
Intersection of Two Checkpoints: Could Inhibiting the DNA Damage Response Checkpoint Rescue Immune Checkpoint-Refractory Cancer?

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Immunotherapy: Progress and Problems
1.1. Clinical Efficacy of Immune Checkpoint Inhibitors
1.2. Limitations of Current Immune Checkpoint Inhibitors
1.3. New Approaches to Improve the Efficacy of ICI and Enhance Anti-Tumor Immunity
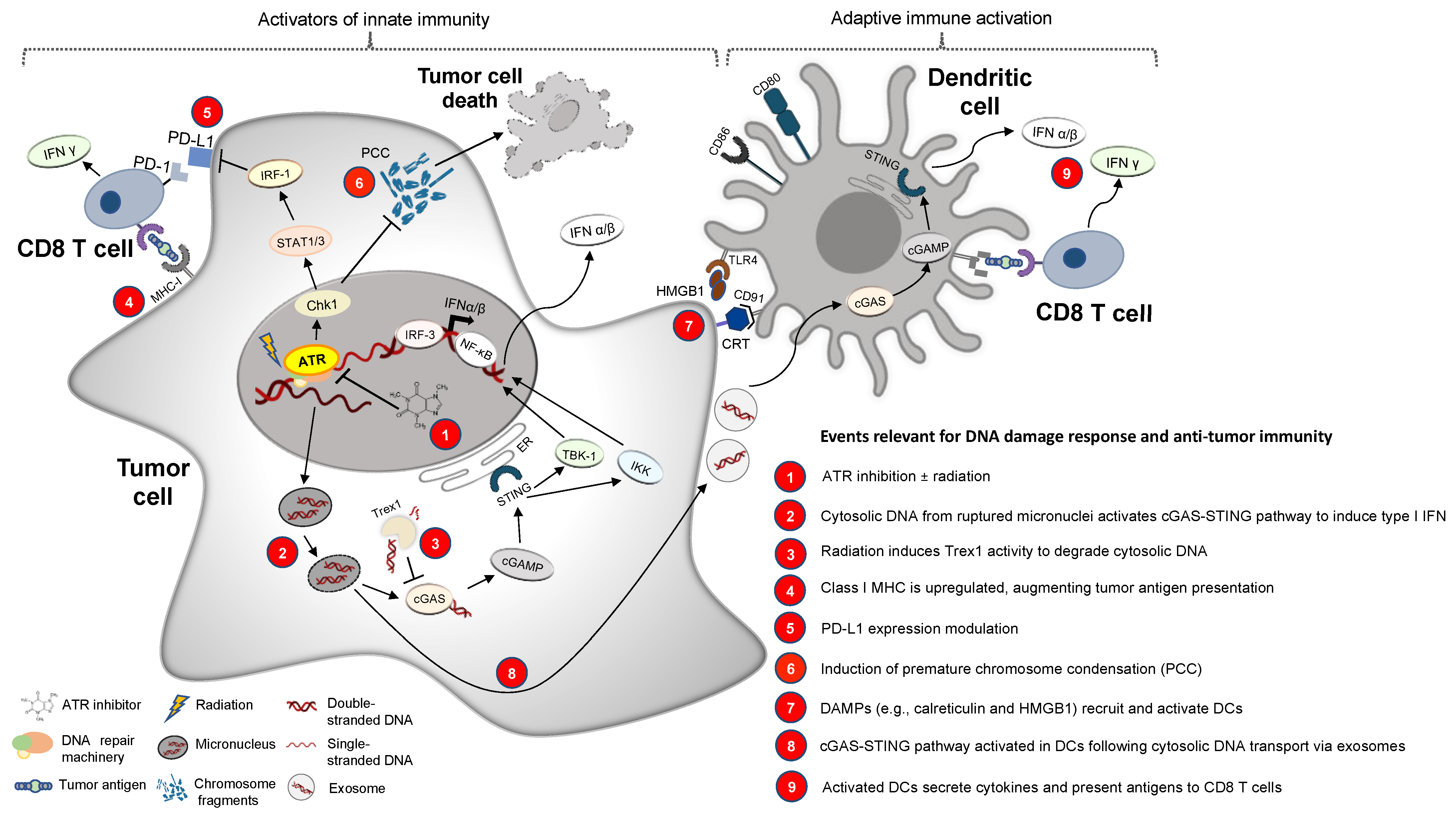
2. Crosstalk between the DNA Damage Response and Innate and Adaptive Immunity
3. Clinical Experience with DNA Damage Response Pathway Inhibitors
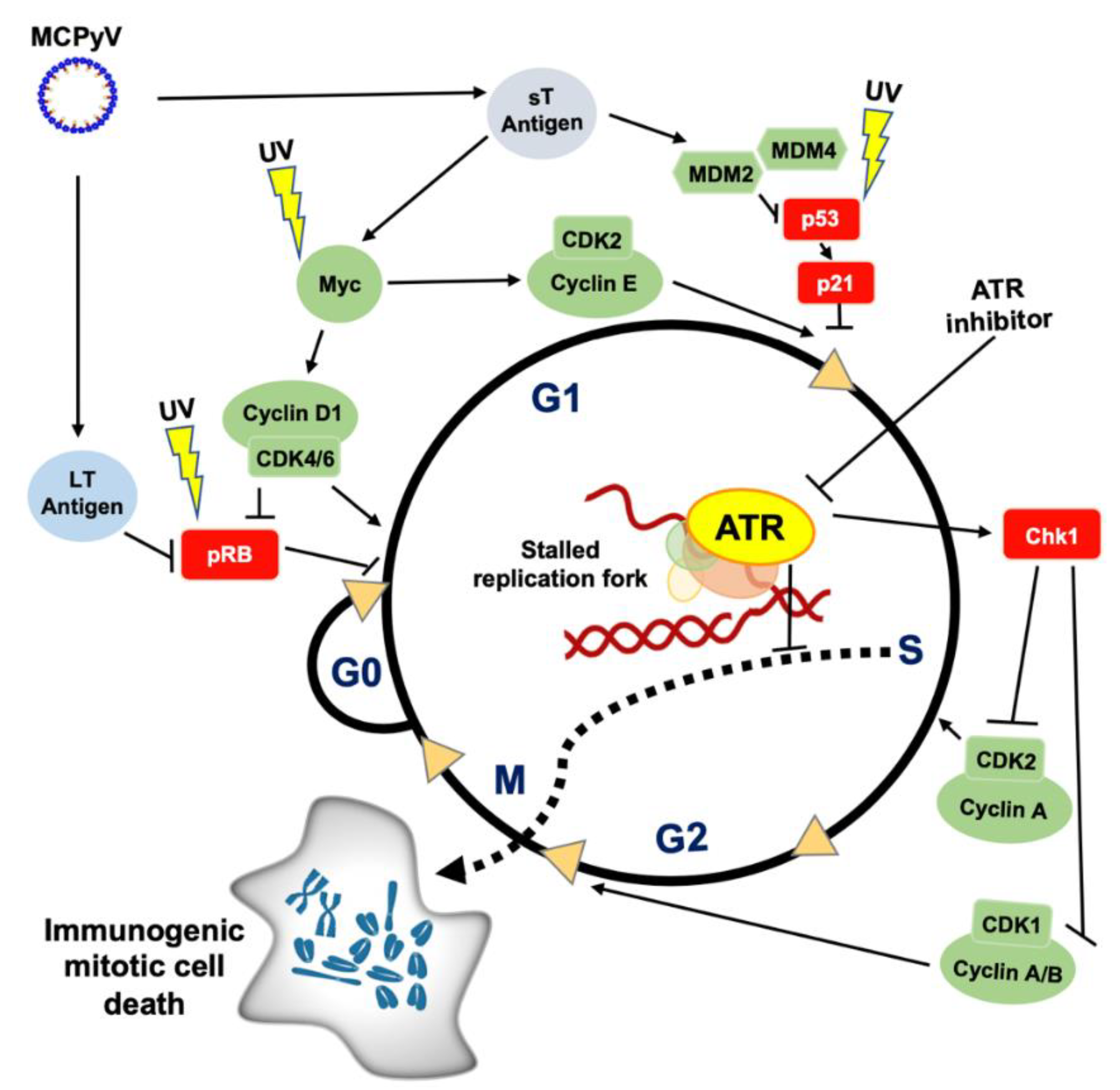
4. Rationale for “Double-Checkpoint Inhibition” in Merkel Cell Carcinoma
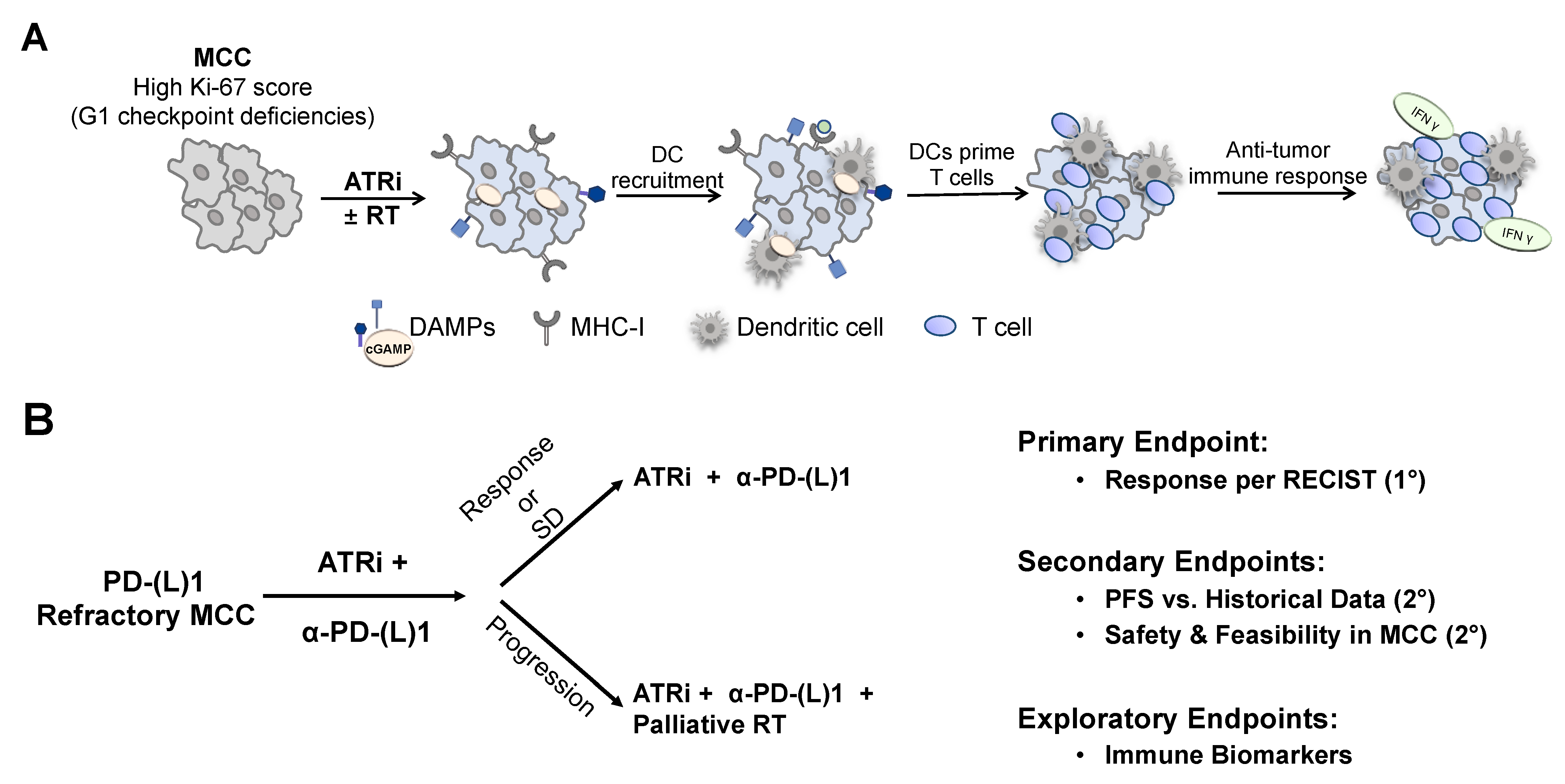
5. Clinical Trials Including Patients with Merkel Cell Carcinoma
6. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- Oaknin, A.; Tinker, A.V.; Gilbert, L.; Samouelian, V.; Mathews, C.; Brown, J.; Barretina-Ginesta, M.P.; Moreno, V.; Gravina, A.; Abdeddaim, C.; et al. Clinical Activity and Safety of the Anti-Programmed Death 1 Monoclonal Antibody Dostarlimab for Patients With Recurrent or Advanced Mismatch Repair-Deficient Endometrial Cancer: A Nonrandomized Phase 1 Clinical Trial. JAMA Oncol. 2020, 6, 1766–1772. [Google Scholar] [CrossRef] [PubMed]
- Galienne, M.; Rodrigues, M. New drug approval: Dostarlimab-second line in advanced MSI endometrial cancer. Bull. Cancer 2021, S0007-4551. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- La-Beck, N.M.; Jean, G.W.; Huynh, C.; Alzghari, S.K.; Lowe, D.B. Immune Checkpoint Inhibitors: New Insights and Current Place in Cancer Therapy. Pharmacotherapy 2015, 35, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.J.; Carlino, M.S. Immune Checkpoint Inhibitor Toxicity. Curr. Oncol. Rep. 2018, 20, 72. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Haslam, A.; Prasad, V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Zhao, B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: Meta-analysis. BMJ 2018, 362, k3529. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.K.; Man, J.; Lord, S.; Cooper, W.; Links, M.; Gebski, V.; Herbst, R.S.; Gralla, R.J.; Mok, T.; Yang, J.C. Clinical and Molecular Characteristics Associated With Survival Among Patients Treated With Checkpoint Inhibitors for Advanced Non-Small Cell Lung Carcinoma: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 210–216. [Google Scholar] [CrossRef]
- Yun, S.; Vincelette, N.D.; Green, M.R.; Wahner Hendrickson, A.E.; Abraham, I. Targeting immune checkpoints in unresectable metastatic cutaneous melanoma: A systematic review and meta-analysis of anti-CTLA-4 and anti-PD-1 agents trials. Cancer Med. 2016, 5, 1481–1491. [Google Scholar] [CrossRef]
- Yang, F.; Markovic, S.N.; Molina, J.R.; Halfdanarson, T.R.; Pagliaro, L.C.; Chintakuntlawar, A.V.; Li, R.; Wei, J.; Wang, L.; Liu, B.; et al. Association of Sex, Age, and Eastern Cooperative Oncology Group Performance Status With Survival Benefit of Cancer Immunotherapy in Randomized Clinical Trials: A Systematic Review and Meta-analysis. JAMA Netw. Open 2020, 3, e2012534. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.G.; Lahman, M.C.; Chapuis, A.G.; Brownell, I. Immunotherapy for skin cancer. Int. Immunol. 2019, 31, 465–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazarika, M.; Chuk, M.K.; Theoret, M.R.; Mushti, S.; He, K.; Weis, S.L.; Putman, A.H.; Helms, W.S.; Cao, X.; Li, H.; et al. U.S. FDA Approval Summary: Nivolumab for Treatment of Unresectable or Metastatic Melanoma Following Progression on Ipilimumab. Clin. Cancer Res. 2017, 23, 3484–3488. [Google Scholar] [CrossRef] [Green Version]
- Barone, A.; Hazarika, M.; Theoret, M.R.; Mishra-Kalyani, P.; Chen, H.; He, K.; Sridhara, R.; Subramaniam, S.; Pfuma, E.; Wang, Y.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Patients with Unresectable or Metastatic Melanoma. Clin. Cancer Res. 2017, 23, 5661–5665. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Lebbé, C.; Meyer, N.; Mortier, L.; Marquez-Rodas, I.; Robert, C.; Rutkowski, P.; Menzies, A.M.; Eigentler, T.; Ascierto, P.A.; Smylie, M.; et al. Evaluation of Two Dosing Regimens for Nivolumab in Combination With Ipilimumab in Patients With Advanced Melanoma: Results From the Phase IIIb/IV CheckMate 511 Trial. J. Clin. Oncol.. 2019, 37, 867–875. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.S.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbe, C.; Milella, M.; Brownell, I.; et al. Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after >/=1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J. Immunother. Cancer 2018, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, S.P.; Bhatia, S.; Brohl, A.S.; Hamid, O.; Mehnert, J.M.; Terheyden, P.; Shih, K.C.; Brownell, I.; Lebbe, C.; Lewis, K.D.; et al. Avelumab in patients with previously treated metastatic Merkel cell carcinoma: Long-term data and biomarker analyses from the single-arm phase 2 JAVELIN Merkel 200 trial. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Durable Tumor Regression and Overall Survival in Patients With Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy. J. Clin. Oncol. 2019, 37, 693–702. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Cemiplimab in locally advanced basal cell carcinoma after hedgehog inhibitor therapy: An open-label, multi-centre, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 848–857. [CrossRef]
