
Metformin and Niclosamide Synergistically Suppress Wnt and YAP in APC-Mutated Colorectal Cancer

 , , , , ,
, , , , ,  , and
, and 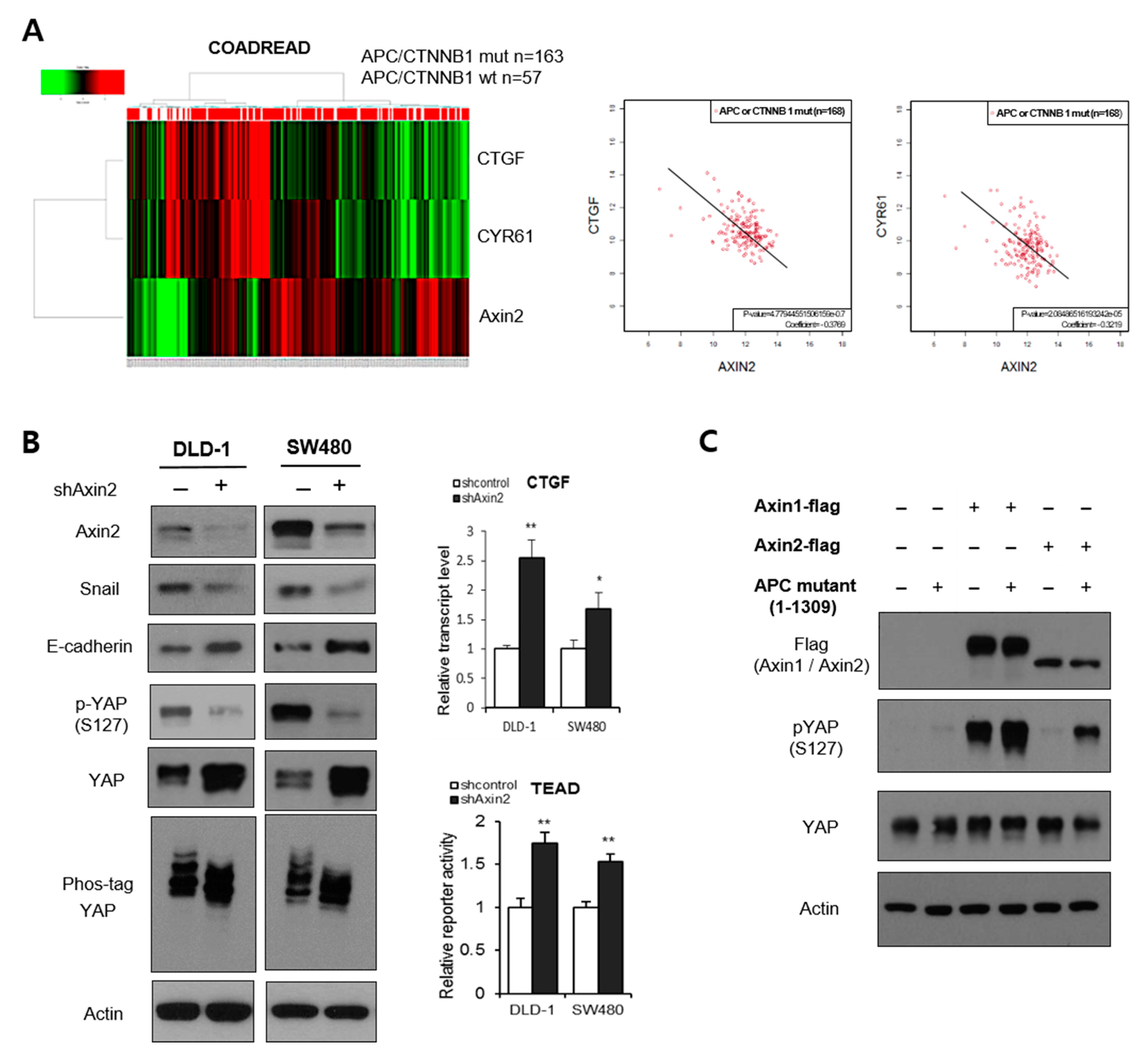{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Immunoblot Analysis
2.3. Cell Migration Assay
2.4. Quantitative Real Time-PCR and Reporter Assay
2.5. Immunofluorescence
2.6. Gene Expression Analysis of Clinical Samples
2.7. Culture of Patient-Derived Organoids from Colon Cancer and FAP Patients
2.8. Tumor Sphere Culture
2.9. Flow-Cytometric Analysis for Cancer Stem Cell Markers
2.10. APC-MIN Mouse Model for FAP and DSS-Induced Colon Cancer
2.11. Immunohistochemistry for In Vivo Samples
2.12. In Vivo Xenograft Assay
2.13. Statistical Analysis
3. Results
3.1. Axin2 Potentiates the Hippo Pathway in APC-Mutant Colorectal Cells
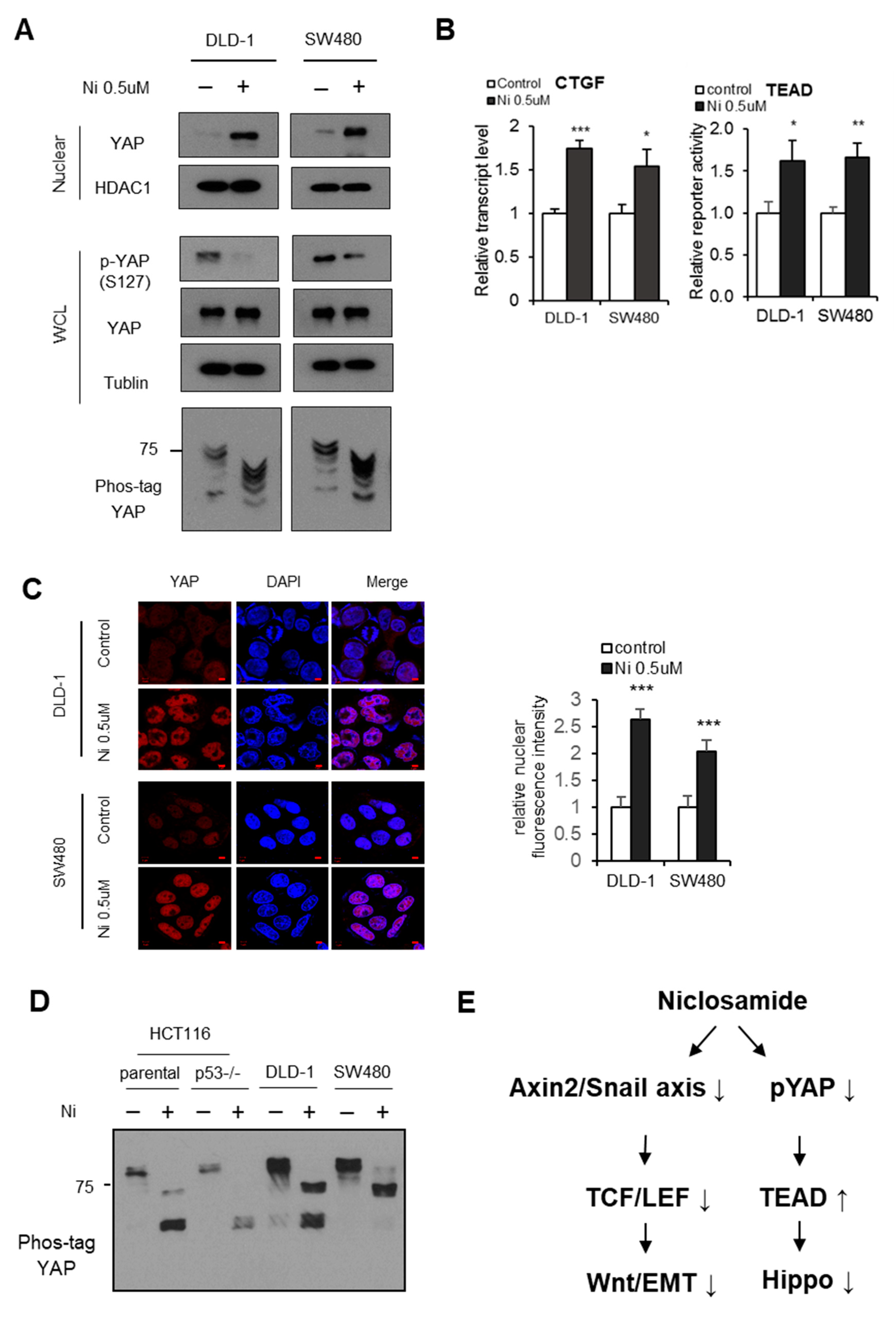
3.2. Niclosamide Suppresses the Canonical Wnt and Hippo Pathways in APC-Mutant Cells
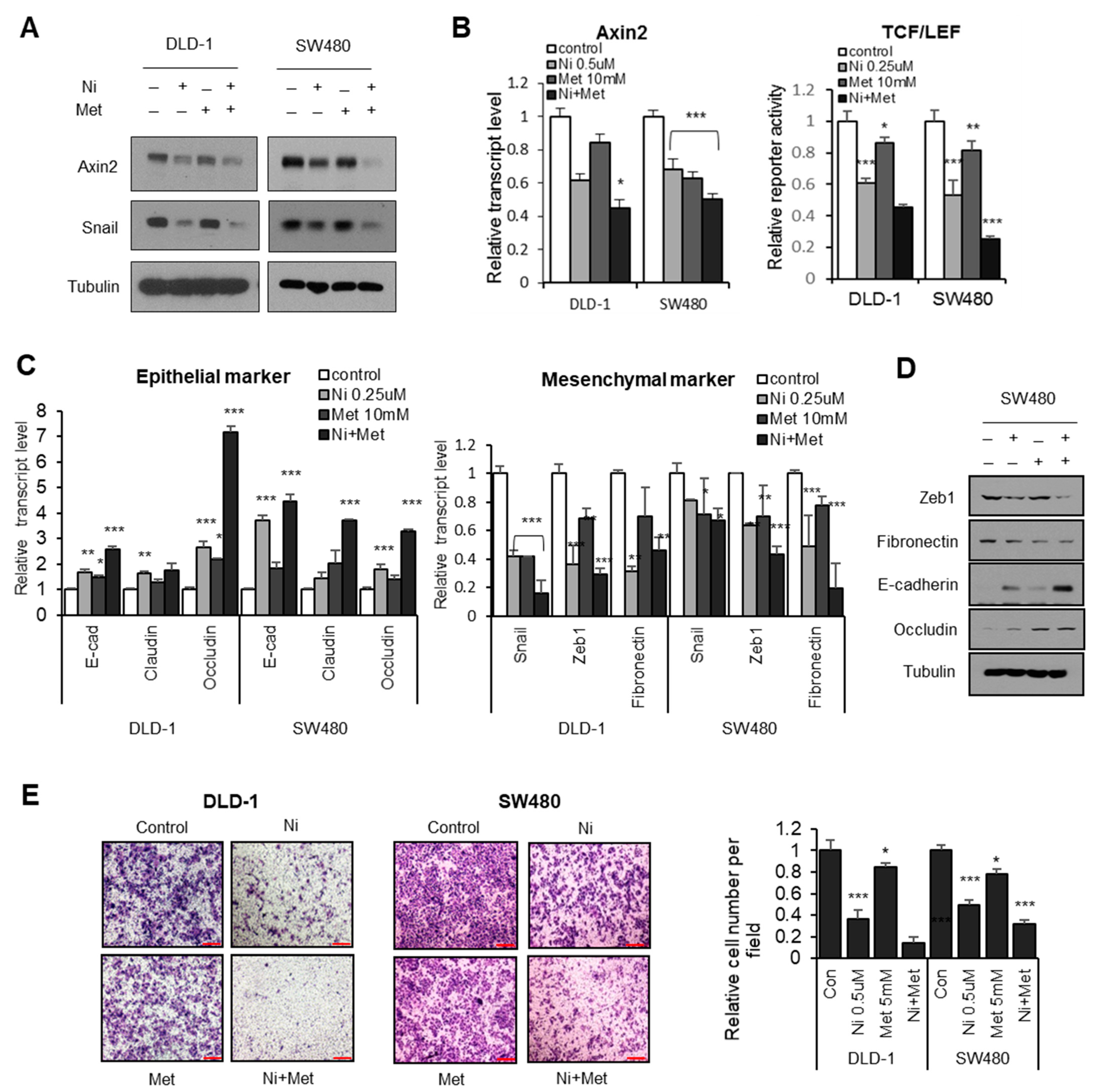
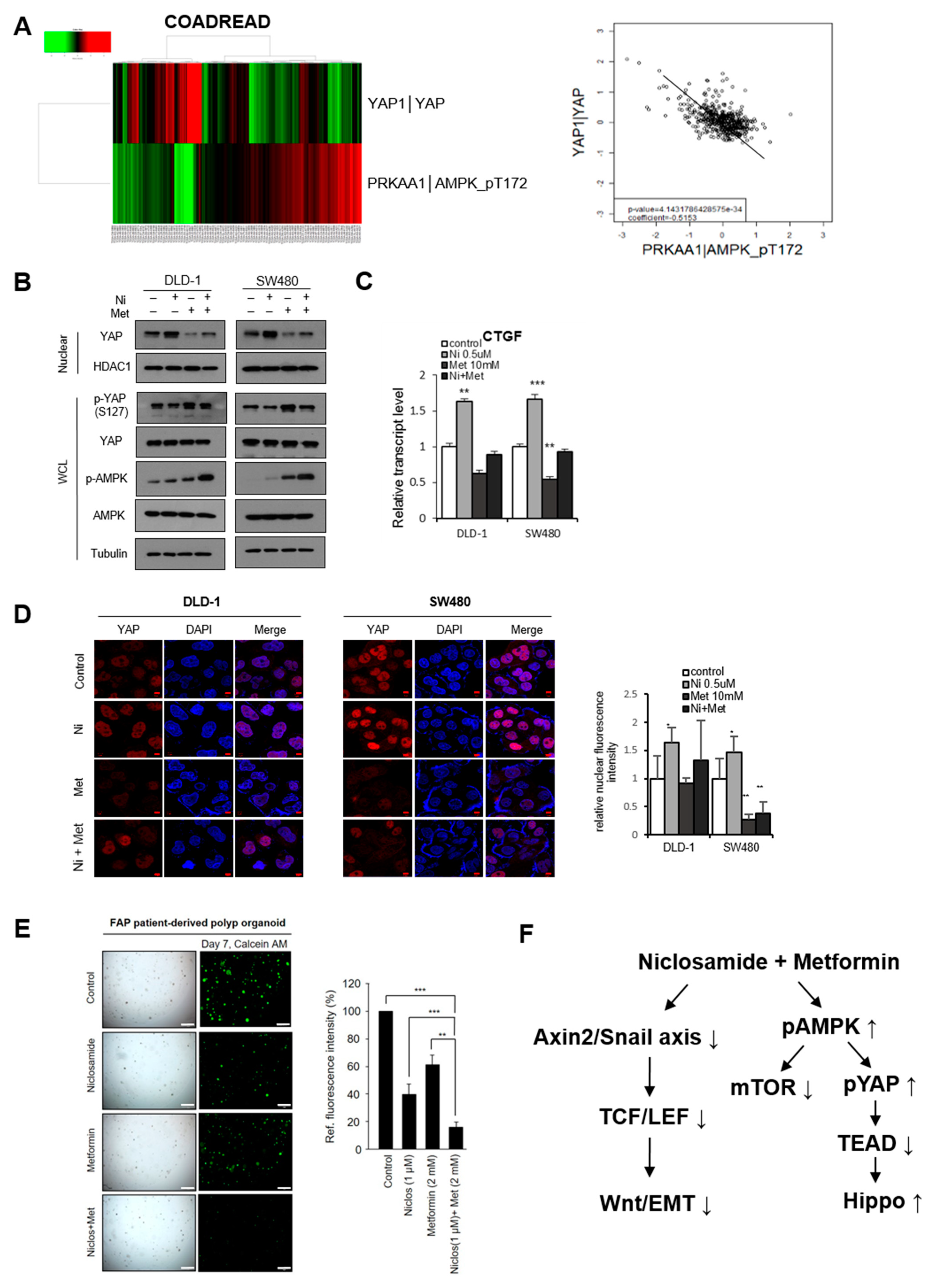
3.3. AMPK-Activator Metformin Attenuates Niclosamide-Mediated Hippo Suppression
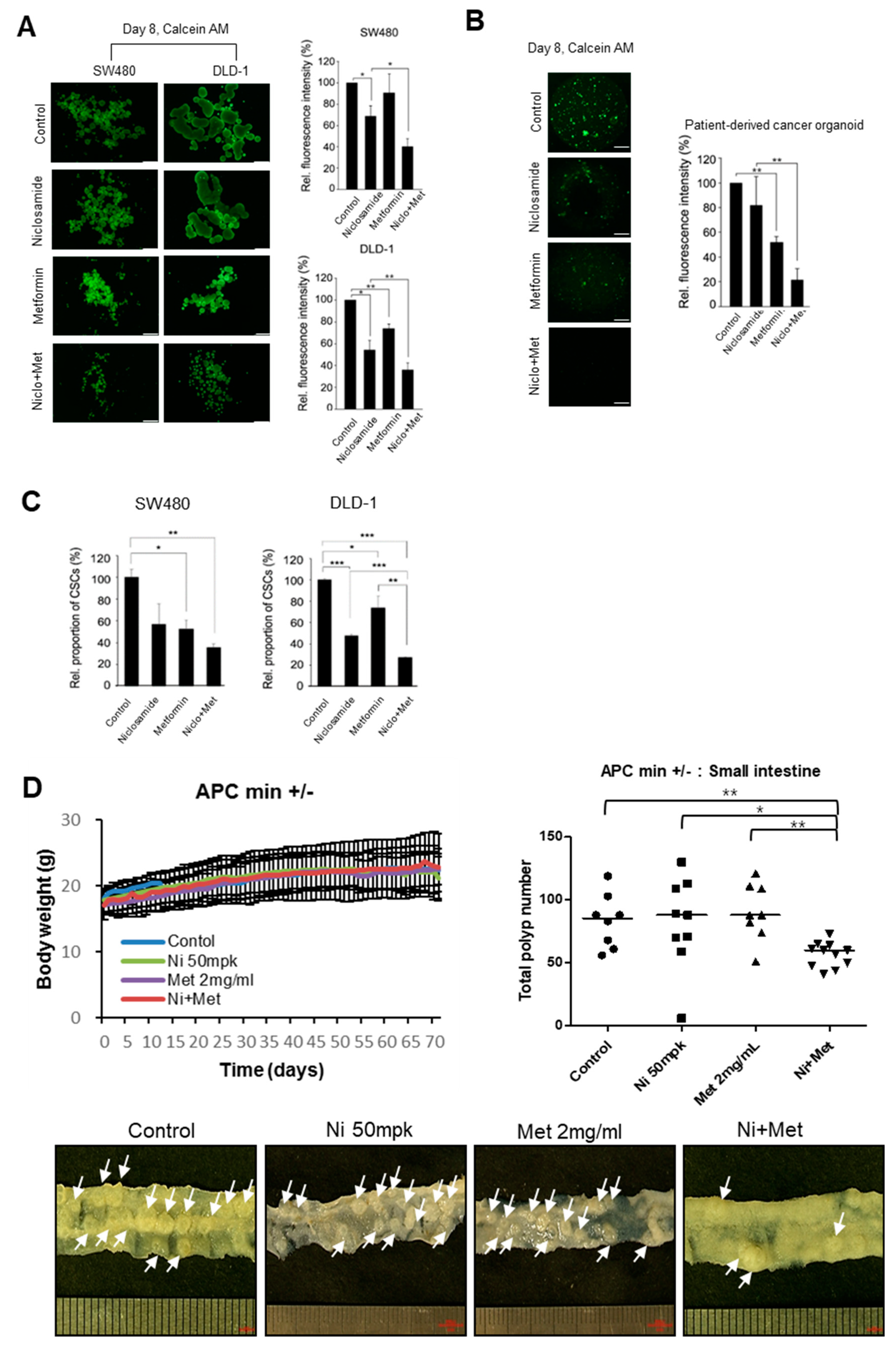
3.4. The Combination of Niclosamide and Metformin Suppresses Tumor Organoids and the In Vivo Tumor Potential of CRC
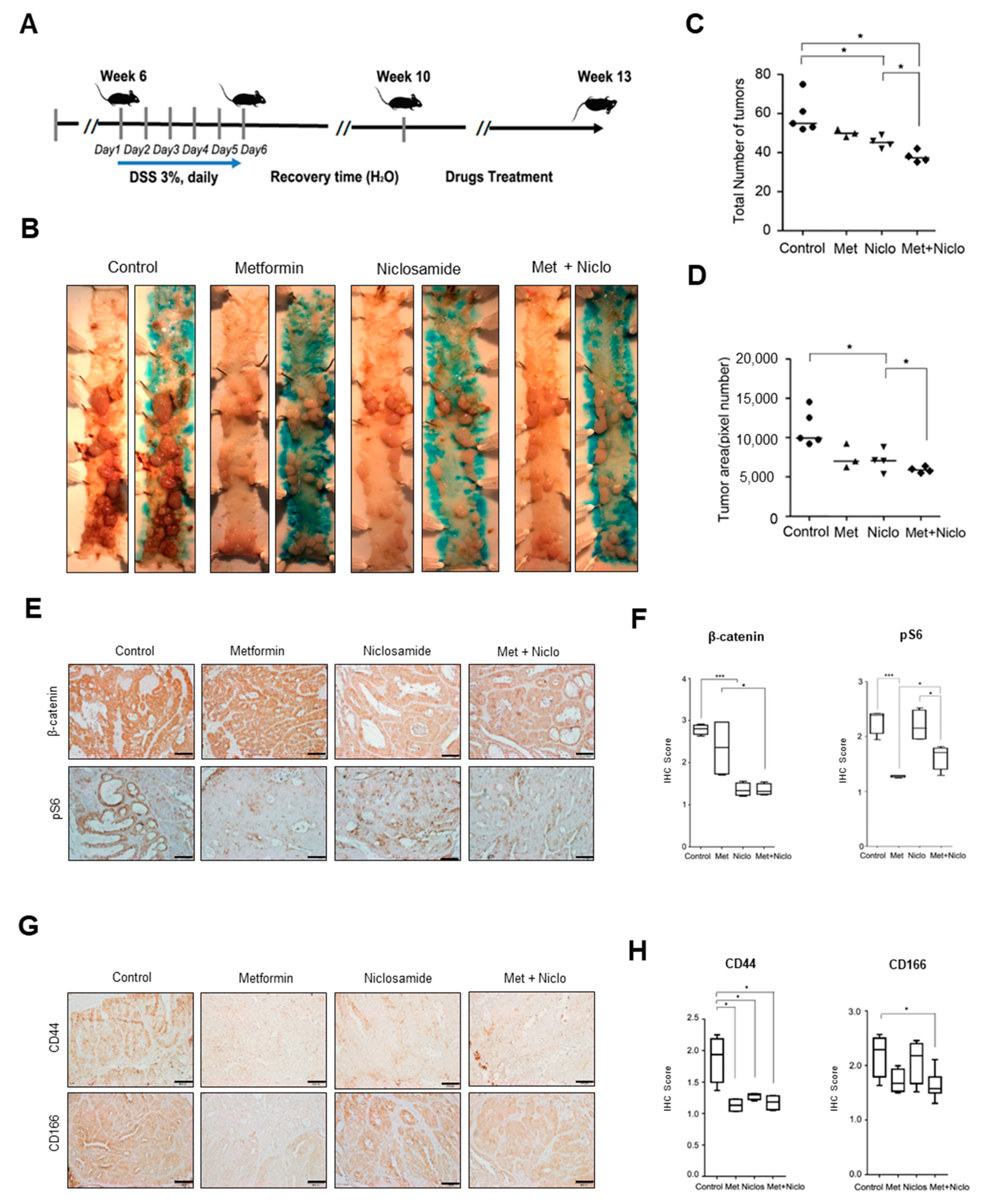
3.5. Combination of Niclosamide and Metformin Efficiently Suppresses Cancer Progression in APC-MIN-DSS Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Wiesmann, M.; Rohan, M.; Chan, V.; Jefferson, A.B.; Guo, L.; Sakamoto, D.; Caothien, R.H.; Fuller, J.H.; Reinhard, C.; et al. Elevated expression of axin2 and hnkd mrna provides evidence that wnt/beta -catenin signaling is activated in human colon tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 14973–14978. [Google Scholar] [CrossRef] [Green Version]
