
Real-World Evaluation of Universal Germline Screening for Cancer Treatment-Relevant Pharmacogenes

 ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Study Population and Pharmacogenomic Landscape
2.2. Clinical Impact
2.2.1. Adverse Effects
2.2.2. Efficacy
3. Discussion
4. Materials and Methods
4.1. Study Design and Data Sources
4.2. Study Population
4.3. Sequencing Methods
4.4. Pharmacogenomic Variant Interpretation and Reporting of Results
4.5. Assessment of Potential and Actual Risk for Pharmacogenomic Adverse Outcomes
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Prior Presentation
Conflicts of Interest
Appendix A

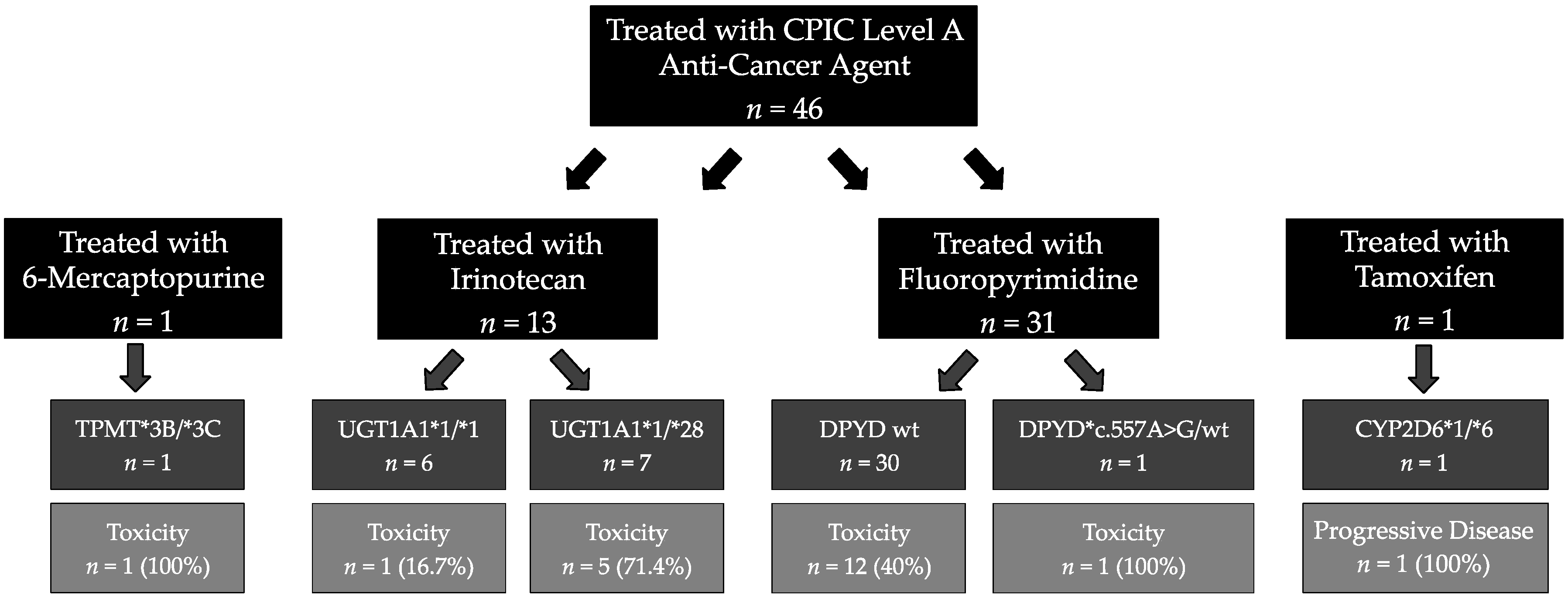{kind=link}
| Gene | Detectable Variants |
|---|---|
| CACNA1S | All pathogenic/likely pathogenic variants a |
| CFTR | *c.1652G>A |
| CYP2C19 | *3, *5, *7, *10 |
| CYP2C9 | *3, *5, *6 |
| CYP2D6 | *6, *8, *14 |
| CYP3A5 | *2, *4, *6, *7, *8 |
| CYP4F2 | *3 |
| DPYD | *2A, *HapB3, *3, *7, *8, *10, *12, *13, *c.557A>G, *c.2846A>T |
| G6PD | Class I deficiency |
| G6PD | Class II deficiency |
| G6PD | Class III deficiency |
| HLA-B | *57:01, *15:02, *58:01 b |
| RYR1 | All pathogenic/likely pathogenic variants a |
| SLCO1B1 | *5, *15/*17 c |
| TPMT | *2, *3A, *3B, *3C, *4, *11, *14, *15, *23, *29, *41 |
| UGT1A1 | *6, *27, *28, *36, *37 |
| VKORC1 | *1173C>T (in linkage with c.-1639G>A) |
Appendix B
| Relevant Genotype | Primary Site | Stage | Regimen | Cycles | Notes |
|---|---|---|---|---|---|
| UGT1A1: Irinotecan Intermediate Metabolizers | |||||
| *1/*28 | Colon | IIA | FOLFIRI + bevacizumab | 14 | No documented toxicity |
| *1/*28 | Colon | IIB | FOLFIRI | 3 | Toxicity-unrelated discontinuation |
| *1/*28 | Colon | IVA | FOLFIRI + panitumumab | 38 | Toxicity-related cycle delay |
| *1/*28 | Rectum | IVA | FOLFIRI | 4 | Toxicity-related dose reduction |
| *1/*28 | Pancreas | IB | FOLFIRINOX | 15 | Toxicity-related dose reduction |
| *1/*28 | Pancreas | III | Liposomal irinotecan + 5-FU | 2 | Toxicity-related discontinuation |
| *1/*28 | Rectum | IIA | XELIRI | 1 | Toxicity-related hospitalization, death |
| UGT1A1: Irinotecan Normal Metabolizers | |||||
| *1/*1 | Rectum | IV | FOLFIRI Irinotecan + cetuximab | 6 8 | No documented toxicity No documented toxicity |
| *1/*1 | Colon | IIIC | FOLFIRI | 8 | No documented toxicity |
| *1/*1 | Colon | IVA | XELIRI | 7 | No documented toxicity |
| *1/*1 | Colon | IIIB | XELIRI | 11 | No documented toxicity |
| *1/*1 | Pancreas | III | FOLFIRINOX | 3 | Toxicity-unrelated discontinuation |
| *1/*1 | Pancreas | IIB | FOLFIRINOX | 12 | Toxicity-related dose reduction |
