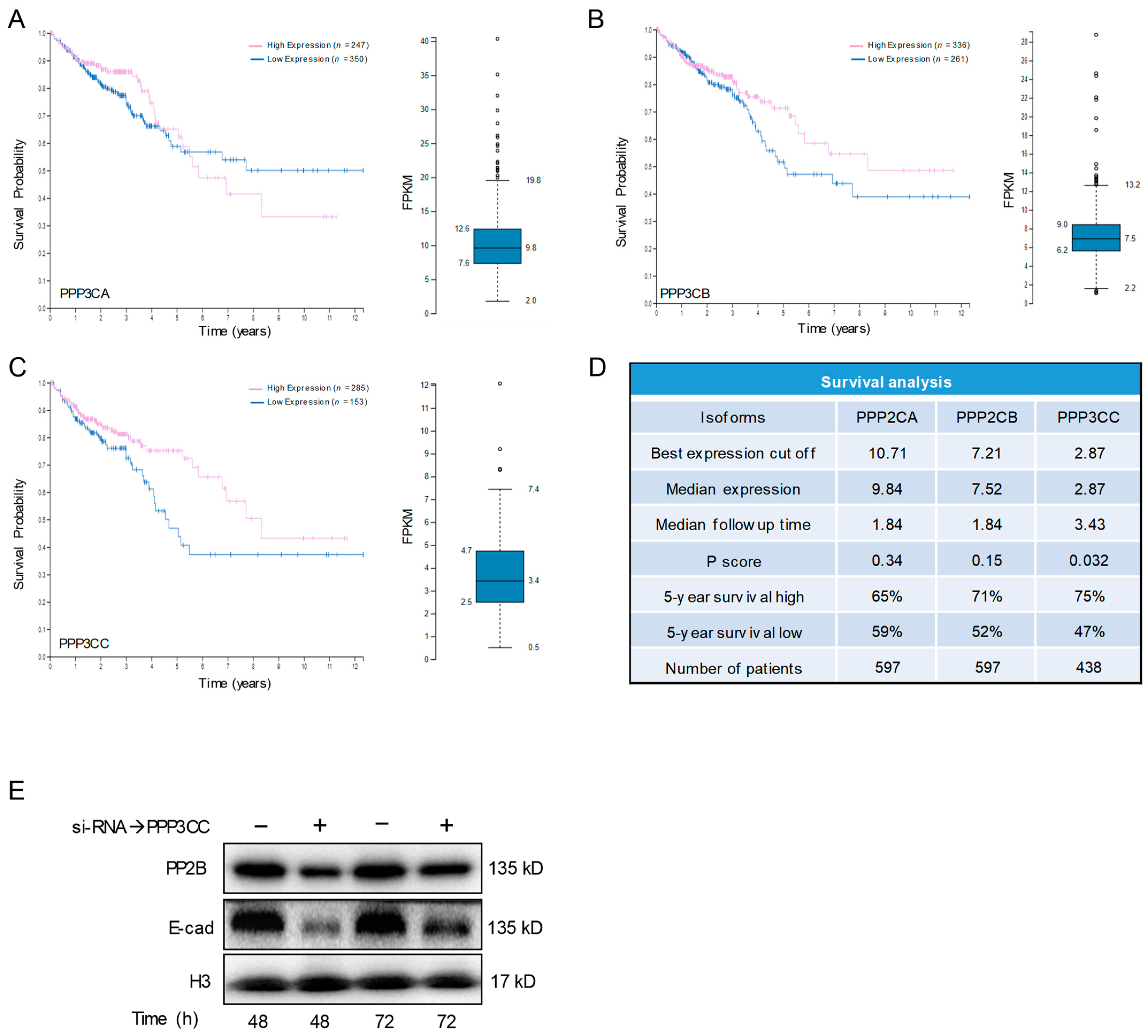
3.6. PP2B-γ Catalytic Isoform Mediates the Inhibitory Effect of Kv11.1 Activation on TGFβ Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Electrophysiology
Appendix A.2. RT-PCR Analysis and si-RNA
- Primers:
- FASN For: 5′-TACGACTACGGCCCTCATTT-3′
- FASN Rev: 5′-CCATGAAGCTCACCCAGTTATC-3′
- CDKNA1 For: 5′-GTCACTGTCTTGTACCCTTGTG-3′
- CDKNA1 Rev: 5′-GGCGTTTGGSGTGGTAGAAA-3′
- GADD45A For: 5′-AGGACGACGACAGAGATG-3′
- GADD45A Rev: 5′-CTGACGCGCAGGATGTT-3′
- SNAIL For 5′-TTCTCTAGGCCCTGGCTGCTACAA-3′
- SNAIL Rev 5′-TCTCTGACATCTGAGTGGGTCTGGA-3′;
- SLUG For 5′-CTGGTCAAGAAGCATTTCAACGCC-3′
- SLUG Rev 5′-AAAGAGGAGAGAGGCCATTGGGTA-3′;
- TWIST For 5′-GGAGTCCGCAGTCTTACGAG-3′
- TWIST Rev 5′-TCTGGAGGACCTGGTAGAGG-3′;
- ZEB1 For 5′-CTCCAGAATGTAATCGCATGTG-3′
- ZEB1 Rev 3′-TGAGAAAACCTTATAGAACAGTGTG-5′;
- Β-ACTIN For 5′-CTACGTCGCCCTGGACTTCGAGC-3′
- Β-ACTIN Rev 5′-GATGGAGCCGCCGATCCACACGG-3′.