- Leung, J.Y.; Kolligs, F.T.; Wu, R.; Zhai, Y.; Kuick, R.; Hanash, S.; Cho, K.R.; Fearon, E.R. Activation of axin2 expression by beta-catenin-t cell factor. A feedback repressor pathway regulating wnt signaling. J. Biol. Chem. 2002, 277, 21657–21665. [Google Scholar] [CrossRef] [Green Version]
- Lustig, B.; Jerchow, B.; Sachs, M.; Weiler, S.; Pietsch, T.; Karsten, U.; van de Wetering, M.; Clevers, H.; Schlag, P.M.; Birchmeier, W.; et al. Negative feedback loop of wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol. Cell Biol. 2002, 22, 1184–1193. [Google Scholar] [CrossRef] [Green Version]
- Yook, J.I.; Li, X.Y.; Ota, I.; Hu, C.; Kim, H.S.; Kim, N.H.; Cha, S.Y.; Ryu, J.K.; Choi, Y.J.; Kim, J.; et al. A wnt-axin2-gsk3beta cascade regulates snail1 activity in breast cancer cells. Nat. Cell Biol. 2006, 8, 1398–1406. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Brabletz, T.; Fearon, E.; Willis, A.L.; Hu, C.Y.; Li, X.Y.; Weiss, S.J. Canonical wnt suppressor, axin2, promotes colon carcinoma oncogenic activity. Proc. Natl. Acad. Sci. USA 2012, 109, 11312–11317. [Google Scholar] [CrossRef] [Green Version]
- Yochum, G.S. Axin2: Tumor suppressor, oncogene or both in colorectal cancer? J. Cancer Sci. Ther. 2012, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-J.; Jan, J.-T.; Chen, C.-M.; Hsieh, H.-P.; Hwang, D.-R.; Liu, H.-W.; Liu, C.-Y.; Huang, H.-W.; Chen, S.-C.; Hong, C.-F.; et al. Inhibition of severe acute respiratory syndrome coronavirus replication by niclosamide. Antimicrob. Agents Chemother. 2004, 48, 2693–2696. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.W.; Yap, Y.L. Old drugs as lead compounds for a new disease? Binding analysis of sars coronavirus main proteinase with hiv, psychotic and parasite drugs. Bioorganic Med. Chem. 2004, 12, 2517–2521. [Google Scholar] [CrossRef]