| DPYD: Intermediate Fluoropyrimidine Metabolizers 1 | |||||
| *c.557A>G/wt | Stomach | IB | FOLFOX | 1 | Toxicity-related discontinuation |
| DPYD: Normal Fluoropyrimidine Metabolizers 2 | |||||
| wt | Stomach | III | FOLFOX | 10 | No documented toxicity |
| wt | Gastric | IV | XELOX | 4 | No documented toxicity |
| wt | Gastric | IIA | FLOT | 4 | No documented toxicity |
| wt | Rectum | IIIC | Capecitabine + RT Capecitabine | 25 d 7 | No documented toxicity No documented toxicity |
| wt | Colon | IIIC | XELOX | 6 | No documented toxicity |
| wt | Rectum | II | XELOX | 4 | No documented toxicity |
| wt | Colon | IIIB | FOLFOX | 12 | No documented toxicity |
| wt | Rectum | I | Capecitabine + RT | 45 d | No documented toxicity |
| wt | Colon | IV | FOLFOX + bevacizumab | 10 | No documented toxicity |
| wt | Pancreas | III | Gemcitabine + capecitabine | 8 | No documented toxicity |
| wt | Pancreas | IIIB | Gemcitabine + capecitabine | 6 | No documented toxicity |
| wt | Colon | IIIC | FOLFIRI | 8 | No documented toxicity |
| wt | Colon | IVA | XELIRI | 7 | No documented toxicity |
| wt | Colon | IIIB | XELIRI | 11 | No documented toxicity |
| wt | Rectum | IV | FOLFIRI | 6 | Toxicity-unrelated dose reduction |
| wt | Esophagus | III | FOLFOX Cisplatin + capecitabine | 12 2 | No documented toxicity Toxicity-unrelated discontinuation |
| wt | Pancreas | IB | Gemcitabine + capecitabine | 1 | Toxicity-unrelated discontinuation |
| wt | Pancreas | III | FOLFIRINOX | 3 | Toxicity-unrelated discontinuation |
| wt | Colon | IIA | FOLFOX | 10 | Toxicity-related dose reduction |
| wt | Colon | IIB | FOLFOX | 7 | Toxicity-related dose reduction, cycle delay |
| wt | Colon | IIIB | FOLFOX + bevacizumab | 11 | Toxicity-related dose reduction |
| wt | Esophagus | IVC | FLOT | 4 | Toxicity-related dose reduction |
| wt | Stomach | II | FLOT XELOX | 3 2 | Toxicity-related discontinuation Toxicity-related discontinuation |
| wt | Esophagus | IIIB | XELOX FOLFOX | 3 6 | Toxicity-related discontinuation Toxicity-related dose reduction |
| wt | Colon | IIIA | XELOX | 4 | Toxicity-related dose reduction |
| wt | Colon | IV | FOLFOX XELOX | 1 10 | Toxicity-related discontinuation Toxicity-related dose reduction |
| wt | Colon | III | Capecitabine | 1 | Toxicity-related discontinuation |
| wt | Colon | IVA | XELIRI | 1 | Toxicity-related discontinuation |
| wt | Stomach | II | XELOX FOLFOX | 2 8 | Toxicity-related hospitalization, discontinuation Toxicity-related dose reduction, cycle delay |
| wt | Colon | IIIB | FOLFOX + bevacizumab | 5 | Toxicity-related discontinuation, hospitalization |
