
A Novel Immunomodulatory 27-Gene Signature to Predict Response to Neoadjuvant Immunochemotherapy for Primary Triple-Negative Breast Cancer

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
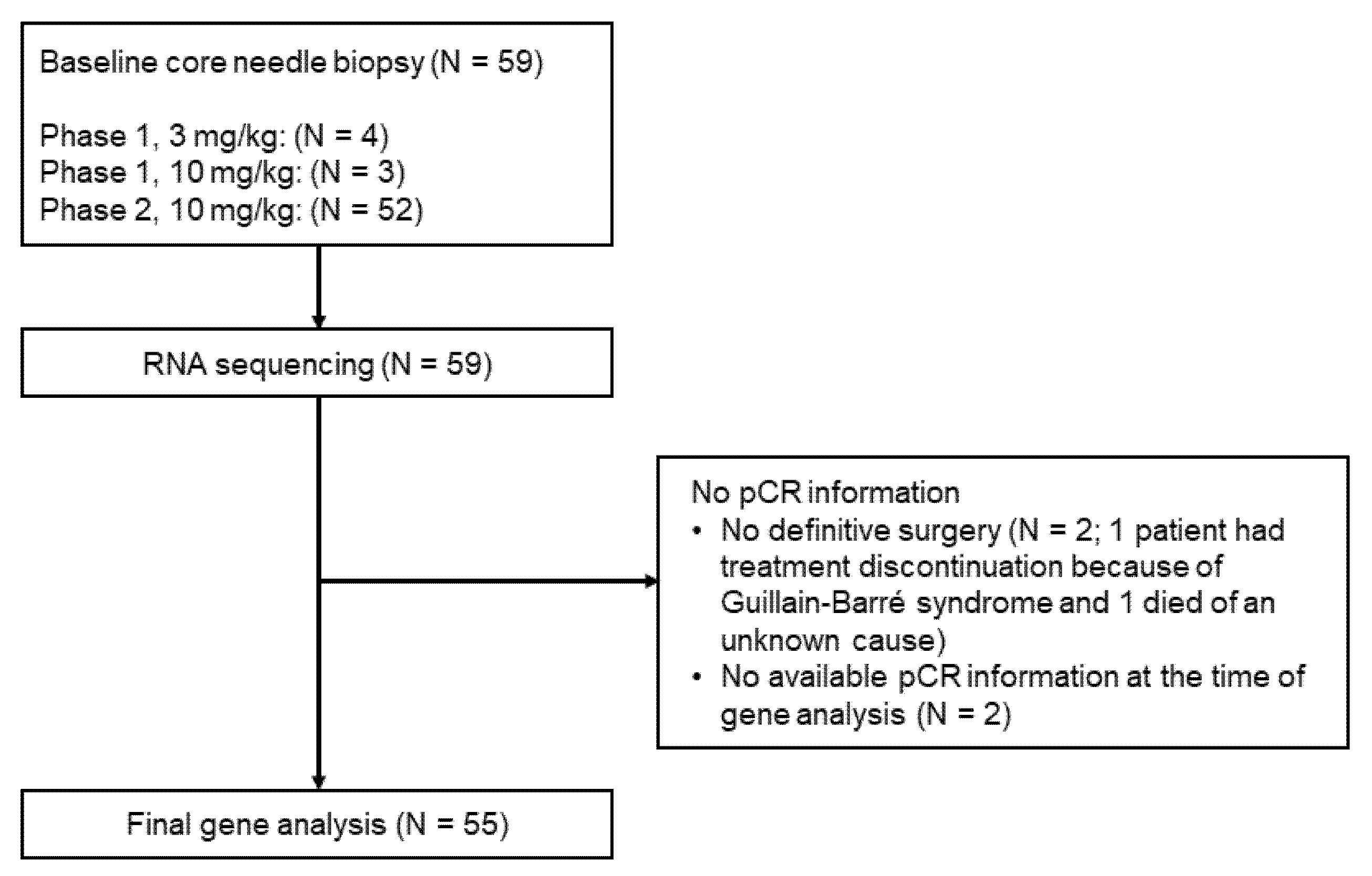
2.1. Design of the Clinical Trial and Sample Acquisition
2.2. RNA Sequencing
2.3. Evaluation of the 27-Gene IO Signature
2.4. Evaluation of PD-L1 Expression by IHC
2.5. Exploration of the Baseline Immune Landscape of the TME
2.6. Statistical Considerations
3. Results
3.1. Predictive Power of the 101-Gene Model and 27-Gene IO Signature
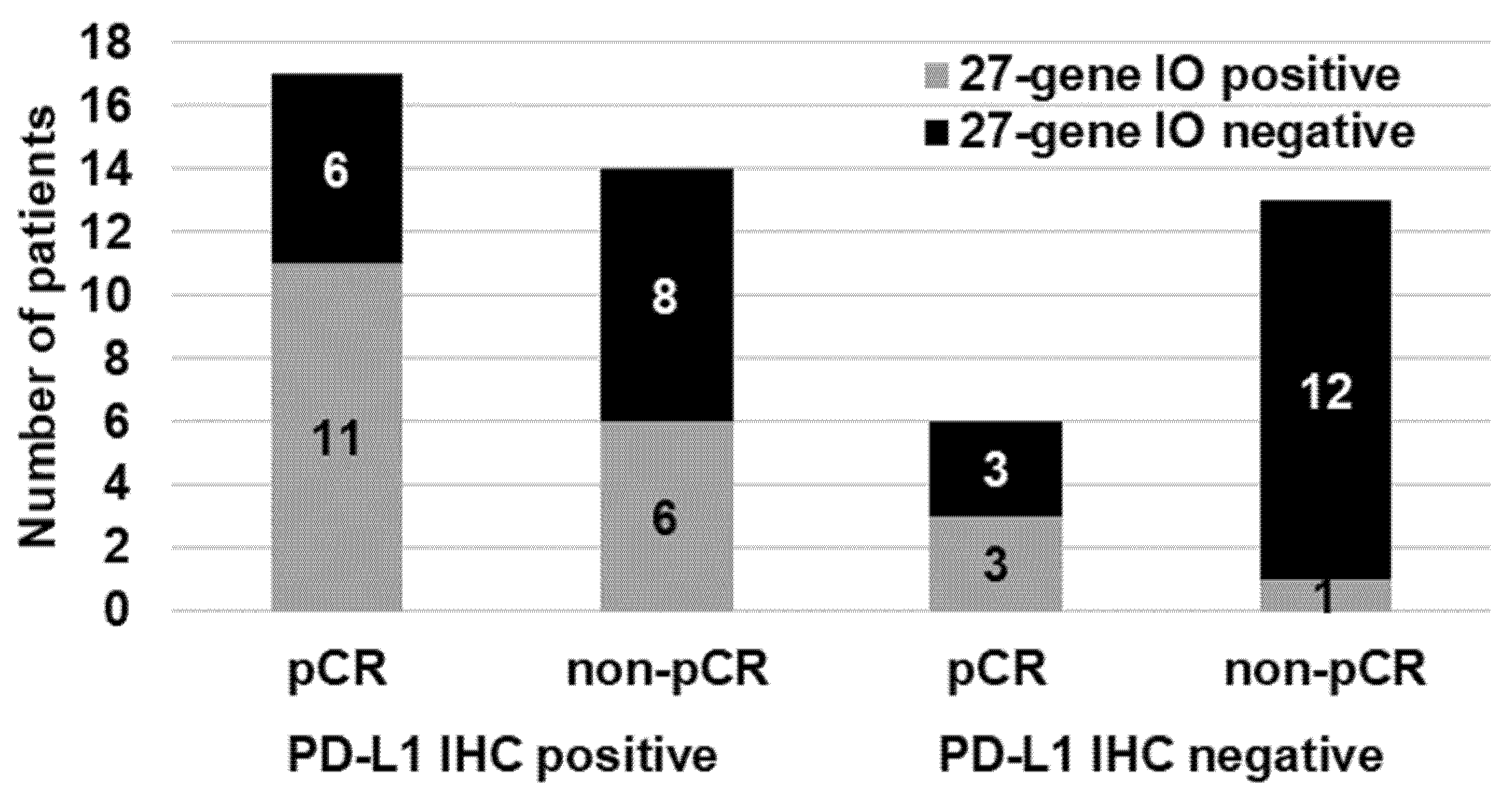
3.2. Predictive Accuracy of the PD-L1 IHC Results and the 27-Gene IO Signature
3.3. Predictive Power of the 27-Gene IO Signature in the BL1 and BL2 Groups
3.4. Tumor Immune Landscape in IM Signature and Non-IM Signature by Deconvolution
CD8+ and CD4+ T Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. IMpassion031: Results from a phase III study of neoadjuvant (neoadj) atezolizumab + chemotherapy in early triple-negative breast cancer (TNBC). Ann. Oncol. 2020, 31, S1142–S1215. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Emens, L.A.; Adams, S.; Barrios, C.H.; Diéras, V.; Iwata, H.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Winer, E.P.; Patel, S.; et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. 2021, 32, 983–993. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; et al. VP7-2021: KEYNOTE-522: Phase III study of neoadjuvant pembrolizumab + chemotherapy vs. placebo + chemotherapy, followed by adjuvant pembrolizumab vs. placebo for early-stage TNBC. Ann. Oncol. 2021, 32, 1198–1200. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invesetig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Ring, B.Z.; Hout, D.R.; Morris, S.W.; Lawrence, K.; Schweitzer, B.L.; Bailey, D.B.; Lehmann, B.D.; Pietenpol, J.A.; Seitz, R.S. Generation of an algorithm based on minimal gene sets to clinically subtype triple negative breast cancer patients. BMC Cancer 2016, 16, 143. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Nielsen, T.J.; Ring, B.Z.; Seitz, R.S.; Hout, D.R.; Schweitzer, B.L. A novel immuno-oncology algorithm measuring tumor microenvironment to predict response to immunotherapies. Heliyon 2021, 7, e06438. [Google Scholar] [CrossRef]