- Sack, U.; Walther, W.; Scudiero, D.; Selby, M.; Kobelt, D.; Lemm, M.; Fichtner, I.; Schlag, P.M.; Shoemaker, R.H.; Stein, U. Novel effect of antihelminthic niclosamide on s100a4-mediated metastatic progression in colon cancer. J. Natl. Cancer Inst. 2011, 103, 1018–1036. [Google Scholar] [CrossRef] [Green Version]
- Londoño-Joshi, A.I.; Arend, R.C.; Aristizabal, L.; Lu, W.; Samant, R.S.; Metge, B.J.; Hidalgo, B.; Grizzle, W.E.; Conner, M.; Forero-Torres, A.; et al. Effect of niclosamide on basal-like breast cancers. Mol. Cancer Ther. 2014, 13, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Arend, R.C.; Londono-Joshi, A.I.; Samant, R.S.; Li, Y.; Conner, M.; Hidalgo, B.; Alvarez, R.D.; Landen, C.N.; Straughn, J.M.; Buchsbaum, D.J. Inhibition of wnt/beta-catenin pathway by niclosamide: A therapeutic target for ovarian cancer. Gynecol. Oncol. 2014, 134, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hu, Z.; Sun, S.-Y.; Chen, Z.G.; Owonikoko, T.K.; Sica, G.L.; Ramalingam, S.S.; Curran, W.J.; Khuri, F.R.; Deng, X. Niclosamide overcomes acquired resistance to erlotinib through suppression of stat3 in non-small cell lung cancer. Mol. Cancer Ther. 2013, 12, 2200–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieland, A.; Trageser, D.; Gogolok, S.; Reinartz, R.; Höfer, H.; Keller, M.; Leinhaas, A.; Schelle, R.; Normann, S.; Klaas, L.; et al. Anticancer effects of niclosamide in human glioblastoma. Clin. Cancer Res. 2013, 19, 4124–4136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Lu, Z.; Ding, K.; Li, J.; Du, X.; Chen, C.; Sun, X.; Wu, Y.; Zhou, J.; Pan, J. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: Inactivation of the nf-κb pathway and generation of reactive oxygen species. Cancer Res. 2010, 70, 2516–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osada, T.; Chen, M.; Yang, X.Y.; Spasojevic, I.; Vandeusen, J.B.; Hsu, D.; Clary, B.M.; Clay, T.M.; Chen, W.; Morse, M.A.; et al. Antihelminth compound niclosamide downregulates wnt signaling and elicits antitumor responses in tumors with activating apc mutations. Cancer Res. 2011, 71, 4172–4182. [Google Scholar] [CrossRef] [Green Version]
- Tomizawa, M.; Shinozaki, F.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Sueishi, M.; Yoshida, T. Niclosamide suppresses hepatoma cell proliferation via the wnt pathway. Onco Targets Ther. 2013, 6, 1685–1693. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.Y.; Kim, N.H.; Lee, K.; Cha, Y.H.; Yang, J.H.; Cha, S.Y.; Cho, E.S.; Lee, Y.; Cha, J.S.; Cho, H.S.; et al. Niclosamide is a potential therapeutic for familial adenomatosis polyposis by disrupting axin-gsk3 interaction. Oncotarget 2017, 8, 31842–31855. [Google Scholar] [CrossRef] [Green Version]
- Burock, S.; Daum, S.; Keilholz, U.; Neumann, K.; Walther, W.; Stein, U. Phase ii trial to investigate the safety and efficacy of orally applied niclosamide in patients with metachronous or sychronous metastases of a colorectal cancer progressing after therapy: The nikolo trial. BMC Cancer 2018, 18, 297. [Google Scholar] [CrossRef]
- Xu, J.; Shi, P.-Y.; Li, H.; Zhou, J. Broad spectrum antiviral agent niclosamide and its therapeutic potential. ACS Infect. Dis. 2020, 6, 909–915. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, J.S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.S.; Guan, K.L. Cellular energy stress induces ampk-mediated regulation of yap and the hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hu, T.; Xu, Z.; Lin, Z.; Zhang, Z.; Feng, T.; Zhu, L.; Rong, Y.; Shen, H.; Luk, J.M.; et