
Early TP53 Alterations Shape Gastric and Esophageal Cancer Development
by
 , ,
, ,
Pranshu Sahgal
1,2,3 ,
,
Brandon M. Huffman
1 ,
,
Deepa T. Patil
4,
Walid K. Chatila
5,6,
Rona Yaeger
7,
James M. Cleary
1,3,8 and
Nilay S. Sethi
1,2,3,8,* 1
Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02215, USA
2
Cancer Program, Broad Institute of Massachusetts Institute of Technology (MIT) and Harvard University, Cambridge, MA 02142, USA
3
Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02215, USA
4
Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02215, USA
5
Tri-Institutional Program in Computational Biology and Medicine, Weill Cornell Medical College, New York, NY 10021, USA
6
Marie-Josée and Henry R. Kravis Center for Molecular Oncology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
7
Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
8
Gastrointestinal Cancer Center, Dana-Farber Cancer Institute, Boston, MA 02215, USA
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(23), 5915; https://doi.org/10.3390/cancers13235915
Submission received: 6 October 2021
/
Revised: 22 November 2021
/
Accepted: 23 November 2021
/
Published: 24 November 2021
(This article belongs to the Special Issue The Role of p53 Family in Cancer)
Abstract
:Simple Summary
Recent evidence establishes that gastric and esophageal (GE) adenocarcinomas are similar cancers at cellular, genomic, and epigenomic levels. Human GE adenocarcinomas develop TP53 mutations at early stages of malignant progression. This contrasts with other gastrointestinal adenocarcinomas, including sporadic colorectal and pancreatic adenocarcinomas, where TP53 alterations occur late. Exposure of the esophagus and stomach to environmental risk factors contributes to the selection of early TP53 mutations and subsequent chromosomal instability, which then lead to activation of mitogen and cell cycle pathways in GE adenocarcinomas by way of focal amplifications rather than mutations. While early TP53 mutations enable GE adenocarcinoma development, they also expose therapeutic vulnerabilities that should be prime for targeted therapy directed against the DNA damage response.
Abstract
Gastric and esophageal (GE) adenocarcinomas are the third and sixth most common causes of cancer-related mortality worldwide, accounting for greater than 1.25 million annual deaths. Despite the advancements in the multi-disciplinary treatment approaches, the prognosis for patients with GE adenocarcinomas remains poor, with a 5-year survival of 32% and 19%, respectively, mainly due to the late-stage diagnosis and aggressive nature of these cancers. Premalignant lesions characterized by atypical glandular proliferation, with neoplastic cells confined to the basement membrane, often precede malignant disease. We now appreciate that premalignant lesions also carry cancer-associated mutations, enabling disease progression in the right environmental context. A better understanding of the premalignant-to-malignant transition can help us diagnose, prevent, and treat GE adenocarcinoma. Here, we discuss the evidence suggesting that alterations in TP53 occur early in GE adenocarcinoma evolution, are selected for under environmental stressors, are responsible for shaping the genomic mechanisms for pathway dysregulation in cancer progression, and lead to potential vulnerabilities that can be exploited by a specific class of targeted therapy.
1. Gastric and Esophageal Adenocarcinomas Are Similar Cancers
Cellular, molecular, genetic, and epigenomic analyses of adenocarcinomas of the stomach and esophagus demonstrate that these cancers are highly related. Gastric and esophageal adenocarcinomas appear to arise from a shared tissue of origin. A mouse model of Barrett’s esophagus (BE), widely considered the precursor lesion to esophageal adenocarcinoma, showed that Lgr5+ gastric cardia stem cells are potentially the cells of origin [1]. Furthermore, esophageal adenocarcinomas display chromatin profiles that mirror those found in gastric tissue (H3K9me3, H3K4me1, H3K4me3, H3K9ac, H3K36me3) rather than in normal esophageal tissue [2]. Beyond the common ancestry of these cancers, deeper evaluation of human gastric and esophageal adenocarcinomas demonstrates shared molecular features. DNA methylation, mRNA and miRNA expression, and somatic copy-number alterations (SCNA) of esophageal adenocarcinomas are almost identical to gastric adenocarcinoma, and quite distinct from esophageal squamous cell carcinoma [3,4,5,6]. While a comprehensive molecular analysis of gastrointestinal adenocarcinomas revealed five molecular subtypes that largely transcended anatomic boundaries [7], the similarity in molecular composition of GE adenocarcinomas is largely due to the high proportion of the chromosomal instability (CIN) subtype, which accounts for more than half of gastric adenocarcinomas and almost 90% of esophageal adenocarcinomas. By appreciating the cellular and molecular resemblance between gastric and esophageal adenocarcinomas, not only can we generate better models for the premalignant and malignant states, but we can also think about treating patients with these cancers in similar ways.
2. TP53 Alteration Is an Early Event in GE Premalignancy
TP53 is the most frequently mutated gene across all cancers, especially advanced metastatic disease [8]. Among GE adenocarcinomas, the frequency of TP53 mutations is particularly high and enriched in the CIN subtype, which is characterized by increased aneuploidy within the tumor cells [7,9]. While the high rate of recurrent TP53 mutations is a common finding amongst all gastrointestinal adenocarcinomas, the timing of its occurrence in the malignant progression has significant, and biologically important, variation. Unlike other gastrointestinal adenocarcinomas, including sporadic colorectal and pancreatic adenocarcinomas, human GE adenocarcinomas develop TP53 mutations early in neoplasia, often found in premalignant lesions [10,11]. If not directly altered, other factors that regulate TP53 are also dysregulated, such as MDM2 and WWOX, the latter of which is a tumor suppressor found in a common chromosomal fragile site, and a regulator of the DNA damage response, whose protein expression is absent in up to 65% of gastric adenocarcinomas [12,13,14,15].
Clinically, a major challenge in the management of BE and gastric intestinal metaplasia, the respective premalignant lesions of GE cancers, is predicting when a precursor lesion will develop into adenocarcinoma. The frequency of progression from BE to esophageal adenocarcinoma is quite low [16]; the rates of gastric intestinal metaplasia progression to cancer are challenging to measure [17] but are thought to be infrequent. While TP53 mutations are known to be early events, genomic analysis of clinical samples of BE and gastric intestinal metaplasia have demonstrated that the frequency of TP53 mutations is actually low (~2%) [18]. Despite the low prevalence of TP53 mutations in premalignant lesions, their presence seems to preferentially influence progression to a malignant state. Using a paired-sampling approach, in which BE lesions and esophageal adenocarcinoma from the same patient were subjected to whole exome sequencing, it was shown that premalignant and malignant lesions from an individual patient shared a specific TP53 mutation more often than other tumor suppressors [19]. Another study that utilized whole genome sequencing of 23 paired human BE and esophageal adenocarcinoma samples showed that the degree of aneuploidy increases in the progression to cancer, suggesting that early inactivation of TP53 might be a critical step in allowing the development of aneuploidy [20]. Furthermore, a separate study demonstrated that 46% of BE lesions that progress to high grade dysplasia or esophageal adenocarcinoma harbor TP53 mutations, whereas BE lesions that did not progress, only had TP53 mutations 5% of the time [21]. These data suggest that the presence of TP53 mutations in premalignant GE lesions, even nondysplastic tissue, is a biomarker that predicts a high risk for malignant progression.