- Liu, D.; Jenkins, R.W.; Sullivan, R.J. Mechanisms of Resistance to Immune Checkpoint Blockade. Am. J. Clin. Dermatol. 2019, 20, 41–54. [Google Scholar] [CrossRef]
- Valero, C.; Lee, M.; Hoen, D.; Weiss, K.; Kelly, D.W.; Adusumilli, P.S.; Paik, P.K.; Plitas, G.; Ladanyi, M.; Postow, M.A.; et al. Pretreatment neutrophil-to-lymphocyte ratio and mutational burden as biomarkers of tumor response to immune checkpoint inhibitors. Nat. Commun. 2021, 12, 729. [Google Scholar] [CrossRef] [PubMed]
- Rogado, J.; Romero-Laorden, N.; Sanchez-Torres, J.M.; Ramos-Levi, A.M.; Pacheco-Barcia, V.; Ballesteros, A.I.; Arranz, R.; Lorenzo, A.; Gullon, P.; Garrido, A.; et al. Effect of excess weight and immune-related adverse events on the efficacy of cancer immunotherapy with anti-PD-1 antibodies. Oncoimmunology 2020, 9, 1751548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Y.; Wu, Z.; Wang, N.; Yang, Z.; Li, Y.; Xu, B.; Sun, M. Association between body mass index and survival outcomes for cancer patients treated with immune checkpoint inhibitors: A systematic review and meta-analysis. J. Transl. Med. 2020, 18, 235. [Google Scholar] [CrossRef]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef]
- Subbiah, V.; Solit, D.B.; Chan, T.A.; Kurzrock, R. The FDA approval of pembrolizumab for adult and pediatric patients with tumor mutational burden (TMB) >/=10: A decision centered on empowering patients and their physicians. Ann. Oncol. 2020, 31, 1115–1118. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Kato, K.; Sun, J.-M.; Shah, M.A.; Enzinger, P.C.; Adenis, A.; Doi, T.; Kojima, T.; Metges, J.-P.; Li, Z.; Kim, S.-B.; et al. LBA8_PR Pembrolizumab plus chemotherapy versus chemotherapy as first-line therapy in patients with advanced esophageal cancer: The phase 3 KEYNOTE-590 study. Ann. Oncol. 2020, 31, S1192–S1193. [Google Scholar] [CrossRef]
- Dosset, M.; Joseph, E.L.; Rivera Vargas, T.; Apetoh, L. Modulation of Determinant Factors to Improve Therapeutic Combinations with Immune Checkpoint Inhibitors. Cells 2020, 9, 1727. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Waxman, D.J. Immunogenic chemotherapy: Dose and schedule dependence and combination with immunotherapy. Cancer Lett. 2018, 419, 210–221. [Google Scholar] [CrossRef]
- Formenti, S.C.; Rudqvist, N.P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef]
- Bang, A.; Schoenfeld, J.D. Immunotherapy and radiotherapy for metastatic cancers. Ann. Palliat. Med. 2019, 8, 312–325. [Google Scholar] [CrossRef]
- Kordbacheh, T.; Honeychurch, J.; Blackhall, F.; Faivre-Finn, C.; Illidge, T. Radiotherapy and anti-PD-1/PD-L1 combinations in lung cancer: Building better translational research platforms. Ann. Oncol. 2018, 29, 301–310. [Google Scholar] [CrossRef]
- Demaria, S.; Guha, C.; Schoenfeld, J.; Morris, Z.; Monjazeb, A.; Sikora, A.; Crittenden, M.; Shiao, S.; Khleif, S.; Gupta, S.; et al. Radiation dose and fraction in immunotherapy: One-size regimen does not fit all settings, so how does one choose? J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Shaverdian, N.; Lisberg, A.E.; Bornazyan, K.; Veruttipong, D.; Goldman, J.W.; Formenti, S.C.; Garon, E.B.; Lee, P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: A secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017, 18, 895–903. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, W.; Li, N.; Neri, S.; Sharma, A.; Jiang, W.; Lin, S.H. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front. Pharmacol. 2018, 9, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Le, T.Q.; Massarelli, E.; Hendifar, A.E.; Tuli, R. Radiation therapy and PD-1/PD-L1 blockade: The clinical development of an evolving anticancer combination. J. Immunother. Cancer 2018, 6, 46. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Kiess, A.P. Time to Debunk an Urban Myth? The “Abscopal Effect” With Radiation and Anti–PD-1. J. Clin. Oncol. 2021, 39, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Wong, J.S.L.; Kwok, G.G.W.; Tang, V.; Li, B.C.W.; Leung, R.; Chiu, J.; Ma, K.W.; She, W.H.; Tsang, J.; Lo, C.M.; et al. Ipilimumab and nivolumab/pembrolizumab in advanced hepatocellular carcinoma refractory to prior immune checkpoint inhibitors. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Geraud, A.; Gougis, P.; Vozy, A.; Anquetil, C.; Allenbach, Y.; Romano, E.; Funck-Brentano, E.; Moslehi, J.J.; Johnson, D.B.; Salem, J.E. Clinical Pharmacology and Interplay of Immune Checkpoint Agents: A Yin-Yang Balance. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 85–112. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2018, 174, 1031–1032. [Google Scholar] [CrossRef]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Quhal, F.; Mori, K.; Bruchbacher, A.; Resch, I.; Mostafaei, H.; Pradere, B.; Schuettfort, V.M.; Laukhtina, E.; Egawa, S.; Fajkovic, H.; et al. First-line Immunotherapy-based Combinations for Metastatic Renal Cell Carcinoma: A Systematic Review and Network Meta-analysis. Eur. Urol. Oncol. 2021. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Sznol, M.; Hu-Lieskovan, S.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Di Gravio, D.; Huang, B.; Gambhire, D.; Chen, Y.; et al. Phase Ib Study of Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5349–5357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.K.; Konstantinopoulos, P.A. Combined PARP and Immune Checkpoint Inhibition in Ovarian Cancer. Trends. Cancer 2019, 5, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Post, C.C.B.; Westermann, A.M.; Bosse, T.; Creutzberg, C.L.; Kroep, J.R. PARP and PD-1/PD-L1 checkpoint inhibition in recurrent or metastatic endometrial cancer. Crit. Rev. Oncol. Hematol. 2020, 152, 102973. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, A.; Mezni, E.; Bertucci, F. Combining poly(ADP-ribose) polymerase inhibitors and immune checkpoint inhibitors in breast cancer: Rationale and preliminary clinical results. Curr. Opin. Oncol. 2020, 32, 585–593. [Google Scholar] [CrossRef]
- Paluch-Shimon, S.; Cardoso, F. PARP inhibitors coming of age. Nat. Rev. Clin. Oncol. 2021, 18, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Lanz, M.C.; Dibitetto, D.; Smolka, M.B. DNA damage kinase signaling: Checkpoint and repair at 30 years. EMBO J. 2019, 38, e101801. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Klingseisen, A.; Jackson, A.P. Mechanisms and pathways of growth failure in primordial dwarfism. Genes Dev. 2011, 25, 2011–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Jerzak, K.J.; Mancuso, T.; Eisen, A. Ataxia-telangiectasia gene (ATM) mutation heterozygosity in breast cancer: A narrative review. Curr. Oncol. 2018, 25, e176–e180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Os, N.J.H.; Roeleveld, N.; Weemaes, C.M.; Jongmans, M.C.; Janssens, G.O.; Taylor, A.M.; Hoogerbrugge, N.; Willemsen, M.A. Health risks for ataxia-telangiectasia mutated heterozygotes: A systematic review, meta-analysis and evidence-based guideline. Clin. Genet. 2016, 90, 105–117. [Google Scholar] [CrossRef]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef] [Green Version]
- van der Burg, M.; van Dongen, J.J.; van Gent, D.C. DNA-PKcs deficiency in human: Long predicted, finally found. Curr. Opin. Allergy Clin. Immunol. 2009, 9, 503–509. [Google Scholar] [CrossRef]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Barlow, C.; Hirotsune, S.; Paylor, R.; Liyanage, M.E.M.; Collins, F.; Shiloh, Y.; Crawley, J.N.; Ried, T.; Tagle, D.e.a. Atm-Deficient Mice: A Paradigm of Ataxia Telangiectasia. Cell 1996, 86, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Espejel, S.; Martin, M.; Klatt, P.; Martin-Caballero, J.; Flores, J.M.; Blasco, M.A. Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice. EMBO Rep. 2004, 5, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, K.A.; Mullany, S.; Thomas, B.; Chien, J.; Loewen, R.; Shridhar, V.; Cliby, W.A. Heterozygous ATR mutations in mismatch repair-deficient cancer cells have functional significance. Cancer Res. 2005, 65, 7091–7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dylgjeri, E.; McNair, C.; Goodwin, J.F.; Raymon, H.K.; McCue, P.A.; Shafi, A.A.; Leiby, B.E.; de Leeuw, R.; Kothari, V.; McCann, J.J.; et al. Pleiotropic Impact of DNA-PK in Cancer and Implications for Therapeutic Strategies. Clin. Cancer Res. 2019, 25, 5623–5637. [Google Scholar] [CrossRef]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef]
- Brandsma, I.; Fleuren, E.D.G.; Williamson, C.T.; Lord, C.J. Directing the use of DDR kinase inhibitors in cancer treatment. Expert. Opin. Investig. Drugs 2017, 26, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Minchom, A.; Aversa, C.; Lopez, J. Dancing with the DNA damage response: Next-generation anti-cancer therapeutic strategies. Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- Tu, X.; Kahila, M.M.; Zhou, Q.; Yu, J.; Kalari, K.R.; Wang, L.; Harmsen, W.S.; Yuan, J.; Boughey, J.C.; Goetz, M.P.; et al. ATR Inhibition Is a Promising Radiosensitizing Strategy for Triple-Negative Breast Cancer. Mol. Cancer Ther. 2018, 17, 2462–2472. [Google Scholar] [CrossRef] [Green Version]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [Green Version]