al. Targeting hippo pathway by specific interruption of yap-tead interaction using cyclic yap-like peptides. FASEB J. 2015, 29, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakiuchi-Kiyota, S.; Schutten, M.M.; Zhong, Y.; Crawford, J.J.; Dey, A. Safety considerations in the development of hippo pathway inhibitors in cancers. Front. Cell Dev. Biol. 2019, 7, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Kim, N.H.; Cho, E.S.; Yang, J.H.; Cha, Y.H.; Kang, H.E.; Yun, J.S.; Cho, S.B.; Lee, S.-H.; Paclikova, P.; et al. Dishevelled has a yap nuclear export function in a tumor suppressor context-dependent manner. Nat. Commun. 2018, 9, 2301. [Google Scholar] [CrossRef] [Green Version]
- Varghese, F.; Bukhari, A.B.; Malhotra, R.; De, A. Ihc profiler: An open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS ONE 2014, 9, e96801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Lei, Q.-Y.; Guan, K.-L. The hippo-yap pathway: New connections between regulation of organ size and cancer. Curr. Opin. Cell Biol. 2008, 20, 638–646. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.-G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Dong, X.; Mai, M.; Seelan, R.S.; Taniguchi, K.; Krishnadath, K.K.; Halling, K.C.; Cunningham, J.M.; Boardman, L.A.; Qian, C.; et al. Mutations in axin2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/tcf signalling. Nat. Genet. 2000, 26, 146–147. [Google Scholar] [CrossRef]
- Aylon, Y.; Michael, D.; Shmueli, A.; Yabuta, N.; Nojima, H.; Oren, M. A positive feedback loop between the p53 and lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006, 20, 2687–2700. [Google Scholar] [CrossRef] [Green Version]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of yap and taz in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. Ampk