While colorectal and GE adenocarcinomas exhibit CIN, the characteristic pattern of aneuploidy observed in these cancers is notably different. GE adenocarcinomas display high intensity amplifications involving narrower genomic regions, whereas CRC tumors manifest broader, lower intensity amplifications [7]. For example, high-level focal amplifications of genes like MET, FGFR2, and HER2 are commonly seen in GE adenocarcinomas, whereas colorectal adenocarcinomas more typically exhibit low-level amplification of multiple genes neighboring each other on a chromosome. It can, therefore, be inferred that TP53 alterations may have occurred earlier in the evolution of GE cancers, enabling the development of high-level focal amplification in oncogenes that are critical to the development of the cancer. GE precursor lesions that develop high-level focal amplification of essential oncogenes have more proficient growth and are positively selected over precursor lesions that lack amplification of these oncogenes (Figure 1, Table 1). In other words, neoplastic cells with CIN that undergo stochastic disruption of critical genes within an amplicon die, whereas those that preserve oncogenes survive and thrive, propagating selection of malignant cells with narrower amplicons, broadcasting desired genes that support cancer progression. Later, we will discuss how known oncogenes in cancer-promoting molecular pathways are likely to be amplified in GE cancer rather than mutated in a gain-of-function fashion, which is typically the predominant mechanism of alteration for oncogenes in CRC (e.g., KRAS). Together, these data suggest that the more fragmented aneuploidy observed in GE cancers may be another indication that TP53 alteration is an early event in disease evolution.
3. Context Matters: Environmental Conditions Contribute to Selection of Early TP53 Alterations
Why are early TP53 mutations selected for in upper, relative to lower, GI adenocarcinomas? Is it because gastric and esophageal cells are more susceptible to TP53 mutations than colorectal cells, implicating the cell of origin as the culprit? Or rather, is it the environmental context of the upper, compared to the lower, GI tract that selects for TP53 mutations? An examination of sporadic versus colitis-associated cancer (CAC) argues that there is a substantial contribution from environmental context. CAC arises in the setting of inflammatory bowel disease (IBD): the colon and other parts of the GI tract are subject to relapsing bouts of inflammation. As opposed to sporadic colorectal cancer (CRC), which is typically initiated by somatic alterations in WNT pathway tumor suppressor APC, followed by alterations in KRAS and SMAD4, CAC demonstrates a distinct pattern of genomic alterations notable for early TP53 mutations [45,46,47], and a significantly lower frequency of APC mutations [48,49]. In fact, nondysplastic tissue from patients with IBD, arising from chronic inflammation of the gastrointestinal tract, often demonstrates TP53 alterations [50]. A recent study analyzed molecular alterations in low-grade dysplastic lesions arising within and outside segments of colon affected by ulcerative colitis, as well as sporadic adenomas from non-IBD patients, and showed that, while all three cohorts harbored mutations in APC and CTNNB1, TP53 mutations were only seen in lesions within areas of known colitis, albeit at low frequencies [51]. These data indicate that environmental context plays a critical role in the selection of genome alterations that lead to premalignant and eventually malignant disease.
Premalignant lesions that develop in the setting of IBD also display more CIN than sporadic adenomas [52]. Phylogenetic analysis of genomic changes in dysplasia and cancer from patients with colitis suggests that copy-number alterations begin to accrue in non-dysplastic bowel; the transition to low-grade/high grade dysplasia, however, often involves a punctuated increase in copy-number alterations [53]. Whole-genome sequencing of colonic crypts from patients with IBD and healthy controls revealed that the number of copy-number variants and retrotranspositions were associated with IBD duration, and that accumulation of structural variants in patients with IBD often exhibited an episodic nature, consistent with rapid accrual in the transition from mucosa to high-grade dysplastic lesions [54]. Furthermore, similar to GE adenocarcinomas in which a greater degree of focal CIN and gene amplifications drive cancer pathogenesis, CAC also displays more fragmented genomes, relative to sporadic CRC [55]. Chronic inflammation is a hallmark in the development of both GE adenocarcinoma and CAC, and strikingly, both cancers display early TP53 mutations, followed by high-level focal amplification of essential oncogenes.
The esophagus and the stomach are exposed to dietary contents that either directly imbue carcinogenic properties or indirectly lead to byproducts with carcinogenic attributes, such as nitrosamines, as well as bile reflux, which collectively contribute to the development of premalignant lesions [56,57,58]. Apart from dietary carcinogens, Helicobacter pylori infection is also a well-established risk factor for gastric cancer development [59]. Many human studies have demonstrated a relationship between Helicobacter-associated gastric neoplasia and the development of TP53 mutations [59,60,61,62,63,64,65]. We suspected that these exogenous exposures provide selective pressures for the emergence of mutant TP53 clones. To test this hypothesis, we recently developed an integrative mouse model that combines disease-relevant exposures, such as dietary carcinogens, with tissue-specific TP53 alterations to study the development of GE premalignancy [66]. In this mouse model, we found that inducible p53 inactivation in the stomach alone did not lead to premalignant lesions; by contrast, p53 inactivation combined with dietary carcinogens led to the expansion of p53 mutant clones and a greater burden of premalignant lesions. The mechanism underlying the cooperation of environmental risk factors and early genomic alterations in GE premalignancy deserves further investigation.
Multiple signaling pathways were upregulated in premalignant p53-deleted gastric lesions of the integrated mouse model that developed in the setting of dietary carcinogen exposure [66]. Among the dysregulated pathways, WNT signaling was notably activated, as indicated by gene expression profiling of derivative organoids, and immunohistochemical staining of downstream mediators in gastric premalignant lesions. These results may explain the relatively lower frequency of activating WNT pathway alterations in GE cancers, compared to sporadic CRC. Supporting this notion, a study involving human gastric cancer organoids demonstrated that co-alteration of TP53 and CDH1 promoted WNT signaling, independent of WNT agonist R-spondin [67]. The mechanism underlying WNT activation without frequent genomic alterations in GE adenocarcinomas requires further investigation. By contrast, WNT signaling appears to be downregulated in CAC; for example, a study looking at β-catenin staining demonstrated a greater proportion of tumors with low levels of nuclear beta-catenin in CAC compared to CRC [53]. Investigative human and mouse models that incorporate TP53 mutations and disease-relevant inflammation in the lower gastrointestinal tract may provide insight into the WNT-independent drivers of CAC.
4. Early TP53 Mutations Shape the Method of Genomic Alterations in Cancer-Promoting Pathways
As discussed above, early TP53 mutations enable the development of a type of chromosomal instability that yields a more fragmented, aneuploid cancer genome with characteristic focal amplifications in GE and CAC, compared to sporadic CRC. GE tumors with focal CIN are associated with genome doubling and poor prognosis [7,68]. Of note, these tumors also select for a distinct genomic pattern of pathway dysregulation, manifesting with high-level focal amplifications of unmutated genes involved in MAPK signaling (ERBB2, KRAS, VEGFA) and cell cycle regulation (CCNE1, CDK6) [4,69]. Similarly, CAC often displays amplifications in ERBB2, FGFR1/2, and MYC [48]. In contrast, sporadic tumors arising from the colon and rectum more typically alter these same pathways by oncogenic mutations. For example, when altered, KRAS is almost exclusively mutated in sporadic CRC, whereas an unmutated version is more commonly amplified, rather than mutated, in GE adenocarcinomas [4,20].