- Dillon, M.T.; Boylan, Z.; Smith, D.; Guevara, J.; Mohammed, K.; Peckitt, C.; Saunders, M.; Banerji, U.; Clack, G.; Smith, S.A.; et al. PATRIOT: A phase I study to assess the tolerability, safety and biological effects of a specific ataxia telangiectasia and Rad3-related (ATR) inhibitor (AZD6738) as a single agent and in combination with palliative radiation therapy in patients with solid tumours. Clin. Transl. Radiat. Oncol. 2018, 12, 16–20. [Google Scholar] [CrossRef]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Pires, I.M.; Olcina, M.M.; Anbalagan, S.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; McKenna, W.G.; Hammond, E.M. Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br. J. Cancer 2012, 107, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunne, V.; Ghita, M.; Small, D.M.; Coffey, C.B.M.; Weldon, S.; Taggart, C.C.; Osman, S.O.; McGarry, C.K.; Prise, K.M.; Hanna, G.G.; et al. Inhibition of ataxia telangiectasia related-3 (ATR) improves therapeutic index in preclinical models of non-small cell lung cancer (NSCLC) radiotherapy. Radiother. Oncol. 2017, 124, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.L.; et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciszewski, W.M.; Tavecchio, M.; Dastych, J.; Curtin, N.J. DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res. Treat. 2014, 143, 47–55. [Google Scholar] [CrossRef]
- Timme, C.R.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The DNA-PK Inhibitor VX-984 Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and as Orthotopic Xenografts. Mol. Cancer Ther. 2018, 17, 1207–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilger, D.; Seymour, L.W.; Jackson, S.P. Interfaces between cellular responses to DNA damage and cancer immunotherapy. Genes Dev. 2021, 35, 602–618. [Google Scholar] [CrossRef] [PubMed]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [Green Version]
- Chao, H.H.; Karagounis, I.V.; Thomas, C.; Francois, N.B.; Facciabene, A.; Koumenis, C.; Maity, A. Combination of CHEK1/2 inhibition and ionizing radiation results in abscopal tumor response through increased micronuclei formation. Oncogene 2020, 39, 4344–4357. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.T.E.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Tubbs, A.; Zhang, C.; Tang, M.; Sridharan, S.; Wang, C.; Jiang, D.; Su, D.; Zhang, H.; Chen, Z.; et al. ATR inhibition potentiates ionizing radiation-induced interferon response via cytosolic nucleic acid-sensing pathways. EMBO J. 2020, 39, e104036. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.T.; Barker, H.E.; Pedersen, M.; Hafsi, H.; Bhide, S.A.; Newbold, K.L.; Nutting, C.M.; McLaughlin, M.; Harrington, K.J. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol. Cancer Ther. 2017, 16, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Hartlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kroger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Ma, F.; Herrup, K. Accumulation of Cytoplasmic DNA Due to ATM Deficiency Activates the Microglial Viral Response System with Neurotoxic Consequences. J. Neurosci. 2019, 39, 6378–6394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Green, M.D.; Lang, X.; Lazarus, J.; Parsels, J.D.; Wei, S.; Parsels, L.A.; Shi, J.; Ramnath, N.; Wahl, D.R.; et al. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res. 2019, 79, 3940–3951. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, L.; Wang, C.; Zhao, W.; Ju, Z.; Zhang, W.; Shen, J.; Peng, Y.; An, C.; Luu, Y.T.; et al. Inhibition of the ATM/Chk2 axis promotes cGAS/STING signaling in ARID1A-deficient tumors. J. Clin. Investig. 2020, 130, 5951–5966. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, T.; Zhao, J.; Xia, H.; Xie, J.; Guo, Y.; Zhong, L.; Li, M.; Yang, Q.; Peng, C.; et al. DNA-PK deficiency potentiates cGAS-mediated antiviral innate immunity. Nat. Commun. 2020, 11, 6182. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Yang, Y.G.; Lindahl, T.; Barnes, D.E. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 2007, 131, 873–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krushkal, J.; Silvers, T.; Reinhold, W.C.; Sonkin, D.; Vural, S.; Connelly, J.; Varma, S.; Meltzer, P.S.; Kunkel, M.; Rapisarda, A.; et al. Epigenome-wide DNA methylation analysis of small cell lung cancer cell lines suggests potential chemotherapy targets. Clin. Epigenetics 2020, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- Zebertavage, L.K.; Alice, A.; Crittenden, M.R.; Gough, M.J. Transcriptional Upregulation of NLRC5 by Radiation Drives STING- and Interferon-Independent MHC-I Expression on Cancer Cells and T Cell Cytotoxicity. Sci. Rep. 2020, 10, 7376. [Google Scholar] [CrossRef]