| TPMT: Poor Mercaptopurine Metabolizer | |||||
| *3B/*3C | ALL | N/A | POMP | 17 | Toxicity-related dose reduction |
| CYP2D6: Intermediate Tamoxifen Metabolizer | |||||
| *1/*6 | Breast | IV | Tamoxifen 20 mg daily | 8 mo | Progressive disease; proceeded to surgical castration |
Appendix C
| Regimen | Dosing |
|---|---|
| FOLFIRI | Irinotecan 180 mg/m2, leucovorin 400 mg/m2, 5-FU 400 mg/m2 bolus, then 2400 mg/m2 CI every 14 d |
| FOLFIRINOX | Irinotecan 180 mg/m2, leucovorin 400 mg/m2, oxaliplatin 85 mg/m2, 5-FU 400 mg/m2 bolus, then 2400 mg/m2 CI every 14 d |
| Liposomal irinotecan + 5-FU | Liposomal irinotecan 70 mg/m2, leucovorin 400 mg/m2, 5-FU 2400 mg/m2 CI every 14 d |
| XELIRI | Irinotecan 80–200 mg/m2 and capecitabine 800–1000 mg/m2 twice daily every 21 d |
| Irinotecan + cetuximab | Irinotecan 180 mg/m2 and cetuximab 250–400 mg/m2 every 14 d |
| FOLFOX | Leucovorin 400 mg/m2, oxaliplatin 85 mg/m2, 5-FU 2400 mg/m2 CI +/− 400 mg/m2 bolus every 14 d |
| XELOX | Oxaliplatin 130 mg/m2 and capecitabine 850–1000 mg/m2 twice daily every 21 d or Oxaliplatin 85 mg/m2 and capecitabine 850–1000 mg/m2 twice daily every 14 d |
| FLOT | Leucovorin 200 mg/m2, docetaxel 50 mg/m2, oxaliplatin 85 mg/m2, 5-FU 2600 mg/m2 CI every 14 d |
| Capecitabine + RT | 825 mg/m2 twice daily during radiation treatments |
| Capecitabine | 850 mg/m2 twice daily every 14–28 d |
| Gemcitabine + capecitabine | Gemcitabine 1000 mg/m2 weekly and capecitabine 650 mg/m2 twice daily every 21 d or Gemcitabine 1000 mg/m2 weekly and capecitabine 830 mg/m2 twice daily every 14 d |
| Cisplatin + capecitabine | Cisplatin 80 mg/m2 and capecitabine 1000 mg/m2 twice daily every 21 d |
| POMP | Vincristine 1.4 mg/m2, mercaptopurine 60 mg/m2, methotrexate 20 mg/m2, prednisone 30 mg/m2 every 28 d |
Appendix D
| Pharmacogene | Drug | Indication |
|---|---|---|
| CYP2D6 | Tamoxifen | Hormone-mediated cancers |
| DPYD | Capecitabine Fluorouracil | Chemotherapy; see Table 3 Chemotherapy; see Table 3 |
| TPMT | Mercaptopurine Thioguanine | Leukemia, Chron disease/ulcerative colitis, other autoimmune disease Leukemia |
| UGT1A1 | Irinotecan | Chemotherapy; see Table 3 |
References
- Vivot, A.; Boutron, I.; Ravaud, P.; Porcher, R. Guidance for pharmacogenomic biomarker testing in labels of FDA-approved drugs. Genet. Med. 2015, 17, 733–738. [Google Scholar] [CrossRef] [Green Version]
- United States Food and Drug Administration Table of Pharmacogenomic Biomarkers in Drug Labeling. Available online: https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling (accessed on 14 April 2021).