Tumors overexpressing wild-type KRAS appear to be more resistant to MAPK inhibition, based on preclinical evidence. One mechanism indicates that KRAS-amplified tumors augment signaling from upstream receptors [70] and, therefore, are prone to feedback reactivation in the setting of downstream MAPK pathway blockade, effectively attenuating the activity of MAPK inhibitors that target MEK and ERK. Another explanation for the observation that KRAS-amplified GE adenocarcinomas are resistant to conventional MAPK pathway blockade is due to a potent adaptive response involving MEK signaling [71]. This adaptive response to MAPK inhibitors is diminished by combining pharmacological targeting of both SHP2 and MEK. While the mechanism for the increased efficacy of the SHP2 and MEK inhibitor combination is not clear, it is possible that SHP2 blocks RTK feedback loops, which lead to resistance in MEK inhibitor monotherapy [71]. These preclinical data provide rationale for utilization of MEK inhibitor/SHP2 inhibitor combinations in KRAS-amplified GE adenocarcinomas, which are currently being tested in multiple clinical trials. Overall, these data indicate that the early onset of CIN in GE adenocarcinoma and CAC leads to a distinct genomic pattern of cancer pathway alterations by selectively amplifying MAPK and cell cycle genes, as opposed to mutating them, which may carry implications for an alternative treatment approach.
5. Therapeutic Vulnerabilities Imparted by Early TP53 Mutations
Among the molecular subtypes of GE adenocarcinomas, CIN is the largest and most heterogeneous, accounting for more than 50% of gastric cancers and almost all esophageal adenocarcinomas [3,9]. The shared genomic background of gastric and esophageal adenocarcinomas has led to the clinical recognition that these cancers should be considered similar, and hence, patients are often referred to as having “gastroesophageal adenocarcinoma”. Furthermore, the shared biology of gastric and esophageal adenocarcinoma has led to clinical trials being designed so that both populations are included, since the response rates to targeted and immunotherapies are often similar. CIN tumors harbor recurrent TP53 mutations, display marked aneuploidy, and often select for amplification in receptor tyrosine kinase pathways (e.g., EGFR and ERBB2). While these methods of pathway activation promote cancer progression, CIN may also yield therapeutic vulnerabilities [72]. Genomic instability from CIN leads to the accumulation of genomic aberrations, which promotes rapid cell division and hampers regulatory mechanisms designed to control the cell cycle. These events can generate significant pressure on the DNA replication process, with resultant stalled or collapsed DNA replication forks, which is termed replicative stress. In response to replicative stress, compensatory proteins in the DNA damage response (DDR) pathway, such as ATR and CHK1, become activated to try to stabilize the DNA replication forks. Activation of these checkpoints in response to DNA damage and replication stress may, therefore, generate new sensitivities in specific genomic contexts. p53-deficient cancer cells have exhibited a selective sensitivity to inhibition of CHK1 when treated with cytotoxic agents or gamma-radiation [73]. Upstream of CHK1, ATR inhibition imparted selective toxicity in ATM- and p53-deficient cancer cells [74].
In addition to p53 deficiency, genomic alterations of other genes involved in cell cycle regulation may further increase sensitivity to inhibitors of cell cycle checkpoints. For example, amplification of CCNE1 and MYC, as well as FBXW7 mutations, are known to increase replicative stress. Hence, these alterations may be biomarkers of increased sensitivity to therapeutic strategies utilizing ATR, CHK1, and WEE1 inhibitors. Ongoing preclinical research is aimed at developing additional biomarkers that could predict sensitivity to ATR, CHK1, and WEE1 inhibitors [9,66,75]. Although ovarian cancer harbors TP53 mutations in over 95% of cases, the CHK1/2 inhibitor prexasertib demonstrated a modest clinical response in a phase II clinical trial of BRCA wild-type ovarian cancers [76,77,78]. DDR inhibitors have yielded modest tumor responses in early phase clinical trials, except for a handful of cases that exhibit exquisite sensitivity, suggesting that predictive biomarkers will help identify patient populations that will benefit the most [79,80]. Other than the PARP inhibitor trials, there is a paucity of data available in GE adenocarcinoma regarding sensitivity to DDR inhibitors, despite their therapeutic potential; therefore, we need a deeper understanding of DDR pathway inhibitors in p53 mutant models of GE cancer.
The success of PARP inhibitors in targeting homologous recombination deficiency in other malignancies has generated interest in exploring whether PARP inhibitors could be effective in GE adenocarcinomas. While inactivating BRCA1/2 and PALB2 mutations are relatively rare, ATM loss occurs in up to 15–20% of gastric adenocarcinomas [81], although this may be an overestimation. In the GOLD randomized phase III clinical trial, treatment of advanced gastric cancer patients with the PARP inhibitor olaparib and paclitaxel did not meet its primary endpoint of improving survival in the overall study population nor in patients with ATM-deficient tumors in the second-line setting [82]. In this trial, ATM deficiency was defined immunohistochemically as observing ATM staining in less than 25% of tumor cell nuclei. While targeting ATM deficiency with PARP inhibition has, thus far, been unsuccessful, recently, a phase I trial of BAY-1895344 ATR inhibitor monotherapy showed encouraging efficacy in ATM-deficient tumors [83]. In this trial, an objective radiological response was observed in three solid tumor patients with ATM loss (defined as observing ATM staining in less than 1% of tumor cell nuclei) (Figure 2). Based on these results, better predictive and functional biomarkers of p53 mutant GE cancers that are sensitive to DDR pathway inhibitors are required for effective translation.
6. Conclusions
In this perspective article, we provided multiple lines of evidence that GE neoplasia harbor early TP53 mutations, and that these lesions have a distinct pattern of genomic evolution en route to malignancy. Further work is needed to identify additional high-risk early genomic events that lead to the development of GE adenocarcinomas, as this may enhance our ability to risk stratify patients with cancer precursor lesions, such as BE. Models of GE adenocarcinomas and CAC will benefit from the incorporation of early TP53 mutations and relevant environmental exposures to better capture complex features of cancer initiation and progression. The early onset and evolution of CIN in GE cancer enabled by initiating TP53 mutations promote amplification of unaltered genomic loci associated with MAPK and cell cycle pathways, providing an adaptive fitness advantage. Focal CIN also imparts potential therapeutic vulnerabilities, especially to DDR pathway inhibitors that are currently being tested in preclinical and clinical settings. We hope that through a better understanding of disease mechanisms, we will be able to define the population of patients with gastric and esophageal adenocarcinomas that will benefit from DDR pathway inhibitors.