- Sharma, A.; Bode, B.; Wenger, R.H.; Lehmann, K.; Sartori, A.A.; Moch, H.; Knuth, A.; Boehmer, L.; Broek, M. gamma-Radiation promotes immunological recognition of cancer cells through increased expression of cancer-testis antigens in vitro and in vivo. PLoS ONE 2011, 6, e28217. [Google Scholar] [CrossRef]
- Wan, S.; Pestka, S.; Jubin, R.G.; Lyu, Y.L.; Tsai, Y.C.; Liu, L.F. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS ONE 2012, 7, e32542. [Google Scholar] [CrossRef]
- Tsai, A.K.; Khan, A.Y.; Worgo, C.E.; Wang, L.L.; Liang, Y.; Davila, E. A Multikinase and DNA-PK Inhibitor Combination Immunomodulates Melanomas, Suppresses Tumor Progression, and Enhances Immunotherapies. Cancer Immunol. Res. 2017, 5, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.L.; Yang, R.Y.; Li, C.W.; Chen, M.K.; Shao, B.; Hsu, J.M.; Chan, L.C.; Yang, Y.; Hsu, J.L.; Lai, Y.J.; et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am. J. Cancer Res. 2018, 8, 1307–1316. [Google Scholar]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Huang, Y.; Xiao, Y.; Zhu, Z.; Shen, M.; Zhou, P.; Guo, Z.; Wang, J.; Wang, H.; Dai, W.; et al. ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Nghiem, P.; Park, P.K.; Kim, Y.; Vaziri, C.; Schreiber, S.L. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc. Natl. Acad. Sci. USA 2001, 98, 9092–9097. [Google Scholar] [CrossRef] [Green Version]
- Rybaczek, D.; Kowalewicz-Kulbat, M. Premature chromosome condensation induced by caffeine, 2-aminopurine, staurosporine and sodium metavanadate in S-phase arrested HeLa cells is associated with a decrease in Chk1 phosphorylation, formation of phospho-H2AX and minor cytoskeletal rearrangements. Histochem. Cell Biol. 2011, 135, 263–280. [Google Scholar] [CrossRef] [Green Version]
- Zuazua-Villar, P.; Rodriguez, R.; Gagou, M.E.; Eyers, P.A.; Meuth, M. DNA replication stress in CHK1-depleted tumour cells triggers premature (S-phase) mitosis through inappropriate activation of Aurora kinase B. Cell Death Dis. 2014, 5, e1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Harding, S.M.; Natesan, R.; Tian, L.; Benci, J.L.; Li, W.; Minn, A.J.; Asangani, I.A.; Greenberg, R.A. Cell Cycle Checkpoints Cooperate to Suppress DNA- and RNA-Associated Molecular Pattern Recognition and Anti-Tumor Immune Responses. Cell Rep. 2020, 32, 108080. [Google Scholar] [CrossRef]
- Kiraly, G.; Simonyi, A.S.; Turani, M.; Juhasz, I.; Nagy, G.; Banfalvi, G. Micronucleus formation during chromatin condensation and under apoptotic conditions. Apoptosis 2017, 22, 207–219. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Combes, E.; Andrade, A.F.; Tosi, D.; Michaud, H.A.; Coquel, F.; Garambois, V.; Desigaud, D.; Jarlier, M.; Coquelle, A.; Pasero, P.; et al. Inhibition of Ataxia-Telangiectasia Mutated and RAD3-Related (ATR) Overcomes Oxaliplatin Resistance and Promotes Antitumor Immunity in Colorectal Cancer. Cancer Res. 2019, 79, 2933–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.H.; Wu, Z.Q.; Qian, D.; Zaorsky, N.G.; Qiu, M.H.; Cheng, J.J.; Jiang, C.; Wang, J.; Zeng, X.L.; Liu, C.L.; et al. Ablative Hypofractionated Radiation Therapy Enhances Non-Small Cell Lung Cancer Cell Killing via Preferential Stimulation of Necroptosis In Vitro and In Vivo. Int. J. Radiat. Oncol. Biol. Phys. 2018, 101, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Moriya, T.; Kitagawa, K.; Hayakawa, Y.; Hemmi, H.; Kaisho, T.; Ueha, S.; Ikebuchi, R.; Yasuda, I.; Nakanishi, Y.; Honda, T.; et al. Immunogenic tumor cell death promotes dendritic cell migration and inhibits tumor growth via enhanced T cell immunity. iScience 2021, 24. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from Irradiated Cancer Cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Yokoi, A.; Villar-Prados, A.; Oliphint, P.A.; Zhang, J.; Song, X.; De Hoff, P.; Morey, R.; Liu, J.; Roszik, J.; Clise-Dwyer, K.; et al. Mechanisms of nuclear content loading to exosomes. Sci. Adv. 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Li, L.; Lemos, H.; Chandler, P.R.; Pacholczyk, G.; Baban, B.; Barber, G.N.; Hayakawa, Y.; McGaha, T.L.; Ravishankar, B.; et al. Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. J. Immunol. 2013, 191, 3509–3513. [Google Scholar] [CrossRef] [Green Version]
- Golden, E.B.; Apetoh, L. Radiotherapy and immunogenic cell death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Dobler, C.; Jost, T.; Hecht, M.; Fietkau, R.; Distel, L. Senescence Induction by Combined Ionizing Radiation and DNA Damage Response Inhibitors in Head and Neck Squamous Cell Carcinoma Cells. Cells 2020, 9, 2012. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [Green Version]