- Clinical Pharmacogenetics Implementation Consortium. Available online: https://cpicpgx.org (accessed on 17 December 2020).
- Relling, M.V.; Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther. 2011, 89, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.; Arnold, S.; Weiss, H.L.; Wu, J.; Durbin, E.B.; Miller, R.; Kolesar, J.M. Pharmacogenomic potential in advanced cancer patients. Am. J. Health Syst. Pharm. 2019, 76, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Drozda, K.; Pacanowski, M.A. Clinical trial designs to support clinical utility of pharmacogenomic testing. Pharmacotherapy 2017, 37, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Kolesar, J.M.; Vermeulen, L.C. Precision medicine: Opportunities for health-system pharmacists. Am. J. Health Syst. Pharm. 2021, 78, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Anagnostou, V.; Lytle, K.; Parpart-Li, S.; Nesselbush, M.; Riley, D.R.; Shukla, M.; Chesnick, B.; Kadan, M.; Papp, E.; et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci. Transl. Med. 2015, 7, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol. 2016, 2, 104–111. [Google Scholar] [CrossRef]
- Hertz, D.L.; McLeod, H.L. Integrated patient and tumor genetic testing for individualized cancer therapy. Clin. Pharmacol. Ther. 2016, 99, 143–146. [Google Scholar] [CrossRef]
- Hertz, D.L.; Glatz, A.; Pasternak, A.L.; Lonigro, R.J.; Vats, P.; Wu, Y.M.; Anderson, B.; Rabban, E.; Mora, E.; Frank, K.; et al. Integration of germline pharmacogenetics into a tumor sequencing program. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Gammal, R.S.; Court, M.H.; Haidar, C.E.; Iwuchukwu, O.F.; Gaur, A.H.; Alvarellos, M.; Guillemette, C.; Lennox, J.L.; Whirl-Carrillo, M.; Brummel, S.S.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for UGT1A1 and atazanavir prescribing. Clin. Pharmacol. Ther. 2016, 99, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.S.; Seligson, N.D.; Bottiglieri, S.; Carballido, E.; Cueto, A.D.; Imanirad, I.; Levine, R.; Parker, A.S.; Swain, S.M.; Tillman, E.M.; et al. UGT1A1 Guided cancer therapy: Review of the evidence and considerations for clinical implementation. Cancers (Basel) 2021, 13, 1566. [Google Scholar] [CrossRef] [PubMed]
- Amstutz, U.; Henricks, L.M.; Offer, S.M.; Barbarino, J.; Schellens, J.H.M.; Swen, J.J.; Klein, T.E.; McLeod, H.L.; Caudle, K.E.; Diasio, R.B.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin. Pharmacol. Ther. 2018, 103, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Schwab, M.; Whirl-Carrillo, M.; Suarez-Kurtz, G.; Pui, C.H.; Stein, C.M.; Moyer, A.M.; Evans, W.E.; Klein, T.E.; Antillon-Klussmann, F.G.; et al. Clinical Pharmacogenetics Implementation Consortium guideline for thiopurine dosing based on TPMT and NUDT15 genotypes: 2018 update. Clin. Pharmacol. Ther. 2019, 105, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.A.; Shah, B.; Advani, A.; Aoun, P.; Boyer, M.W.; Burke, P.W.; DeAngelo, D.J.; Dinner, S.; Fathi, A.T.; Gauthier, J.; et al. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology: Acute Lymphoblastic Leukemia. Available online: https://www.nccn.org/professionals/physician_gls/pdf/all.pdf (accessed on 20 May 2021).