- E-CAD For: 5′-AAGGTGACAGAGCCTCTGGAT-3′
- E-CAD Rev: 5′-CGTCTGTGGCTGTGACCT-3′
- N-CAD For: 5′-CGAGCCGCCTGCGCTGCCAC-3′
- N-CAD Rev: 5′-CGCTGCTCTCCGCTCCCCGC-3′
Appendix A.3. Wound-Healing Assay
Appendix A.4. Protein Extraction and Western Blotting
Appendix A.5. Flow Cytometry Analysis
Appendix A.6. Cell Spheroid Formation and Drug Treatment on Agarose-Microwells
Appendix A.7. Cell Apoptosis Assay
References
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Lastraioli, E.; Lottini, T.; Bencini, L.; Bernini, M.; Arcangeli, A. hERG1 Potassium Channels: Novel Biomarkers in Human Solid Cancers. Biomed. Res. Int. 2015, 2015, 896432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, V.R.; Perez-Neut, M.; Kaja, S.; Gentile, S. Voltage-gated ion channels in cancer cell proliferation. Cancers 2015, 7, 849–875. [Google Scholar] [CrossRef] [PubMed]
- Urrego, D.; Tomczak, A.P.; Zahed, F.; Stuhmer, W.; Pardo, L.A. Potassium channels in cell cycle and cell proliferation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, F.; Li, H.; Du, W.; Huang, S. Role of hERG1 K+ channels in leukemia cells as a positive regulator in SDF-1a-induced proliferation. Hematology 2011, 16, 177–184. [Google Scholar] [CrossRef]
- Beech, D.J.; Cheong, A. Potassium channels at the beginnings of cell proliferation. J. Physiol. 2006, 570, 1. [Google Scholar] [CrossRef] [Green Version]
- Gentile, S. hERG1 potassium channel in cancer cells: A tool to reprogram immortality. Eur. Biophys. J. 2016, 45, 649–655. [Google Scholar] [CrossRef]
- Wang, K.; Klionsky, D.J. Mitochondria removal by autophagy. Autophagy 2011, 7, 297–300. [Google Scholar] [CrossRef]
- Breuer, E.K.; Fukushiro-Lopes, D.; Dalheim, A.; Burnette, M.; Zartman, J.; Kaja, S.; Wells, C.; Campo, L.; Curtis, K.J.; Romero-Moreno, R.; et al. Potassium channel activity controls breast cancer metastasis by affecting beta-catenin signaling. Cell Death Dis. 2019, 10, 180. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Y.; Cao, L.; Han, H.; Wang, J.; Yang, B.; Nattel, S.; Wang, Z. HERG K+ channel, a regulator of tumor cell apoptosis and proliferation. Cancer Res. 2002, 62, 4843–4848. [Google Scholar]
- Kannankeril, P.; Roden, D.M.; Darbar, D. Drug-induced long QT syndrome. Pharmacol. Rev. 2010, 62, 760–781. [Google Scholar] [CrossRef] [Green Version]
- Fukushiro-Lopes, D.F.; Hegel, A.D.; Rao, V.; Wyatt, D.; Baker, A.; Breuer, E.K.; Osipo, C.; Zartman, J.J.; Burnette, M.; Kaja, S.; et al. Preclinical study of a Kv11.1 potassium channel activator as antineoplastic approach for breast cancer. Oncotarget 2018, 9, 3321–3337. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Li, W. Epithelial-mesenchymal transition in human cancer: Comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol. Ther. 2015, 150, 33–46. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investg. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 2019, 12, aav5183. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Vogelmann, R.; Nguyen-Tat, M.D.; Giehl, K.; Adler, G.; Wedlich, D.; Menke, A. TGFbeta-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. J. Cell Sci. 2005, 118, 4901–4912. [Google Scholar] [CrossRef] [Green Version]