modulates hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Gadalla, A.E.; Olsen, G.S.; Hardie, D.G. The antidiabetic drug metformin activates the amp-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 2002, 51, 2420–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindupur, S.K.; González, A.; Hall, M.N. The opposing actions of target of rapamycin and amp-activated protein kinase in cell growth control. Cold Spring Harb. Perspect. Biol. 2015, 7, a019141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cave, D.D.; Hernando-Momblona, X.; Sevillano, M.; Minchiotti, G.; Lonardo, E. Nodal-induced l1cam/cxcr4 subpopulation sustains tumor growth and metastasis in colorectal cancer derived organoids. Theranostics 2021, 11, 5686–5699. [Google Scholar] [CrossRef]
- Pearson, R.D.; Hewlett, E.L. Niclosamide therapy for tapeworm infections. Ann. Intern. Med. 1985, 102, 550–551. [Google Scholar] [CrossRef]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, X.; Xu, H.; Shi, X.; Zhao, J.; Yang, M.; Zhang, L.; Jin, X.; Hu, Y.; Li, X.; et al. Niclosamide inhibits cell growth and enhances drug sensitivity of hepatocellular carcinoma cells via stat3 signaling pathway. J. Cancer 2018, 9, 4150–4155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, P.K.; Roberts, M.J.; Arend, R.C.; Samant, R.S.; Buchsbaum, D.J. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer Lett. 2014, 349, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arend, R.C.; Londono-Joshi, A.I.; Gangrade, A.; Katre, A.A.; Kurpad, C.; Li, Y.; Samant, R.S.; Li, P.K.; Landen, C.N.; Yang, E.S.; et al. Niclosamide and its analogs are potent inhibitors of wnt/beta-catenin, mtor and stat3 signaling in ovarian cancer. Oncotarget 2016, 7, 86803–86815. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, Q.; Wang, G.; Huang, Y.; Mao, X.; Zhang, Y.; Wang, X. Inhibition of wnt/beta-catenin by anthelmintic drug niclosamide effectively targets growth, survival, and angiogenesis of retinoblastoma. Am. J. Transl. Res. 2017, 9, 3776–3786. [Google Scholar]