- Clinical Pharmacogenetics Implementation Consortium: Genes/Drugs. Available online: https://cpicpgx.org/genes-drugs/ (accessed on 14 December 2020).
- Benson, A.B., III; Venook, A.P.; Al-Hawary, M.M.; Arain, M.A.; Chen, Y.-J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Farkas, L.; et al. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology: Colon Cancer. Available online: https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf (accessed on 30 October 2020).
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Behrman, S.W.; Benson, A.B., III; Cardin, D.B.; Chiorean, E.G.; Chung, V.; Czito, B.; Del Chiaro, M.; et al. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology: Pancreatic Adenocarcinoma. Version 2.2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/pancreatic.pdf (accessed on 1 September 2021).
- Ajani, J.A.; D’Amico, T.A.; Bentrem, D.J.; Chao, J.; Collier, S.; Corvera, C.; Das, P.; Denlinger, C.S.; Enzinger, P.C.; Enzler, T.; et al. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology: Gastric Cancer. Version 4.2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/gastric.pdf (accessed on 1 September 2021).
- Ajani, J.A.; D’Amico, T.A.; Bentrem, D.J.; Chao, J.; Collier, S.; Corvera, C.; Das, P.; Denlinger, C.S.; Enzinger, P.C.; Enzler, T.; et al. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology: Esophageal and Esophagogastric Junction Cancer. Version 4.2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/esophageal.pdf (accessed on 1 September 2021).
- O’Dwyer, P.J.; Catalano, R.B. Uridine diphosphate glucuronosyltransferase (UGT) 1A1 and irinotecan: Practical pharmacogenomics arrives in cancer therapy. J. Clin. Oncol. 2006, 24, 4534–4538. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, A.; Sun, X.; Liu, L.; Zhu, Y.; Zhang, T.; Jia, J.; Tan, S.; Wu, J.; Wang, X.; et al. Multicenter, randomized, phase III trial of neoadjuvant chemoradiation with capecitabine and irinotecan guided by UGT1A1 status in patients with locally advanced rectal cancer. J. Clin. Oncol. 2020, 38, 4231–4239. [Google Scholar] [CrossRef]
- Henricks, L.M.; Opdam, F.L.; Beijnen, J.H.; Cats, A.; Schellens, J.H.M. DPYD genotype-guided dose individualization to improve patient safety of fluoropyrimidine therapy: Call for a drug label update. Ann. Oncol. 2017, 28, 2915–2922. [Google Scholar] [CrossRef]
- Henricks, L.M.; van Merendonk, L.N.; Meulendijks, D.; Deenen, M.J.; Beijnen, J.H.; de Boer, A.; Cats, A.; Schellens, J.H.M. Effectiveness and safety of reduced-dose fluoropyrimidine therapy in patients carrying the DPYD*2A variant: A matched pair analysis. Int. J. Cancer 2019, 144, 2347–2354. [Google Scholar] [CrossRef]
- Kondo, Y.; Nakajima, M.M.; Nakajima, G.; Hayashi, K. When should anti-cancer treatment be ended? Why and when to discontinue palliative chemotherapy. J. Clin. Oncol. 2018, 36, 41. [Google Scholar] [CrossRef]
- Zanuso, V.; Fregoni, V.; Gervaso, L. Side effects of adjuvant chemotherapy and their impact on outcome in elderly breast cancer patients: A cohort study. Future Sci. OA 2020, 6, Fso617. [Google Scholar] [CrossRef] [PubMed]
- Hershman, D.L.; Shao, T.; Kushi, L.H.; Buono, D.; Tsai, W.Y.; Fehrenbacher, L.; Kwan, M.; Gomez, S.L.; Neugut, A.I. Early discontinuation and non-adherence to adjuvant hormonal therapy are associated with increased mortality in women with breast cancer. Breast Cancer Res. Treat. 2011, 126, 529–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, C.; Byrne, S.; Ahmed, G.; Kenny, A.; Gallagher, J.; Harvey, H.; O’Farrell, E.; Bird, B. Cost implications of reactive versus prospective testing for dihydropyrimidine dehydrogenase deficiency in patients with colorectal cancer: A single-institution experience. Dose Response 2018, 16, 1559325818803042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortejoso, L.; García-González, X.; García, M.I.; García-Alfonso, P.; Sanjurjo, M.; López-Fernández, L.A. Cost-effectiveness of screening for DPYD polymorphisms to prevent neutropenia in cancer patients treated with fluoropyrimidines. Pharmacogenomics 2016, 17, 979–984. [Google Scholar] [CrossRef]