Author Contributions
Conceptualization, N.S.S.; writing—original draft preparation, P.S. and N.S.S.; writing—review and editing, P.S., B.M.H., D.T.P., W.K.C., R.Y., J.M.C., and N.S.S.; visualization, P.S. and J.M.C.; supervision, N.S.S.; funding acquisition, N.S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Institutes of Health Cancer Center Core Grant P30 CA008748 to R.Y. and W.K.C. and American Gastroenterology Association Augustyn Award in Digestive Cancer and the Degregorio Family Foundation Award to N.S.S.
Acknowledgments
We thank Adam J. Bass for insightful discussions, many of which shaped the way we think about gastroesophageal cancer.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; DiPrete, B.; Betz, K.S.; et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polak, P.; Karlić, R.; Koren, A.; Thurman, R.; Sandstrom, R.; Lawrence, M.S.; Reynolds, A.; Rynes, E.; Vlahoviček, K.; Stamatoyannopoulos, J.A.; et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nat. Cell Biol. 2015, 518, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Analysis Working Group: Asan University; BC Cancer Agency; Brigham and Women’s Hospital; Broad Institute; Brown University; Case Western Reserve University; Dana-Farber Cancer Institute; Duke University; Greater Poland Cancer Centre; et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Dulak, A.M.; Schumacher, S.E.; van Lieshout, J.; Imamura, Y.; Fox, C.; Shim, B.; Ramos, A.H.; Saksena, G.; Baca, S.C.; Baselga, J.; et al. Gastrointestinal Adenocarcinomas of the Esophagus, Stomach, and Colon Exhibit Distinct Patterns of Genome Instability and Oncogenesis. Cancer Res. 2012, 72, 4383–4393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.W.; Karakasheva, T.A.; Lee, D.-J.; Lee, J.-S.; Long, Q.; Bass, A.J.; Wong, K.K.; Rustgi, A.K. Comparative transcriptomes of adenocarcinomas and squamous cell carcinomas reveal molecular similarities that span classical anatomic boundaries. PLoS Genet. 2017, 13, e1006938. [Google Scholar] [CrossRef]
- Sukawa, Y.; Yamamoto, H.; Nosho, K.; Kunimoto, H.; Suzuki, H.; Adachi, Y.; Nakazawa, M.; Nobuoka, T.; Kawayama, M.; Mikami, M.; et al. Alterations in the human epidermal growth factor receptor 2-phosphatidylinositol 3-kinase-v-Akt pathway in gastric cancer. World J. Gastroenterol. 2012, 18, 6577–6586. [Google Scholar] [CrossRef]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 33, 721–735.e728. [Google Scholar] [CrossRef] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nat. Cell Biol. 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Guo, H.; Lin, C.; Yang, L.; Wang, X. Enrichment and characterization of cancer stem-like cells from a cervical cancer cell line. Mol. Med. Rep. 2014, 9, 2117–2123. [Google Scholar] [CrossRef] [Green Version]
- Van De Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.-S.; Hsu, L.-J.; Lin, Y.-S.; Lai, F.-J.; Sheu, H.-M. WW domain-containing oxidoreductase: A candidate tumor suppressor. Trends Mol. Med. 2007, 13, 12–22. [Google Scholar] [CrossRef]
- Aqeilan, R.I.; Kuroki, T.; Pekarsky, Y.; Albagha, O.; Trapasso, F.; Baffa, R.; Huebner, K.; Edmonds, P.; Croce, C.M. Loss of WWOX Expression in Gastric Carcinoma. Clin. Cancer Res. 2004, 10, 3053–3058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Mare, S.; Husanie, H.; Iancu, O.; Abu-Odeh, M.; Evangelou, K.; Lovat, F.; Volinia, S.; Gordon, J.; Amir, G.; Stein, J.; et al. WWOX and p53 Dysregulation Synergize to Drive the Development of Osteosarcoma. Cancer Res. 2016, 76, 6107–6117. [Google Scholar] [CrossRef] [Green Version]
- Abdeen, S.K.; Aqeilan, R.I. Decoding the link between WWOX and p53 in aggressive breast cancer. Cell Cycle 2019, 18, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Rustgi, A.K.; El-Serag, H.B. Esophageal Carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef]
- Giroux, V.; Rustgi, A.K. Metaplasia: Tissue injury adaptation and a precursor to the dysplasia–cancer sequence. Nat. Rev. Cancer 2017, 17, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.K.; Ramnarayanan, K.; Zhu, F.; Srivastava, S.; Xu, C.; Tan, A.L.K.; Lee, M.; Tay, S.; Das, K.; Xing, M.; et al. Genomic and Epigenomic Profiling of High-Risk Intestinal Metaplasia Reveals Molecular Determinants of Progression to Gastric Cancer. Cancer Cell 2018, 33, 137–150.e135. [Google Scholar] [CrossRef] [Green Version]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef] [Green Version]
- Ross-Innes, C.S.; Becq, J.; Warren, A.; Cheetham, R.K.; Northen, H.; O’Donovan, M.; Malhotra, S.; Di Pietro, M.; Ivakhno, S.; He, M.; et al. Whole-genome sequencing provides new insights into the clonal architecture of Barrett’s esophagus and esophageal adenocarcinoma. Nat. Genet. 2015, 47, 1038–1046. [Google Scholar] [CrossRef]