- Lu, W.; Lin, C.; Roberts, M.J.; Waud, W.R.; Piazza, G.A.; Li, Y. Niclosamide suppresses cancer cell growth by inducing wnt co-receptor lrp6 degradation and inhibiting the wnt/beta-catenin pathway. PLoS ONE 2011, 6, e29290. [Google Scholar] [CrossRef]
- Wang, L.; Shi, S.; Guo, Z.; Zhang, X.; Han, S.; Yang, A.; Wen, W.; Zhu, Q. Overexpression of yap and taz is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS ONE 2013, 8, e65539. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dong, Q.; Zhang, Q.; Li, Z.; Wang, E.; Qiu, X. Overexpression of yes-associated protein contributes to progression and poor prognosis of non-small-cell lung cancer. Cancer Sci. 2010, 101, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, N.; Zheng, Y.; de Wilde, R.F.; Maitra, A.; Pan, D. The hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 2010, 24, 2383–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Su, L.; Ou, Q. Yes-associated protein promotes tumour development in luminal epithelial derived breast cancer. Eur. J. Cancer 2012, 48, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Maitra, A.; Anders, R.A.; Taketo, M.M.; Pan, D. Beta-catenin destruction complex-independent regulation of hippo-yap signaling by apc in intestinal tumorigenesis. Genes Dev. 2015, 29, 1493–1506. [Google Scholar] [CrossRef] [Green Version]