- Ranganath, H.; Jain, A.; Smith, J.; Ryder, J.; Chaudry, A.; Miller, E.; Hare, F.; Valasareddy, P.; Seitz, R.; Hout, D.; et al. One year progression free survival in lung cancer patients treated with immune checkpoint inhibitors is significantly associated with a novel immunomodulatory signature but not PD L1 staining. In Proceedings of the Society for Immunotherapy of Cancer (SITC) Annual Meeting, National Harbor, MA, USA, 6–10 November 2019. [Google Scholar]
- Ahmed, F.S.; Gaule, P.; McGuire, J.; Patel, K.; Blenman, K.; Pusztai, L.; Rimm, D.L. PD-L1 Protein Expression on Both Tumor Cells and Macrophages are Associated with Response to Neoadjuvant Durvalumab with Chemotherapy in Triple-negative Breast Cancer. Clin. Cancer Res. 2020, 26, 5456–5461. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, B.D.; Pietenpol, J.A.; Hortobagyi, G.N.; et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res. 2013, 19, 5533–5540. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Ericsson, P.I.; Stovgaard, E.S.; Sua, L.F.; Reisenbichler, E.; Kos, Z.; Carter, J.M.; Michiels, S.; Le Quesne, J.; Nielsen, T.O.; Laenkholm, A.V.; et al. The path to a better biomarker: Application of a risk management framework for the implementation of PD-L1 and TILs as immuno-oncology biomarkers in breast cancer clinical trials and daily practice. J. Pathol. 2020, 250, 667–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Immunotherapy Slows TNBC Progression. Cancer Discov. 2015, 5, 570. [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy—New insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Mani, N.L.; Schalper, K.A.; Hatzis, C.; Saglam, O.; Tavassoli, F.; Butler, M.; Chagpar, A.B.; Pusztai, L.; Rimm, D.L. Quantitative assessment of the spatial heterogeneity of tumor-infiltrating lymphocytes in breast cancer. Breast Cancer Res. 2016, 18, 78. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| APOD | GZMB | PSMB9 |
| ASPN | HTRA1 | PTGDS |
| CCL5 | IDO1 | RARRES3 |
| CD52 | IL23A | RTP4 |
| COL2A1 | ITM2A | S100A8 |