- Stachler, M.D.; Camarda, N.D.; Deitrick, C.; Kim, A.; Agoston, A.T.; Odze, R.D.; Hornick, J.L.; Nag, A.; Thorner, A.R.; Ducar, M.; et al. Detection of Mutations in Barrett’s Esophagus Before Progression to High-Grade Dysplasia or Adenocarcinoma. Gastroenterology 2018, 155, 156–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Shitara, K.; Bang, Y.-J.; Iwasa, S.; Sugimoto, N.; Ryu, M.-H.; Sakai, D.; Chung, H.-C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N. Engl. J. Med. 2020, 382, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.; Bang, Y.-J.; Qin, S.K.; Chung, H.C.; Xu, J.M.; Park, J.O.; Jeziorski, K.; Shparyk, Y.; Hoff, P.M.; Sobrero, A.; et al. Lapatinib in Combination With Capecitabine Plus Oxaliplatin in Human Epidermal Growth Factor Receptor 2–Positive Advanced or Metastatic Gastric, Esophageal, or Gastroesophageal Adenocarcinoma: TRIO-013/LOGiC—A Randomized Phase III Trial. J. Clin. Oncol. 2016, 34, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Suenaga, M.; Hatake, K.; Takahashi, S.; Yokoyama, M.; Onozawa, Y.; Yamazaki, K.; Hironaka, S.; Hashigami, K.; Hasegawa, H.; et al. Safety, Efficacy and Pharmacokinetics of Neratinib (HKI-272) in Japanese Patients with Advanced Solid Tumors: A Phase 1 Dose-escalation Study. Jpn. J. Clin. Oncol. 2012, 42, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.-C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.-Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Kim, S.T.; Lee, J.; Lee, S.J.; Park, S.H.; Jung, S.-H.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Park, J.O. Prospective phase II trial of pazopanib plus CapeOX (capecitabine and oxaliplatin) in previously untreated patients with advanced gastric cancer. Oncotarget 2016, 7, 24088–24096. [Google Scholar] [CrossRef]
- Moehler, M.; Gepfner-Tuma, I.; Maderer, A.; Thuss-Patience, P.C.; Ruessel, J.; Hegewisch-Becker, S.; Wilke, H.; Al-Batran, S.-E.; Rafiyan, M.-R.; Weißinger, F.; et al. Sunitinib added to FOLFIRI versus FOLFIRI in patients with chemorefractory advanced adenocarcinoma of the stomach or lower esophagus: A randomized, placebo-controlled phase II AIO trial with serum biomarker program. BMC Cancer 2016, 16, 699. [Google Scholar] [CrossRef] [Green Version]
- Ohtsu, A.; Shah, M.A.; Van Cutsem, E.; Rha, S.Y.; Sawaki, A.; Park, S.R.; Lim, H.Y.; Yamada, Y.; Wu, J.; Langer, B.; et al. Bevacizumab in Combination With Chemotherapy As First-Line Therapy in Advanced Gastric Cancer: A Randomized, Double-Blind, Placebo-Controlled Phase III Study. J. Clin. Oncol. 2011, 29, 3968–3976. [Google Scholar] [CrossRef]
- Sun, W.; Powell, M.; O’Dwyer, P.J.; Catalano, P.; Ansari, R.H.; Benson, A.B., III. Phase II Study of Sorafenib in Combination With Docetaxel and Cisplatin in the Treatment of Metastatic or Advanced Gastric and Gastroesophageal Junction Adenocarcinoma: ECOG. J. Clin. Oncol. 2010, 28, 2947–2951. [Google Scholar] [CrossRef]
- Fukuoka, S.; Hara, H.; Takahashi, N.; Kojima, T.; Kawazoe, A.; Asayama, M.; Yoshii, T.; Kotani, D.; Tamura, H.; Mikamoto, Y.; et al. Regorafenib Plus Nivolumab in Patients With Advanced Gastric or Colorectal Cancer: An Open-Label, Dose-Escalation, and Dose-Expansion Phase Ib Trial (REGONIVO, EPOC1603). J. Clin. Oncol. 2020, 38, 2053–2061. [Google Scholar] [CrossRef] [PubMed]
- Pavlakis, N.; Sjoquist, K.M.; Martin, A.J.; Tsobanis, E.; Yip, S.; Kang, Y.-K.; Bang, Y.-J.; Alcindor, T.; O’Callaghan, C.J.; Burnell, M.J.; et al. Regorafenib for the Treatment of Advanced Gastric Cancer (INTEGRATE): A Multinational Placebo-Controlled Phase II Trial. J. Clin. Oncol. 2016, 34, 2728–2735. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.-Y.; Doi, T.; Shirao, K.; Lee, K.-W.; Park, S.R.; Chen, Y.; Yang, L.; Valota, O.; Bang, Y.-J. Phase I Study of Axitinib in Combination with Cisplatin and Capecitabine in Patients with Previously Untreated Advanced Gastric Cancer. Cancer Res. Treat. 2015, 47, 687–696. [Google Scholar] [CrossRef]
- Kawazoe, A.; Fukuoka, S.; Nakamura, Y.; Kuboki, Y.; Wakabayashi, M.; Nomura, S.; Mikamoto, Y.; Shima, H.; Fujishiro, N.; Higuchi, T.; et al. Lenvatinib plus pembrolizumab in patients with advanced gastric cancer in the first-line or second-line setting (EPOC1706): An open-label, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1057–1065. [Google Scholar] [CrossRef]
- Karasic, T.B.; O’Hara, M.H.; Teitelbaum, U.R.; Damjanov, N.; Giantonio, B.J.; D’Entremont, T.S.; Gallagher, M.; Zhang, P.J.; O’Dwyer, P.J. Phase II Trial of Palbociclib in Patients with Advanced Esophageal or Gastric Cancer. Oncologist 2020, 25, e1864–e1868. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.A.; Blaszkowsky, L.S.; Enzinger, P.C.; Ryan, D.P.; Abrams, T.A.; Zhu, A.X.; Temel, J.S.; Schrag, D.; Bhargava, P.; Meyerhardt, J.A.; et al. A multicenter phase II trial of single-agent cetuximab in advanced esophageal and gastric adenocarcinoma. Ann. Oncol. 2011, 22, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Pinto, C.; Di Fabio, F.; Siena, S.; Cascinu, S.; Llimpe, F.R.; Ceccarelli, C.; Mutri, V.; Giannetta, L.; Giaquinta, S.; Funaioli, C.; et al. Phase II study of cetuximab in combination with FOLFIRI in patients with untreated advanced gastric or gastroesophageal junction adenocarcinoma (FOLCETUX study). Ann. Oncol. 2007, 18, 510–517. [Google Scholar] [CrossRef]