- Thompson, A.J.; Newman, W.G.; Elliott, R.A.; Roberts, S.A.; Tricker, K.; Payne, K. The cost-effectiveness of a pharmacogenetic test: A trial-based evaluation of TPMT genotyping for azathioprine. Value Health 2014, 17, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnan, J.R.; Ungar, W.J.; Mathews, M.; Hancock-Howard, R.L.; Rahman, P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr. Blood Cancer 2011, 57, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Pichereau, S.; Le Louarn, A.; Lecomte, T.; Blasco, H.; Le Guellec, C.; Bourgoin, H. Cost-effectiveness of UGT1A1*28 genotyping in preventing severe neutropenia following FOLFIRI therapy in colorectal cancer. J. Pharm. Pharm. Sci. 2010, 13, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Butzke, B.; Oduncu, F.S.; Severin, F.; Pfeufer, A.; Heinemann, V.; Giessen-Jung, C.; Stollenwerk, B.; Rogowski, W.H. The cost-effectiveness of UGT1A1 genotyping before colorectal cancer treatment with irinotecan from the perspective of the German statutory health insurance. Acta. Oncol. 2016, 55, 318–328. [Google Scholar] [CrossRef]
- Obradovic, M.; Mrhar, A.; Kos, M. Cost-effectiveness of UGT1A1 genotyping in second-line, high-dose, once every 3 weeks irinotecan monotherapy treatment of colorectal cancer. Pharmacogenomics 2008, 9, 539–549. [Google Scholar] [CrossRef]
- Wei, X.; Cai, J.; Sun, H.; Li, N.; Xu, C.; Zhang, G.; Sui, Y.; Zhuang, J.; Zheng, B. Cost-effectiveness analysis of UGT1A1*6/*28 genotyping for preventing FOLFIRI-induced severe neutropenia in Chinese colorectal cancer patients. Pharmacogenomics 2019, 20, 241–249. [Google Scholar] [CrossRef]
- Caligiuri, M.A.; Dalton, W.S.; Rodriguez, L.; Sellers, T.; Willman, C.L. Orien. Oncology Issues 2016, 31, 62–66. [Google Scholar] [CrossRef]
- Fenstermacher, D.A.; Wenham, R.M.; Rollison, D.E.; Dalton, W.S. Implementing personalized medicine in a cancer center. Cancer J. 2011, 17, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, W.S.; Sullivan, D.; Ecsedy, J.; Caligiuri, M.A. Patient enrichment for precision-based cancer clinical trials: Using prospective cohort surveillance as an approach to improve clinical trials. Clin. Pharmacol. Ther. 2018, 104, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Taylor, R.; Thielke, R.; Payne, J.; Gonzalez, N.; Conde, J.G. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 2009, 42, 377–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, P.A.; Taylor, R.; Minor, B.L.; Elliott, V.; Fernandez, M.; O’Neal, L.; McLeod, L.; Delacqua, G.; Delacqua, F.; Kirby, J.; et al. The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inform. 2019, 95, 103208. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra, 1st ed.; O’Reilly Media: Sebastopol, CA, USA, 2020. [Google Scholar]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Fairley, S.; Lowy-Gallego, E.; Perry, E.; Flicek, P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2020, 48, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. NCCN Guidelines: Treatment by Cancer Type. Available online: https://www.nccn.org/guidelines/category_1 (accessed on 14 April 2021).
- United States Census Bureau. QuickFacts: Kentucky. Available online: https://www.census.gov/quickfacts/KY (accessed on 14 April 2021).