- Vendetti, F.P.; Karukonda, P.; Clump, D.A.; Teo, T.; Lalonde, R.; Nugent, K.; Ballew, M.; Kiesel, B.F.; Beumer, J.H.; Sarkar, S.N.; et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J. Clin. Investig. 2018, 128, 3926–3940. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.T.; Yeh, C.N.; Chang, Y.C.; Cheng, J.H.; Fang, W.L.; Yeh, Y.C.; Wang, Y.C.; Hsu, D.S.; Wu, C.E.; Lai, J.I.; et al. PRKDC: New biomarker and drug target for checkpoint blockade immunotherapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Kantidze, O.L.; Velichko, A.K.; Luzhin, A.V.; Petrova, N.V.; Razin, S.V. Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs. Trends. Cancer 2018, 4, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Smith, S.A.; Mortimer, P.; Loembe, A.B.; Cho, H.; Kim, K.M.; Smith, C.; Willis, S.; Irurzun-Arana, I.; Berges, A.; et al. Phase I study of ceralasertib (AZD6738), a novel DNA damage repair agent, in combination with weekly paclitaxel in refractory cancer. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Yap, T.A.; O’Carrigan, B.P.M.S.; Penney, M.S.; Lim, J.S.; Brown, J.S.; Luken, M.J.M.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.R.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination with Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Redon, C.E.; Sciuto, L.; Padiernos, E.; Ji, J.; Lee, M.J.; Yuno, A.; Lee, S.; Zhang, Y.; Tran, L.; et al. Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Noorbakhsh, S.I.; Sundaram, R.K.; Kalathil, A.N.; Ganesa, S.; Jia, L.; Breslin, H.; Burgenske, D.M.; Gilad, O.; Sarkaria, J.N.; et al. Temozolomide Sensitizes MGMT-Deficient Tumor Cells to ATR Inhibitors. Cancer Res. 2019, 79, 4331–4338. [Google Scholar] [CrossRef]
- Jucaite, A.; Stenkrona, P.; Cselenyi, Z.; De Vita, S.; Buil-Bruna, N.; Varnas, K.; Savage, A.; Varrone, A.; Johnstrom, P.; Schou, M.; et al. Brain exposure of the ATM inhibitor AZD1390 in humans-a positron emission tomography study. Neuro. Oncol. 2021, 23, 687–696. [Google Scholar] [CrossRef]
- van Bussel, M.T.J.; Awada, A.; de Jonge, M.J.A.; Mau-Sorensen, M.; Nielsen, D.; Schoffski, P.; Verheul, H.M.W.; Sarholz, B.; Berghoff, K.; El Bawab, S.; et al. A first-in-man phase 1 study of the DNA-dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. Br. J. Cancer 2021, 124, 728–735. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A. Molecular Pathogenesis of Merkel Cell Carcinoma. Annu. Rev. Pathol. 2021, 16, 69–91. [Google Scholar] [CrossRef]
- Paulson, K.G.; Iyer, J.G.; Simonson, W.T.; Blom, A.; Thibodeau, R.M.; Schmidt, M.; Pietromonaco, S.; Sokil, M.; Warton, E.M.; Asgari, M.M.; et al. CD8+ lymphocyte intratumoral infiltration as a stage-independent predictor of Merkel cell carcinoma survival: A population-based study. Am. J. Clin. Pathol. 2014, 142, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, S.P.; Russell, J.; Lebbe, C.; Chmielowski, B.; Gambichler, T.; Grob, J.J.; Kiecker, F.; Rabinowits, G.; Terheyden, P.; Zwiener, I.; et al. Efficacy and Safety of First-line Avelumab Treatment in Patients With Stage IV Metastatic Merkel Cell Carcinoma: A Preplanned Interim Analysis of a Clinical Trial. JAMA Oncol. 2018, 4, e180077. [Google Scholar] [CrossRef] [Green Version]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Three-year survival, correlates and salvage therapies in patients receiving first-line pembrolizumab for advanced Merkel cell carcinoma. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Bhatia, S.; Amin, A.; Kudchadkar, R.R.; Sharfman, W.H.; Lebbé, C.; Delord, J.P.; Dunn, L.A.; Shinohara, M.M.; Kulikauskas, R.; et al. Neoadjuvant Nivolumab for Patients With Resectable Merkel Cell Carcinoma in the CheckMate 358 Trial. J. Clin. Oncol. 2020, 38, 2476–2487. [Google Scholar] [CrossRef]
- Lahman, M.C.; Paulson, K.G.; Nghiem, P.T.; Chapuis, A.G. Quality Is King: Fundamental Insights into Tumor Antigenicity from Virus-Associated Merkel Cell Carcinoma. J. Invest. Dermatol. 2021. [Google Scholar] [CrossRef]
- Park, J.W.; Lee, J.K.; Sheu, K.M.; Wang, L.; Balanis, N.G.; Nguyen, K.; Smith, B.A.; Cheng, C.; Tsai, B.L.; Cheng, D.; et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 2018, 362, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, C.; Houben, R.; Baeurle, A.; Ritter, C.; Schrama, D.; Landthaler, M.; Becker, J.C. Activation of the PI3K/AKT pathway in Merkel cell carcinoma. PLoS ONE 2012, 7, e31255. [Google Scholar] [CrossRef]
- Borchert, S.; Czech-Sioli, M.; Neumann, F.; Schmidt, C.; Wimmer, P.; Dobner, T.; Grundhoff, A.; Fischer, N. High-affinity Rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened Merkel cell polyomavirus large T antigens. J. Virol. 2014, 88, 3144–3160. [Google Scholar] [CrossRef] [Green Version]
- Houben, R.; Dreher, C.; Angermeyer, S.; Borst, A.; Utikal, J.; Haferkamp, S.; Peitsch, W.K.; Schrama, D.; Hesbacher, S. Mechanisms of p53 restriction in Merkel cell carcinoma cells are independent of the Merkel cell polyoma virus T antigens. J. Investig. Dermatol. 2013, 133, 2453–2460. [Google Scholar] [CrossRef] [Green Version]