- Waddell, T.; Chau, I.; Cunningham, D.; Gonzalez, D.; Okines, A.F.C.; Wotherspoon, A.; Saffery, C.; Middleton, G.; Wadsley, J.; Ferry, D.; et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): A randomised, open-label phase 3 trial. Lancet Oncol. 2013, 14, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Dragovich, T.; McCoy, S.; Fenoglio-Preiser, C.M.; Wang, J.; Benedetti, J.K.; Baker, A.F.; Hackett, C.B.; Urba, S.G.; Zaner, K.S.; Blanke, C.D.; et al. Phase II Trial of Erlotinib in Gastroesophageal Junction and Gastric Adenocarcinomas: SWOG. J. Clin. Oncol. 2006, 24, 4922–4927. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Rasco, D.; Lee, J.; Rha, S.Y.; Lee, K.-W.; Bang, Y.J.; Bendell, J.; Enzinger, P.; Marina, N.; Xiang, H.; et al. Phase I Escalation and Expansion Study of Bemarituzumab (FPA144) in Patients With Advanced Solid Tumors and FGFR2b-Selected Gastroesophageal Adenocarcinoma. J. Clin. Oncol. 2020, 38, 2418–2426. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Enzinger, P.C.; Kang, Y.-K.; Yamaguchi, K.; Qin, S.; Lee, K.-W.; Oh, S.C.; Li, J.; Turk, H.M.; Teixeira, A.C.; et al. Randomized double-blind placebo-controlled phase 2 study of bemarituzumab combined with modified FOLFOX6 (mFOLFOX6) in first-line (1L) treatment of advanced gastric/gastroesophageal junction adenocarcinoma (FIGHT). J. Clin. Oncol. 2021, 39, 160. [Google Scholar] [CrossRef]
- Lennerz, J.K.; Kwak, E.L.; Ackerman, A.; Michael, M.; Fox, S.B.; Bergethon, K.; Lauwers, G.Y.; Christensen, J.G.; Wilner, K.D.; Haber, D.A.; et al. MET Amplification Identifies a Small and Aggressive Subgroup of Esophagogastric Adenocarcinoma With Evidence of Responsiveness to Crizotinib. J. Clin. Oncol. 2011, 29, 4803–4810. [Google Scholar] [CrossRef] [Green Version]
- Bang, Y.J.; Su, W.C.; Schuler, M.; Nam, D.H.; Lim, W.T.; Bauer, T.M.; Azaro, A.; Poon, R.T.P.; Hong, D.; Lin, C.C.; et al. Phase 1 study of capmatinib in MET-positive solid tumor patients: Dose escalation and expansion of selected cohorts. Cancer Sci. 2020, 111, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falchook, G.S.; Kurzrock, R.; Amin, H.M.; Xiong, W.; Fu, S.; Piha-Paul, S.A.; Janku, F.; Eskandari, G.; Catenacci, D.V.T.; Klevesath, M.B.; et al. First-in-Man Phase I Trial of the Selective MET Inhibitor Tepotinib in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2019, 26, 1237–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentnall, T.A.; Crispin, D.A.; Rabinovitch, P.S.; Haggitt, R.C.; Rubin, C.E.; Stevens, A.C.; Burmer, G.C. Mutations in the p53 gene: An early marker of neoplastic progression in ulcerative colitis. Gastroenterology 1994, 107, 369–378. [Google Scholar] [CrossRef]
- Rabinovitch, P.S.; Dziadon, S.; Brentnall, T.A.; Emond, M.J.; Crispin, D.A.; Haggitt, R.C.; Bronner, M.P. Pancolonic chro-mosomal instability precedes dysplasia and cancer in ulcerative colitis. Cancer Res. 1999, 59, 5148–5153. [Google Scholar] [PubMed]
- Yin, J.; Harpaz, N.; Tong, Y.; Huang, Y.; Laurin, J.; Greenwald, B.D.; Hontanosas, M.; Newkirk, C.; Meltzer, S.J. p53 Point mutations in dysplastic and cancerous ulcerative colitis lesions. Gastroenterology 1993, 104, 1633–1639. [Google Scholar] [CrossRef]
- Yaeger, R.; Shah, M.A.; Miller, V.A.; Kelsen, J.R.; Wang, K.; Heins, Z.J.; Ross, J.S.; He, Y.; Sanford, E.; Yantiss, R.K.; et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct From Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology 2016, 151, 278–287.e276. [Google Scholar] [CrossRef] [Green Version]
- Robles, A.I.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease−Associated Colorectal Cancers. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef] [Green Version]
- Saraggi, D.; Fassan, M.; Mescoli, C.; Scarpa, M.; Valeri, N.; Michielan, A.; D’Incá, R.; Rugge, M. The molecular landscape of colitis-associated carcinogenesis. Dig. Liver Dis. 2017, 49, 326–330. [Google Scholar] [CrossRef]
- Christakis, J.N.A.; Odze, R.; Hamilton, M.; Parrack, P.; Goldblum, J.; Pankaj, A.; Deshpande, V.; Lindeman, N.; Patil, D. Abstracts from USCAP 2021: Gastrointestinal Pathology (311–404). Mod. Pathol. 2021, 34, 397. [Google Scholar] [CrossRef]
- Wanders, L.K.; Cordes, M.; Voorham, Q.; Sie, D.; De Vries, S.D.; D’Haens, G.; de Boer, N.K.H.; Ylstra, B.; Van Grieken, N.C.T.; Meijer, G.A.; et al. IBD-Associated Dysplastic Lesions Show More Chromosomal Instability Than Sporadic Adenomas. Inflamm. Bowel Dis. 2020, 26, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.-M.; Cross, W.; Curtius, K.; Al Bakir, I.; Choi, C.-H.R.; Davis, H.L.; Temko, D.; Biswas, S.; Martinez, P.; Williams, M.J.; et al. Evolutionary history of human colitis-associated colorectal cancer. Gut 2019, 68, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Olafsson, S.; McIntyre, R.E.; Coorens, T.; Butler, T.; Jung, H.; Robinson, P.S.; Lee-Six, H.; Sanders, M.A.; Arestang, K.; Dawson, C.; et al. Somatic Evolution in Non-neoplastic IBD-Affected Colon. Cell 2020, 182, 672–684.e611. [Google Scholar] [CrossRef]
- Chatila, W.K.; (Weill Cornell Medical College, New York, NY, USA); Yaeger, R.; (Memorial Sloan Kettering, New York, NY, USA). Personal communication, 2021.
- Chen, S.-L.; Mo, J.-Z.; Cao, Z.-J.; Chen, X.-Y.; Xiao, S.-D. Effects of bile reflux on gastric mucosal lesions in patients with dyspepsia or chronic gastritis. World J. Gastroenterol. 2005, 11, 2834–2837. [Google Scholar] [CrossRef]
- Sobala, G.M.; O’Connor, H.J.; Dewar, E.P.; King, R.F.; Axon, A.T.; Dixon, M.F. Bile reflux and intestinal metaplasia in gastric mucosa. J. Clin. Pathol. 1993, 46, 235–240. [Google Scholar] [CrossRef]
- Stefaniwsky, A.B.; Tint, G.S.; Speck, J.; Shefer, S.; Salen, G. Ursodeoxycholic acid treatment of bile reflux gastritis. Gastroenterology 1985, 89, 1000–1004. [Google Scholar] [CrossRef]