- Bradley, M.C.; Ferrara, A.; Achacoso, N.; Ehrlich, S.F.; Quesenberry, C.P., Jr.; Habel, L.A. A cohort study of metformin and colorectal cancer risk among patients with diabetes mellitus. Cancer Epidemiol. Biomark. Prev. 2018, 27, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Marks, A.R.; Pietrofesa, R.A.; Jensen, C.D.; Zebrowski, A.; Corley, D.A.; Doubeni, C.A. Metformin use and risk of colorectal adenoma after polypectomy in patients with type 2 diabetes mellitus. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1692–1698. [Google Scholar] [CrossRef] [Green Version]
- Tomimoto, A.; Endo, H.; Sugiyama, M.; Fujisawa, T.; Hosono, K.; Takahashi, H.; Nakajima, N.; Nagashima, Y.; Wada, K.; Nakagama, H.; et al. Metformin suppresses intestinal polyp growth in apcmin/+ mice. Cancer Sci. 2008, 99, 2136–2141. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Z.; Hu, L. The regulatory effects of metformin on the [snail/mir-34]:[zeb/mir-200] system in the epithelial-mesenchymal transition(emt) for colorectal cancer(crc). Eur. J. Pharmacol. 2018, 834, 45–53. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H.E.; Seo, Y.; Yun, J.S.; Song, S.H.; Han, D.; Cho, E.S.; Cho, S.B.; Jeon, Y.; Lee, H.; Kim, H.S.; et al. Metformin and Niclosamide Synergistically Suppress Wnt and YAP in APC-Mutated Colorectal Cancer. Cancers 2021, 13, 3437. https://doi.org/10.3390/cancers13143437
Kang HE, Seo Y, Yun JS, Song SH, Han D, Cho ES, Cho SB, Jeon Y, Lee H, Kim HS, et al. Metformin and Niclosamide Synergistically Suppress Wnt and YAP in APC-Mutated Colorectal Cancer. Cancers. 2021; 13(14):3437. https://doi.org/10.3390/cancers13143437
Chicago/Turabian StyleKang, Hee Eun, Yoojeong Seo, Jun Seop Yun, Sang Hyun Song, Dawool Han, Eunae Sandra Cho, Sue Bean Cho, Yoon Jeon, Ho Lee, Hyun Sil Kim, and et al. 2021. "Metformin and Niclosamide Synergistically Suppress Wnt and YAP in APC-Mutated Colorectal Cancer" Cancers 13, no. 14: 3437. https://doi.org/10.3390/cancers13143437