- Bellomo, C.; Caja, L.; Moustakas, A. Transforming growth factor beta as regulator of cancer stemness and metastasis. Br. J. Cancer 2016, 115, 761–769. [Google Scholar] [CrossRef]
- Brautigan, D.L.; Shenolikar, S. Protein Serine/Threonine Phosphatases: Keys to Unlocking Regulators and Substrates. Annu. Rev. Biochem. 2018, 87, 921–964. [Google Scholar] [CrossRef] [Green Version]
- Low, H.B.; Zhang, Y. Regulatory Roles of MAPK Phosphatases in Cancer. Immune Netw. 2016, 16, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Stebbing, J.; Lit, L.C.; Zhang, H.; Darrington, R.S.; Melaiu, O.; Rudraraju, B.; Giamas, G. The regulatory roles of phosphatases in cancer. Oncogene 2014, 33, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Arcangeli, A.; Rosati, B.; Cherubini, A.; Crociani, O.; Fontana, L.; Ziller, C.; Wanke, E.; Olivotto, M. HERG- and IRK-like inward rectifier currents are sequentially expressed during neuronal development of neural crest cells and their derivatives. Eur. J. Neurosci. 1997, 9, 2596–2604. [Google Scholar] [CrossRef] [PubMed]
- Pohl, M.; Radacz, Y.; Pawlik, N.; Schoeneck, A.; Baldus, S.E.; Munding, J.; Schmiegel, W.; Schwarte-Waldhoff, I.; Reinacher-Schick, A. SMAD4 mediates mesenchymal-epithelial reversion in SW480 colon carcinoma cells. Anticancer Res. 2010, 30, 2603–2613. [Google Scholar]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 2340. [Google Scholar] [CrossRef] [Green Version]
- Aburai, N.; Yoshida, M.; Ohnishi, M.; Kimura, K. Sanguinarine as a potent and specific inhibitor of protein phosphatase 2C in vitro and induces apoptosis via phosphorylation of p38 in HL60 cells. Biosci. Biotechnol. Biochem. 2010, 74, 548–552. [Google Scholar] [CrossRef] [Green Version]
- Chi, H.; Zhang, X.; Chen, X.; Fang, S.; Ding, Q.; Gao, Z. Sanguinarine is an agonist of TRPA1 channel. Biochem. Biophys. Res. Commun. 2021, 534, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Rusnak, F.; Mertz, P. Calcineurin: Form and function. Physiol. Rev. 2000, 80, 1483–1521. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, aan2507. [Google Scholar] [CrossRef] [Green Version]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Kourtidis, A.; Lu, R.; Pence, L.J.; Anastasiadis, P.Z. A central role for cadherin signaling in cancer. Exp. Cell Res. 2017, 358, 78–85. [Google Scholar] [CrossRef]
- Wijnhoven, B.P.; Pignatelli, M. E-cadherin-catenin: More than a “sticky” molecular complex. Lancet 1999, 354, 356–357. [Google Scholar] [CrossRef]
- Montserrat, N.; Gallardo, A.; Escuin, D.; Catasus, L.; Prat, J.; Gutierrez-Avigno, F.J.; Peiro, G.; Barnadas, A.; Lerma, E. Repression of E-cadherin by SNAIL, ZEB1, and TWIST in invasive ductal carcinomas of the breast: A cooperative effort? Hum. Pathol. 2011, 42, 103–110. [Google Scholar] [CrossRef]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef] [PubMed]
- Pradella, D.; Naro, C.; Sette, C.; Ghigna, C. EMT and stemness: Flexible processes tuned by alternative splicing in development and cancer progression. Mol. Cancer 2017, 16, 8. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.N.; Peng, H.; Chen, B.S.; Wang, Y.J.; Wu, P.Y.; Lin, M.W. Potent activation of large-conductance Ca2+-activated K+ channels by the diphenylurea 1,3-bis-[2-hydroxy-5-(trifluoromethyl)phenyl]urea (NS1643) in pituitary tumor (GH3) cells. Mol. Pharmacol. 2008, 74, 1696–1704. [Google Scholar] [CrossRef] [Green Version]