| Characteristic | Patients (n) | % |
|---|---|---|
| Total | 291 | |
| Age (median) | 61 | IQR: 52–68 |
| Race | ||
| Non-Hispanic Black | 14 | 4.8% |
| Non-Hispanic White | 277 | 95.2% |
| Gender | ||
| Female | 147 | 50.5% |
| Male | 139 | 47.8% |
| Non-Binary 1 | 1 | 0.3% |
| Primary Cancer Site | ||
| Colon/rectal | 55 | 18.9% |
| Gynecologic | 49 | 16.8% |
| Head and neck | 36 | 12.4% |
| Brain | 25 | 8.6% |
| Pancreatic | 16 | 5.5% |
| Leukemia/lymphoma | 15 | 5.2% |
| Gastric/gastroesophageal | 15 | 5.2% |
| Kidney and bladder | 14 | 4.8% |
| Lung | 14 | 4.8% |
| Small bowel 2 | 12 | 4.1% |
| Skin | 12 | 4.1% |
| Breast | 7 | 2.4% |
| Other 3 | 21 | 7.2% |
| Cancer Stage | ||
| I | 31 | 10.7% |
| II | 42 | 14.4% |
| III | 93 | 32.0% |
| IV | 78 | 26.8% |
| N/A 4 | 47 | 16.2% |
| Pharmacogenomic Variant | Patients with Variant Allele n (%) | Variant Allele Frequency | Expected Variant Allele Frequency 1 |
|---|---|---|---|
| Any | 263 (90.4%) | ||
| UGT1A1 | |||
| *28 | 0.3093 | 0.3165 | |
| Homozygote | 23 (7.9%) | ||
| Heterozygote | 134 (46.0%) | ||
| Total UGT1A1 | 157 (54.0%) | 0.3093 | 0.3165 |
| DPYD | |||
| *c.2846A>T | 1 (0.3%) | 0.0017 | 0.0037 |
| *2A | 4 (1.4%) | 0.0069 | 0.0079 |
| *HapB3 | 7 (2.4%) | 0.0120 | 0.0237 |
| *c.557A>G | 1 (0.3%) | 0.0017 | 0.0001 |
| *7 | 1 (0.3%) | 0.0017 | 0.0002 |
| Total DPYD | 14 (4.8%) | 0.0240 | 0.0353 |
| TPMT | |||
| *3A | 0.0601 | 0.0343 | |
| Homozygote | 2 (0.7%) | ||
| Heterozygote | 31 (10.7%) | ||
| *3B | 1 (0.3%) | 0.0017 | 0.0027 |
| *3C | 0.0123 | 0.0047 | |
| Homozygote | 1 (0.3%) | ||
| Heterozygote | 3 (1.0%) | ||
| *2 | 2 (0.7%) | 0.0034 | 0.0021 |
| Total TPMT | 40 (14.0%) | 0.0775 | 0.0438 |
| CYP2D6 | |||
| *6 | 5 (1.7%) | 0.0086 | 0.0025 |
| CYP2C9 | |||
| *3 | 37 (12.7%) | 0.0636 | 0.0301 |
| *11 | 2 (0.7%) | 0.0034 | 0.0028 |
| Total CYP2C9 | 39 (13.4%) | 0.0670 | 0.0329 |
| CYP3A5 | |||
| *6 | 3 (1.0%) | 0.0052 | 0.0015 |
| *7 | 2 (0.7%) | 0.0034 | 0.0000 |
| Total CYP3A5 | 5 (1.7%) | 0.0086 | 0.0015 |
| G6PD | |||
| A-202A_376G-III | 1 (0.3%) | 0.0017 | 0.0000–0.0340 2 |
| CYP4F2 | |||
| *3 | 0.2629 | 0.4108 | |
| Homozygote | 21 (7.2%) | ||
| Heterozygote | 111 (38.1%) | ||
| Total CYP4F2 | 132 (45.0%) | ||
| SLCO1B1 | |||
| *15 or *17 3 | 0.1186 | 0.1214 (*15); 0.0519 (*17) | |
| Homozygote | 5 (1.7%) | ||
| Heterozygote | 59 (20.3%) | ||
| *5 | 0.0241 | 0.0224 | |
| Homozygote | 2 (0.7%) | ||
| Heterozygote | 10 (3.4%) | ||
| Total SLCO1B1 | 12 (4.1%) | 0.1427 | 0.1957 |
| VKORC1 | |||
| *1173C>T | 0.1409 | 0.4643 | |
| Homozygote | 11 (3.8%) | ||