- Shimizu, T.; Marusawa, H.; Matsumoto, Y.; Inuzuka, T.; Ikeda, A.; Fujii, Y.; Minamiguchi, S.; Miyamoto, S.; Kou, T.; Sakai, Y.; et al. Accumulation of Somatic Mutations in TP53 in Gastric Epithelium With Helicobacter pylori Infection. Gastroenterology 2014, 147, 407–417.e403. [Google Scholar] [CrossRef]
- Li, J.-H.; Shi, X.-Z.; Lv, S.; Liu, M.; Xu, G.-W. Effect ofHelicobacter pyloriinfection onp53expression of gastric mucosa and adenocarcinoma with microsatellite instability. World J. Gastroenterol. 2005, 11, 4363–4366. [Google Scholar] [CrossRef]
- Petersson, F.; Franzén, L.E.; Borch, K. Characterization of the Gastric Cardia in Volunteers from the General Population. Type of mucosa, Helicobacter pylori infection, inflammation, mucosal proliferative activity, p53 and p21 expression, and relations to gastritis. Dig. Dis. Sci. 2010, 55, 46–53. [Google Scholar] [CrossRef]
- Salih, B.A.; Gucin, Z.; Bayyurt, N. A study on the effect of Helicobacter pylori infection on p53 expression in gastric cancer and gastritis tissues. J. Infect. Dev. Ctries. 2013, 7, 651–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, K.; Kihira, K.; Kawata, H.; Tokumaru, K.; Kumakura, Y.; Ishino, Y.; Kawakami, S.; Inoue, K.; Kojima, T.; Satoh, Y.; et al. p53 Expression in the Gastric Mucosa Before and After Eradication of Helicobacter pylori. Helicobacter 2001, 6, 31–36. [Google Scholar] [CrossRef]
- Teh, M.; Tan, K.B.; Seet, B.L.; Yeoh, K.G. Study of p53 immunostaining in the gastric epithelium of cagA-positive and cagA-negativeHelicobacter pylori gastritis. Cancer 2002, 95, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; El Zaatari, M.; Merchant, J.L. Recapitulating Human Gastric Cancer Pathogenesis: Experimental Models of Gastric Cancer. Adv. Exp. Med. Biol. 2016, 908, 441–478. [Google Scholar] [CrossRef] [Green Version]
- Sethi, N.S.; Kikuchi, O.; Duronio, G.N.; Stachler, M.D.; McFarland, J.M.; Ferrer-Luna, R.; Zhang, Y.; Bao, C.; Bronson, R.; Patil, D.; et al. Early TP53 alterations engage environmental exposures to promote gastric premalignancy in an integrative mouse model. Nat. Genet. 2020, 52, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Nanki, K.; Toshimitsu, K.; Takano, A.; Fujii, M.; Shimokawa, M.; Ohta, Y.; Matano, M.; Seino, T.; Nishikori, S.; Ishikawa, K.; et al. Divergent Routes toward Wnt and R-spondin Niche Independency during Human Gastric Carcinogenesis. Cell 2018, 174, 856–869.e817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, A.; Bandla, S.; Reveiller, M.; Toia, L.; Zhou, Z.; Gooding, W.E.; Kalatskaya, I.; Stein, L.; D’Souza, M.; Litle, V.R.; et al. Early G1 Cyclin-Dependent Kinases as Prognostic Markers and Potential Therapeutic Targets in Esophageal Adenocarcinoma. Clin. Cancer Res. 2011, 17, 4513–4522. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, R.; Yao, Z.; Hyman, D.M.; Hechtman, J.F.; Vakiani, E.; Zhao, H.; Su, W.; Wang, L.; Joelson, A.; Cercek, A.; et al. Mechanisms of Acquired Resistance to BRAF V600E Inhibition in Colon Cancers Converge on RAF Dimerization and Are Sensitive to Its Inhibition. Cancer Res. 2017, 77, 6513–6523. [Google Scholar] [CrossRef] [Green Version]
- Wong, G.S.; Zhou, J.; Bin Liu, J.; Wu, Z.; Xu, X.; Li, T.; Xu, D.; Schumacher, S.E.; Puschhof, J.; McFarland, J.; et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 2018, 24, 968–977. [Google Scholar] [CrossRef]
- McClelland, S.E. Role of chromosomal instability in cancer progression. Endocr.-Relat. Cancer 2017, 24, T23–T31. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Xiao, Z.; Gu, W.-Z.; Xue, J.; Bui, M.H.; Kovar, P.; Li, G.; Wang, G.; Tao, Z.-F.; Tong, Y.; et al. Selective Chk1 inhibitors differentially sensitize p53-deficient cancer cells to cancer therapeutics. Int. J. Cancer 2006, 119, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Deng, N.; Goh, L.K.; Wang, H.; Das, K.; Tao, J.; Tan, I.B.; Zhang, S.; Lee, M.; Wu, J.; Lim, K.H.; et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 2012, 61, 673–684. [Google Scholar] [CrossRef]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.-B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Färkkilä, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Cleary, J.M.; Aguirre, A.J.; Shapiro, G.I.; D’Andrea, A.D. Biomarker-Guided Development of DNA Repair Inhibitors. Mol. Cell 2020, 78, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.-J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cheng, S.-C.; Hendrickson, A.E.W.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Hendrickson, A.E.W.; et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, M.A.; Hodgson, D.; Harbron, C.; Wellings, R.; O’Connor, M.J.; Womack, C.; Yin, X.; Bang, Y.-J.; Im, S.-A.; et al. Concordance of ATM (Ataxia Telangiectasia Mutated) Immunohistochemistry between Biopsy or Metastatic Tumor Samples and Primary Tumors in Gastric Cancer Patients. Pathobiology 2013, 80, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Xu, R.-H.; Chin, K.; Lee, K.-W.; Park, S.H.; Rha, S.Y.; Shen, L.; Qin, S.; Xu, N.; Im, S.-A.; et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1637–1651. [Google Scholar] [CrossRef]
- Yap, T.A.; Tan, D.S.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef] [PubMed]
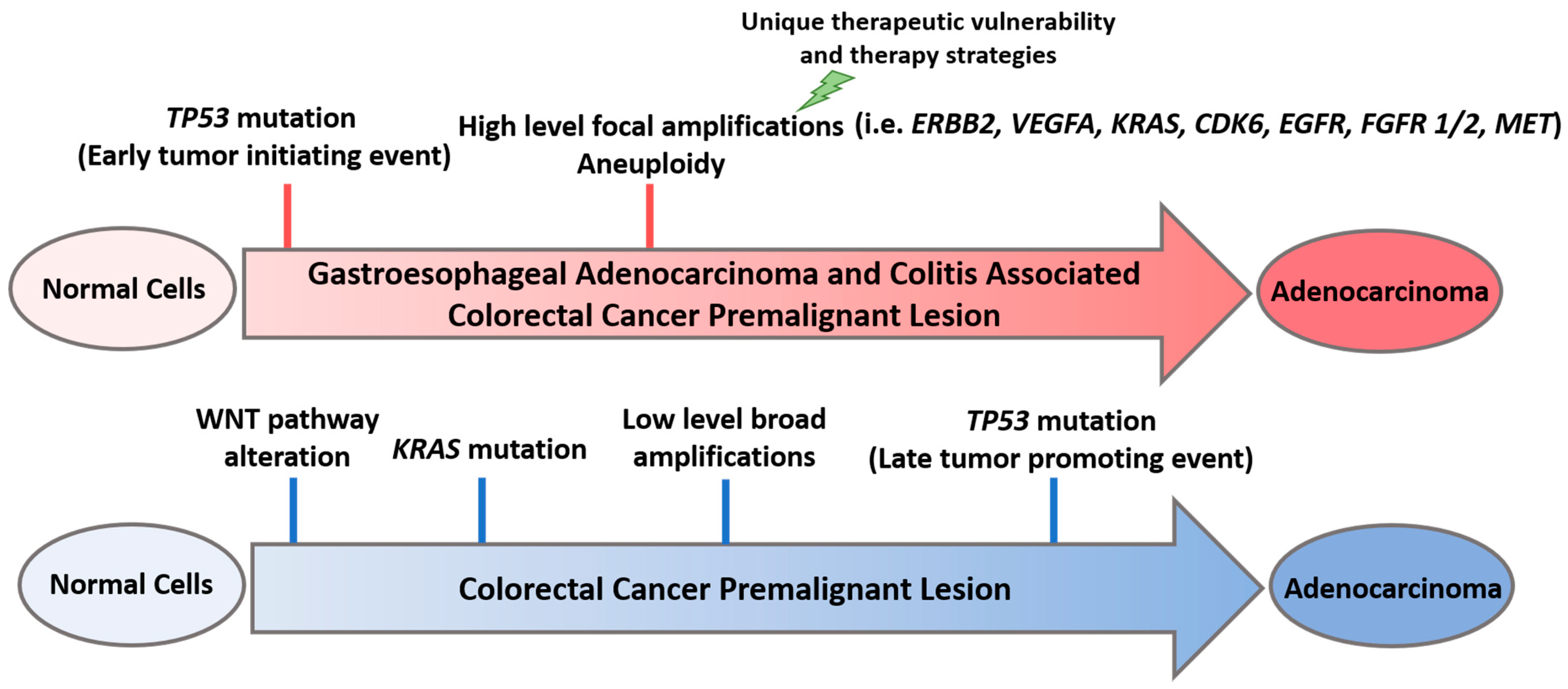
Figure 1.