| CXCL11 | KMO | SFRP1 |
| CXCL13 | KRT16 | SPTLC2 |
| DUSP5 | KYNU | TNFAIP8 |
| FOXC1 | MIA | TNFSF10 |
| Characteristic | Number of Patients (%) |
|---|---|
| All patients | 55 (100) |
| Age, years | |
| ≤40 | 11 (20) |
| 41–50 | 18 (33) |
| 51–69 | 26 (47) |
| Clinical tumor status | |
| cT1 | 18 (33) |
| cT2 | 29 (53) |
| cT3 | 7 (13) |
| Unknown | 1 (2) |
| Clinical nodal status | |
| cN0 | 28 (51) |
| cN1 | 22 (40) |
| cN2 | 1 (2) |
| cN3 | 3 (5) |
| Unknown | 1 (2) |
| pCR | |
| Yes | 25 (45) |
| No | 30 (55) |
| Subtyping Method | Threshold | OR (95% CI) | p | Sensitivity (%) | Specificity (%) | PPV | NPV | PLR | NLR |
|---|---|---|---|---|---|---|---|---|---|
| 101-gene model | 0.17 | 3.14 (0.98–10.9) | 0.054 | 64.7 | 63.2 | 0.44 | 0.80 | 1.76 | 0.56 |
| 0.195 | 3.14 (0.98–10.9) | 0.054 | 64.7 | 63.2 | 0.44 | 0.80 | 1.76 | 0.56 | |
| 0.10 | 3.03 (0.98–10) | 0.054 | 63.2 | 63.9 | 0.48 | 0.77 | 1.75 | 0.58 | |
| 27-gene IO signature | 0.09 | 4.13 (1.36–13.5) | 0.012 | 65.2 | 68.8 | 0.60 | 0.73 | 2.09 | 0.51 |
| PD-L1 IHC | PD-L1: 1% | 2.63 (0.82–9.21) | 0.106 | 73.9 | 48.1 | 0.55 | 0.68 | 1.43 | 0.54 |
| PD-L1+ or 27-gene IO signature | PD-L1: 1% | 5.33 (1.27–22.32) | 0.022 | 87 | 44.4 | 0.57 | 0.80 | 1.57 | 0.29 |
| PD-L1 high or 27-gene IO signature | PD-L1: >50% in tumor and/or >10% in immune cells | 6.53 (1.9–22.5) | 0.003 | 65.4 | 74.2 | 0.68 | 0.72 | 2.53 | 0.47 |
| Variation | Pos vs. Neg for 27-Gene IO Signature | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TIMER | CIBERSORT | xCell | ||||||||
| Immune Cell | Subpopulation | Neg (n = 32) | Pos (n = 23) | p | Neg (n = 32) | Pos (n = 23) | p | Neg (n = 32) | Pos (n = 23) | p |
| CD8+ T cell | 0.43 | Low | High | 0.0072 | Low | High | 5.10 × 10−5 | |||
| CD4+ T cell | General | Low | High | 0.015 | Low | High | 0.00071 | |||
| Resting memory T cell | 0.27 | |||||||||
| Activated memory T cell | Low | High | 1.80 × 10−6 | |||||||
| Follicular helper T cell | 0.5 | |||||||||
| Regulatory T cell | 0.52 | |||||||||
| Macrophage | General | High | Low | 0.029 | Low | High | 0.00041 | |||
| M1 | Low | High | 9.00 × 10−5 | |||||||
| M2 | 0.19 | |||||||||