| Heterozygote | 60 (20.6%) | ||
| Total VKORC1 | 71 (24.4%) | ||
| RYR1 | |||
| c.7042_7044delGAG | 1 (0.3%) | 0.0017 | 0.0000 4 |
| c.14818G>A | 1 (0.3%) | 0.0017 | 0.0000 5 |
| Total RYR1 | 2 (0.6%) | 0.0034 | 0.0000 4,5 |
| Pharmacogene | Anti-Cancer Drug | Malignancy | Patients at Potential Risk (a 2/A 3) | Patients at Actual Risk (b 4/B 5) |
|---|---|---|---|---|
| Toxicity-Associated Pharmacogenes | ||||
| UGT1A1 | Irinotecan | Colon/rectal P | 33/55 | 5/27 1 |
| Pancreas P | 7/16 | 2/6 1 | ||
| Gastric/gastroesophageal P | 4/15 | 0/4 | ||
| Cervix O | 1/1 | 0/1 | ||
| Ovary O | 16/25 | 0/16 | ||
| Hepatobiliary O | 5/7 | 0/4 1 | ||
| Carcinoid/neuroendocrine O | 6/10 | 0/6 | ||
| Small bowel adenocarcinoma O | 0/2 | N/A 6 | ||
| DPYD | Capecitabine | Colon/rectal P | 3/55 | 0/2 1 |
| Pancreas P | 1/16 | 0/1 | ||
| Breast P | 0/7 | N/A | ||
| Ovary O | 2/25 | 0/2 | ||
| Cervix O | 0/1 | N/A | ||
| Anus O | 0/1 | N/A | ||
| Bladder O | 0/3 | N/A | ||
| DPYD | Fluorouracil | Colon/rectal P | 3/55 | 0/2 1 |
| Pancreas P | 1/16 | 1/1 | ||
| Gastric/gastroesophageal P | 1/15 | 1/1 | ||
| Head and neck P | 0/36 | N/A | ||
| Anus O | 0/1 | N/A | ||
| Vulva O | 0/1 | N/A | ||
| Basal cell skin O | 0/1 | N/A | ||
| Squamous cell skin O | 0/4 | N/A | ||
| Bladder O | 0/3 | N/A | ||
| Thyroid O | 0/4 | N/A | ||
| Small bowel adenocarcinoma O | 0/2 | N/A | ||
| TPMT | Mercaptopurine | Acute lymphoblastic leukemia P | 1/3 | 1/1 |
| Efficacy-Associated Pharmacogenes | ||||
| CYP2D6 | Tamoxifen | Breast P | 1/7 | 1/1 |
| Ovary P | 1/25 | 0/1 | ||
| Uterus P | 0/22 | N/A | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hutchcraft, M.L.; Lin, N.; Zhang, S.; Sears, C.; Zacholski, K.; Belcher, E.A.; Durbin, E.B.; Villano, J.L.; Cavnar, M.J.; Arnold, S.M.; et al. Real-World Evaluation of Universal Germline Screening for Cancer Treatment-Relevant Pharmacogenes. Cancers 2021, 13, 4524. https://doi.org/10.3390/cancers13184524
Hutchcraft ML, Lin N, Zhang S, Sears C, Zacholski K, Belcher EA, Durbin EB, Villano JL, Cavnar MJ, Arnold SM, et al. Real-World Evaluation of Universal Germline Screening for Cancer Treatment-Relevant Pharmacogenes. Cancers. 2021; 13(18):4524. https://doi.org/10.3390/cancers13184524
Chicago/Turabian StyleHutchcraft, Megan L., Nan Lin, Shulin Zhang, Catherine Sears, Kyle Zacholski, Elizabeth A. Belcher, Eric B. Durbin, John L. Villano, Michael J. Cavnar, Susanne M. Arnold, and et al. 2021. "Real-World Evaluation of Universal Germline Screening for Cancer Treatment-Relevant Pharmacogenes" Cancers 13, no. 18: 4524. https://doi.org/10.3390/cancers13184524