Early TP53 mutations and high-level focal amplifications in gastroesophageal (GE) adenocarcinoma and colitis-associated colorectal cancer (CAC) premalignant lesion, as opposed to late TP53 mutations and low-level broad amplifications and mutations in colorectal cancer (CRC) premalignant lesion. ERBB2: Erb-B2 receptor tyrosine kinase 2; VEGFA: vascular endothelial growth factor A; KRAS: Kirsten rat sarcoma; CDK6: cyclin dependent kinase 6; EGFR: epidermal growth factor receptor; FGFR1/2: fibroblast growth factor receptor 1/2; MET: mesenchymal epithelial transition factor.
Figure 1.
Early TP53 mutations and high-level focal amplifications in gastroesophageal (GE) adenocarcinoma and colitis-associated colorectal cancer (CAC) premalignant lesion, as opposed to late TP53 mutations and low-level broad amplifications and mutations in colorectal cancer (CRC) premalignant lesion. ERBB2: Erb-B2 receptor tyrosine kinase 2; VEGFA: vascular endothelial growth factor A; KRAS: Kirsten rat sarcoma; CDK6: cyclin dependent kinase 6; EGFR: epidermal growth factor receptor; FGFR1/2: fibroblast growth factor receptor 1/2; MET: mesenchymal epithelial transition factor.

Figure 2.
DNA damage response (DDR) and putative inhibitors of potential clinical importance. Single lightning bolt indicates damage to one strand of DNA. Double lightning bolt indicates damage to both strands of DNA. Upon DNA damage, the pathway is activated, leading to either G1/S phase checkpoint blockade or S phase prolongation and prevention of growth/mitosis. Repair of the DNA strand breaks lead to p53 activation, but in those cells with TP53 alterations, checkpoints are bypassed. Targeting the DDR pathway upstream of p53 will theoretically lead to cell death due to mitotic catastrophe. dsDNA: double stranded DNA; ssDNA: single-stranded DNA; G1/S: restriction checkpoint before DNA synthesis phase in cell cycle; S phase: synthesis phase; G2/M: growth phase checkpoint leading to mitotic phase. * Given the molecular similarities of CHK1 and CHK2, CHK1 inhibitors also have varying degrees of activity against CHK2. dsDNA: double stranded DNA; ssDNA: single-stranded DNA; G1/S: restriction checkpoint before DNA synthesis phase in cell cycle; S phase: synthesis phase; G2/M: growth phase checkpoint leading to mitotic phase.
Figure 2.
DNA damage response (DDR) and putative inhibitors of potential clinical importance. Single lightning bolt indicates damage to one strand of DNA. Double lightning bolt indicates damage to both strands of DNA. Upon DNA damage, the pathway is activated, leading to either G1/S phase checkpoint blockade or S phase prolongation and prevention of growth/mitosis. Repair of the DNA strand breaks lead to p53 activation, but in those cells with TP53 alterations, checkpoints are bypassed. Targeting the DDR pathway upstream of p53 will theoretically lead to cell death due to mitotic catastrophe. dsDNA: double stranded DNA; ssDNA: single-stranded DNA; G1/S: restriction checkpoint before DNA synthesis phase in cell cycle; S phase: synthesis phase; G2/M: growth phase checkpoint leading to mitotic phase. * Given the molecular similarities of CHK1 and CHK2, CHK1 inhibitors also have varying degrees of activity against CHK2. dsDNA: double stranded DNA; ssDNA: single-stranded DNA; G1/S: restriction checkpoint before DNA synthesis phase in cell cycle; S phase: synthesis phase; G2/M: growth phase checkpoint leading to mitotic phase.


{kind=link}
{kind=link}
Table 1.
Potential therapeutic vulnerabilities associated with chromosomal instability and high-level focal amplifications in gastroesophageal (GE) adenocarcinoma. Food and Drug Administration-approved drugs for gastroesophageal cancer are indicated by *. Aside from trastuzumab deruxtecan and bemarituzumab, most agents listed in this table have, thus far, not shown single-agent efficacy clinically, suggesting that combinatorial approaches may be needed [3,9].
Table 1.
Potential therapeutic vulnerabilities associated with chromosomal instability and high-level focal amplifications in gastroesophageal (GE) adenocarcinoma. Food and Drug Administration-approved drugs for gastroesophageal cancer are indicated by *. Aside from trastuzumab deruxtecan and bemarituzumab, most agents listed in this table have, thus far, not shown single-agent efficacy clinically, suggesting that combinatorial approaches may be needed [3,9].
| High Level Focal Amplification | Prevalence in GE Adenocarcinoma | Potential Therapeutic Agent | Trial References |
|---|---|---|---|
| ERBB2 | 24–32% | trastuzumab 3,*, lapatinib 3, neratinib 1, tucatinib 0, trastuzumab deruxtecan 2,* | [22,23,24,25] |
| VEGFA | 28% | VEGF inhibitors (ramucirumab 3,*, Lenvatinib 2) | [26,27,28,29,30,31,32,33,34] |
| KRAS | 13–17% | MEK inhibitors (binimetinib 0, cobimetinib 0), ERK inhibitors 0, RAF inhibitors 0 | - |
| CDK6 | 14% | palbociclib 2, abemaciclib 0, ribociclib 0 | [35] |
| EGFR | 10% | cetuximab 2, panitumumab 3, ABT-806 2 | [36,37,38,39] |
| FGFR1/2 | 8–10% | bemarituzumab 2 | [40,41] |
| MET | 8% | crizotinib 1, capmatinib 1, tepotinib 1 | [42,43,44] |
1,2,3: Phase of clinical trials in gastroesophageal cancer; 0: no published data or clinical trials in patients with gastroesophageal cancer. ERBB2: Erb-B2 receptor tyrosine kinase 2; VEGFA: vascular endothelial growth factor A; KRAS: Kirsten rat sarcoma; CDK6: cyclin dependent kinase 6; EGFR: epidermal growth factor receptor; FGFR1/2: fibroblast growth factor receptor 1/2; MET: mesenchymal epithelial transition factor.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sahgal, P.; Huffman, B.M.; Patil, D.T.; Chatila, W.K.; Yaeger, R.; Cleary, J.M.; Sethi, N.S. Early TP53 Alterations Shape Gastric and Esophageal Cancer Development. Cancers 2021, 13, 5915. https://doi.org/10.3390/cancers13235915
AMA Style
Sahgal P, Huffman BM, Patil DT, Chatila WK, Yaeger R, Cleary JM, Sethi NS. Early TP53 Alterations Shape Gastric and Esophageal Cancer Development. Cancers. 2021; 13(23):5915. https://doi.org/10.3390/cancers13235915
Chicago/Turabian StyleSahgal, Pranshu, Brandon M. Huffman, Deepa T. Patil, Walid K. Chatila, Rona Yaeger, James M. Cleary, and Nilay S. Sethi. 2021. "Early TP53 Alterations Shape Gastric and Esophageal Cancer Development" Cancers 13, no. 23: 5915. https://doi.org/10.3390/cancers13235915
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.