- Park, D.E.; Cheng, J.; Berrios, C.; Montero, J.; Cortes-Cros, M.; Ferretti, S.; Arora, R.; Tillgren, M.L.; Gokhale, P.C.; DeCaprio, J.A. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. USA 2019, 116, 1027–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Park, D.E.; Berrios, C.; White, E.A.; Arora, R.; Yoon, R.; Branigan, T.; Xiao, T.; Westerling, T.; Federation, A.; et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 2017, 13, e1006668. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Waldeck, K.; Vergara, I.A.; Schroder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.J.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-Associated Mutations Underlie the Etiology of MCV-Negative Merkel Cell Carcinomas. Cancer Res. 2015, 75, 5228–5234. [Google Scholar] [CrossRef] [Green Version]
- Cimino, P.J.; Robirds, D.H.; Tripp, S.R.; Pfeifer, J.D.; Abel, H.J.; Duncavage, E.J. Retinoblastoma gene mutations detected by whole exome sequencing of Merkel cell carcinoma. Mod. Pathol. 2014, 27, 1073–1087. [Google Scholar] [CrossRef] [Green Version]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I.; International Workshop on Merkel Cell Carcinoma Research Working Group. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Paulson, K.G.; Lemos, B.D.; Feng, B.; Jaimes, N.; Penas, P.F.; Bi, X.; Maher, E.; Cohen, L.; Leonard, J.H.; Granter, S.R.; et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J. Investig. Dermatol. 2009, 129, 1547–1555. [Google Scholar] [CrossRef] [Green Version]
- Harms, P.W.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.M.; Dhanasekaran, S.M.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res. 2015, 75, 3720–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knepper, T.C.; Montesion, M.; Russell, J.S.; Sokol, E.S.; Frampton, G.M.; Miller, V.A.; Albacker, L.A.; McLeod, H.L.; Eroglu, Z.; Khushalani, N.I.; et al. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5961–5971. [Google Scholar] [CrossRef] [Green Version]
- Horny, K.; Gerhardt, P.; Hebel-Cherouny, A.; Wulbeck, C.; Utikal, J.; Becker, J.C. Mutational Landscape of Virus- and UV-Associated Merkel Cell Carcinoma Cell Lines Is Comparable to Tumor Tissue. Cancers 2021, 13, 649. [Google Scholar] [CrossRef]
- La Rosa, S.; Bonzini, M.; Sciarra, A.; Asioli, S.; Maragliano, R.; Arrigo, M.; Foschini, M.P.; Righi, A.; Maletta, F.; Motolese, A.; et al. Exploring the Prognostic Role of Ki67 Proliferative Index in Merkel Cell Carcinoma of the Skin: Clinico-Pathologic Analysis of 84 Cases and Review of the Literature. Endocr. Pathol. 2020, 31, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Matsushita, M.; Nonaka, D.; Kato, M.; Nagata, K.; Murakami, I.; Hayashi, K. Phosphohistone-H3 (PHH3) is prognostic relevant in Merkel cell carcinomas but Merkel cell polyomavirus is a more powerful prognostic factor than AJCC clinical stage, PHH3, Ki-67 or mitotic indices. Pathol. Int. 2015, 65, 404–409. [Google Scholar] [CrossRef]
- Nielsen, P.S.; Riber-Hansen, R.; Jensen, T.O.; Schmidt, H.; Steiniche, T. Proliferation indices of phosphohistone H3 and Ki67: Strong prognostic markers in a consecutive cohort with stage I/II melanoma. Mod. Pathol. 2013, 26, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [Green Version]
- Fokas, E.; Prevo, R.; Hammond, E.M.; Brunner, T.B.; McKenna, W.G.; Muschel, R.J. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat. Rev. 2014, 40, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, J.G.; Parvathaneni, U.; Gooley, T.; Miller, N.J.; Markowitz, E.; Blom, A.; Lewis, C.W.; Doumani, R.F.; Parvathaneni, K.; Anderson, A.; et al. Single-fraction radiation therapy in patients with metastatic Merkel cell carcinoma. Cancer Med. 2015, 4, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.G.; Tegeder, A.; Willmes, C.; Iyer, J.G.; Afanasiev, O.K.; Schrama, D.; Koba, S.; Thibodeau, R.; Nagase, K.; Simonson, W.T.; et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol. Res. 2014, 2, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- Ritter, C.; Fan, K.; Paulson, K.G.; Nghiem, P.; Schrama, D.; Becker, J.C. Reversal of epigenetic silencing of MHC class I chain-related protein A and B improves immune recognition of Merkel cell carcinoma. Sci. Rep. 2016, 6, 21678. [Google Scholar] [CrossRef] [Green Version]
- Iyer, J.G.; Blom, A.; Doumani, R.; Lewis, C.; Tarabadkar, E.S.; Anderson, A.; Ma, C.; Bestick, A.; Parvathaneni, U.; Bhatia, S.; et al. Response rates and durability of chemotherapy among 62 patients with metastatic Merkel cell carcinoma. Cancer Med. 2016, 5, 2294–2301. [Google Scholar] [CrossRef] [PubMed]
- Chan, I.S.; Bhatia, S.; Kaufman, H.L.; Lipson, E.J. Immunotherapy for Merkel cell carcinoma: A turning point in patient care. J. Immunother. Cancer 2018, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Voog, E.B.P.; Martin, J.P.; Blay, J.Y. Chemotherapy for Patients with Locally Advanced or Metastatic Merkel Cell Carcinoma. Cancer 1999, 85, 2589–2595. [Google Scholar] [CrossRef]