| Dendritic cell | General | Low | High | 8.00 × 10−5 | Low | High | 1.10 × 10−5 | |||
| Resting dendritic cell | 0.24 | |||||||||
| Activated dendritic cell | 0.71 | |||||||||
| Variation | pCR vs. Non-pCR | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TIMER | CIBERSORT | xCell | ||||||||
| Immune Cell | Subpopulation | Non-pCR (n = 30) | pCR (n = 25) | p | Non-pCR (n = 30) | pCR (n = 25) | p | Non-pCR (n = 30) | pCR (n = 25) | p |
| CD8+ T cell | 0.67 | Low | High | 0.089 | Low | High | 0.036 | |||
| CD4+ T cell | General | Low | High | 0.019 | 0.22 | |||||
| Resting memory T cell | 0.14 | |||||||||
| Activated memory T cell | 0.12 | |||||||||
| Follicular helper T cell | mLow | mHigh | 0.072 | |||||||
| Regulatory T cell | 0.64 | |||||||||
| Macrophage | General | mHigh | mLow | 0.077 | 0.79 | |||||
| M1 | Low | High | 0.0018 | |||||||
| M2 | 0.16 | |||||||||
| Dendritic cell | General | mLow | mHigh | 0.1 | mLow | mHigh | 0.048 | |||
| Resting dendritic cell | 0.33 | |||||||||
| Activated dendritic cell | 0.6 | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwase, T.; Blenman, K.R.M.; Li, X.; Reisenbichler, E.; Seitz, R.; Hout, D.; Nielsen, T.J.; Schweitzer, B.L.; Bailey, D.B.; Shen, Y.; et al. A Novel Immunomodulatory 27-Gene Signature to Predict Response to Neoadjuvant Immunochemotherapy for Primary Triple-Negative Breast Cancer. Cancers 2021, 13, 4839. https://doi.org/10.3390/cancers13194839
Iwase T, Blenman KRM, Li X, Reisenbichler E, Seitz R, Hout D, Nielsen TJ, Schweitzer BL, Bailey DB, Shen Y, et al. A Novel Immunomodulatory 27-Gene Signature to Predict Response to Neoadjuvant Immunochemotherapy for Primary Triple-Negative Breast Cancer. Cancers. 2021; 13(19):4839. https://doi.org/10.3390/cancers13194839
Chicago/Turabian StyleIwase, Toshiaki, Kim R. M. Blenman, Xiaotong Li, Emily Reisenbichler, Robert Seitz, David Hout, Tyler J. Nielsen, Brock L. Schweitzer, Daniel B. Bailey, Yichao Shen, and et al. 2021. "A Novel Immunomodulatory 27-Gene Signature to Predict Response to Neoadjuvant Immunochemotherapy for Primary Triple-Negative Breast Cancer" Cancers 13, no. 19: 4839. https://doi.org/10.3390/cancers13194839