- Maluccio, M.; Sharma, V.; Lagman, M.; Vyas, S.; Yang, H.; Li, B.; Suthanthiran, M. Tacrolimus enhances transforming growth factor-beta1 expression and promotes tumor progression. Transplantation 2003, 76, 597–602. [Google Scholar] [CrossRef]
- Hojo, M.; Morimoto, T.; Maluccio, M.; Asano, T.; Morimoto, K.; Lagman, M.; Shimbo, T.; Suthanthiran, M. Cyclosporine induces cancer progression by a cell-autonomous mechanism. Nature 1999, 397, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Lansu, K.; Gentile, S. Potassium channel activation inhibits proliferation of breast cancer cells by activating a senescence program. Cell Death Dis. 2013, 4, e652. [Google Scholar] [CrossRef]
- Perez-Neut, M.; Haar, L.; Rao, V.; Santha, S.; Lansu, K.; Rana, B.; Jones, W.K.; Gentile, S. Activation of hERG3 channel stimulates autophagy and promotes cellular senescence in melanoma. Oncotarget 2016, 7, 21991–22004. [Google Scholar] [CrossRef] [PubMed]
- Perez-Neut, M.; Rao, V.R.; Gentile, S. hERG1/Kv11.1 activation stimulates transcription of p21waf/cip in breast cancer cells via a calcineurin-dependent mechanism. Oncotarget 2016, 7, 58893–58902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Neut, M.; Shum, A.; Cuevas, B.D.; Miller, R.; Gentile, S. Stimulation of hERG1 channel activity promotes a calcium-dependent degradation of cyclin E2, but not cyclin E1, in breast cancer cells. Oncotarget 2015, 6, 1631–1639. [Google Scholar] [CrossRef] [Green Version]
- Crociani, O.; Zanieri, F.; Pillozzi, S.; Lastraioli, E.; Stefanini, M.; Fiore, A.; Fortunato, A.; D’Amico, M.; Masselli, M.; De Lorenzo, E.; et al. hERG1 channels modulate integrin signaling to trigger angiogenesis and tumor progression in colorectal cancer. Sci. Rep. 2013, 3, 3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiore, A.; Carraresi, L.; Morabito, A.; Polvani, S.; Fortunato, A.; Lastraioli, E.; Femia, A.P.; De Lorenzo, E.; Caderni, G.; Arcangeli, A. Characterization of hERG1 channel role in mouse colorectal carcinogenesis. Cancer Med. 2013, 2, 583–594. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eskandari, N.; Senyuk, V.; Moore, J.; Kalik, Z.; Luan, Q.; Papautsky, I.; Moshiri, A.; Bocchetta, M.; Salami, S.A.; Oryan, S.; et al. Molecular Activation of the Kv11.1 Channel Reprograms EMT in Colon Cancer by Inhibiting TGFβ Signaling via Activation of Calcineurin. Cancers 2021, 13, 6025. https://doi.org/10.3390/cancers13236025
Eskandari N, Senyuk V, Moore J, Kalik Z, Luan Q, Papautsky I, Moshiri A, Bocchetta M, Salami SA, Oryan S, et al. Molecular Activation of the Kv11.1 Channel Reprograms EMT in Colon Cancer by Inhibiting TGFβ Signaling via Activation of Calcineurin. Cancers. 2021; 13(23):6025. https://doi.org/10.3390/cancers13236025
Chicago/Turabian StyleEskandari, Najmeh, Vitalyi Senyuk, Jennifer Moore, Zane Kalik, Qiyue Luan, Ian Papautsky, Arfa Moshiri, Maurizio Bocchetta, Seyed Alireza Salami, Shahrbanoo Oryan, and et al. 2021. "Molecular Activation of the Kv11.1 Channel Reprograms EMT in Colon Cancer by Inhibiting TGFβ Signaling via Activation of Calcineurin" Cancers 13, no. 23: 6025. https://doi.org/10.3390/cancers13236025