- Colunga, A.; Pulliam, T.; Nghiem, P. Merkel Cell Carcinoma in the Age of Immunotherapy: Facts and Hopes. Clin Cancer Res. 2018, 24, 2035–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tormoen, G.W.; Crittenden, M.R.; Gough, M.J. Role of the immunosuppressive microenvironment in immunotherapy. Adv. Radiat. Oncol. 2018, 3, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Kwon, M.; Kim, S.T.; Smith, S.; Smith, C.; Mortimer, P.G.; LoembE, B.; Kozarewa, I.; Dean, E.; Lee, J. Phase II study of ceralasertib (AZD6738), in combination with durvalumab in patients with metastatic melanoma who have failed prior anti-PD-1 therapy. J. Clin. Oncol. 2021, 39, 9514. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| DDRi Agent | Chemical Structure, IC50 | Total No. of Trials | Trials with ICI | Phase | ICI Agents in Combination with DDRi |
|---|---|---|---|---|---|
| ATR inhibitors | |||||
| AZD6738/ Ceralasertib Oral, AstraZeneca |  Cell-based IC50 = 74 nM | 33 | 9 | I/II, II | Durvalumab, Cisplatin/Carboplatin + Etoposide + Durvalumab |
| VX-970/M6620/ Berzosertib Intravenous, EMD Serono |  Cell-based IC50 = 19 nM | 23 | 3 | I, I/II | Avelumab, Avelumab + Carboplatin, Carboplatin + Gemcitabine hydrochloride + Pembrolizumab |
| BAY1895344/ Elimusertib Oral, Bayer |  Cell-based IC50 = 36 nM | 8 | 2 | I | Pembrolizumab, Stereotactic body radiation therapy + Pembrolizumab |
| DNA-PK inhibitor | |||||
| M3814/MSC2490484A/ Nedisertib/ Peposertib Oral, EMD Serono |  Cell-based IC50 < 0.5 μM | 14 | 3 | I, I/II | Avelumab ± radiotherapy, Avelumab + Radium-223 Dichloride, Avelumab + Hypo-fractionated radiotherapy |
| Drug Class | Sub-Class | Specific Agents | Phase |
|---|---|---|---|
| Immune checkpoint inhibitors (ICI) | Combination ICI | Ipilimumab (anti-CTLA-4) + Nivolumab (anti-PD-1) | I, II |
| Novel ICI | INCAGN02385 (Lag3), INCAGN02390 (Tim3) | I | |
| New PD-(L)1/CTLA4 mAbs | BT-001 (Treg depleting anti-CTLA-4), Tremelimumab (anti-CTLA-4), retifanlimab (anti-PD1) | I/II, II | |
| Radiotherapy | Radiotherapy + ICI | Ipilimumab/Nivolumab + SBRT; Pembrolizumab + SBRT | II |
| Novel radiosensitizer | NBTXR3 (radioenhancer hafnium oxide nanoparticle) | I | |
| Immune agonists | T cell co-stimulatory agonist | INCAGN01949 (OX40), INCAGN01876 (GITR), Utomilumab (4-1BB) | I, I/II |
| Intralesional innate immune agonists | Poly-ICLC (TLR3), Imiquimod (TLR7), NKTR-262 (TLR7/8), Cavrotolimod (TLR9) | I, I/II | |
| Oncolytic viruses | T-VEC (Herpes virus), Ad-p53 (adenovirus expressing p53) | I, II | |
| Tumor vaccines | IFx-Hu2.0 (DNA vaccine with streptococcal antigen) | I | |
| Cytokines | Bempegaldesleukin (CD122-preferential IL-2 pathway agonist), NT-I7 (IL-7 agonist), N-803 (IL-15 superagonist +ICI or NK cells), SO-C101 (IL-15 superagonist) | I, I/II, II | |
| Adoptive cell therapies | T cells | MCPyV T antigen-specific polyclonal autologous CD8+ T cells | I/II |
| NK cells | Allogeneic NK cell | Case only | |
| Novel agents targeting immunosuppressive TME | NOS inhibitor | L-NG-monomethyl Arginine acetate (L-NMMA) | I |
| Adenosine antagonist | Etrumadenant | I | |
| IDO1 inhibitor | Epacadostat | I/II | |
| Tumor antigen targeted therapy | Somatostatin | Lanreotide (SST analogue), Tidutamab/XmAb18087 (bispecific mAb targeting CD3 and SSTR) | I/II, II |
| Targeted radionuclide | Lutetium-177 DOTATATE (targeting SSTR) | I/II | |
| Antibody–drug conjugates | Anti-DLL3 | Rovalpituzumab tesirine (anti-DLL3 with DNA cross-linking drug) | I |
| Anti-CD56 | huN901-DM1 (anti-CD56 with microtubule inhibitor) | I | |
| Cell cycle/cell death and proliferation pathways | MDM2 inhibitor | KRT-232 | II |
| Anti-Bcl2 | Oblimersen | II | |
| mTOR inhibitors | RAD001, MLN0128 | I, I/II | |
| Anti-angiogenics | Anti-VEGF mAb | Bevazicumab (with atezolizumab) | I/II, II |
| Tyrosine kinase inhibitor | Vatalanib (small molecule targeting VEGF receptors, PDGF receptor beta and c-kit) | I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goff, P.H.; Bhakuni, R.; Pulliam, T.; Lee, J.H.; Hall, E.T.; Nghiem, P. Intersection of Two Checkpoints: Could Inhibiting the DNA Damage Response Checkpoint Rescue Immune Checkpoint-Refractory Cancer? Cancers 2021, 13, 3415. https://doi.org/10.3390/cancers13143415
Goff PH, Bhakuni R, Pulliam T, Lee JH, Hall ET, Nghiem P. Intersection of Two Checkpoints: Could Inhibiting the DNA Damage Response Checkpoint Rescue Immune Checkpoint-Refractory Cancer? Cancers. 2021; 13(14):3415. https://doi.org/10.3390/cancers13143415
Chicago/Turabian StyleGoff, Peter H., Rashmi Bhakuni, Thomas Pulliam, Jung Hyun Lee, Evan T. Hall, and Paul Nghiem. 2021. "Intersection of Two Checkpoints: Could Inhibiting the DNA Damage Response Checkpoint Rescue Immune Checkpoint-Refractory Cancer?" Cancers 13, no. 14: 3415. https://doi.org/10.3390/cancers13143415