
Challenges and Opportunities in the Statistical Analysis of Multiplex Immunofluorescence Data

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
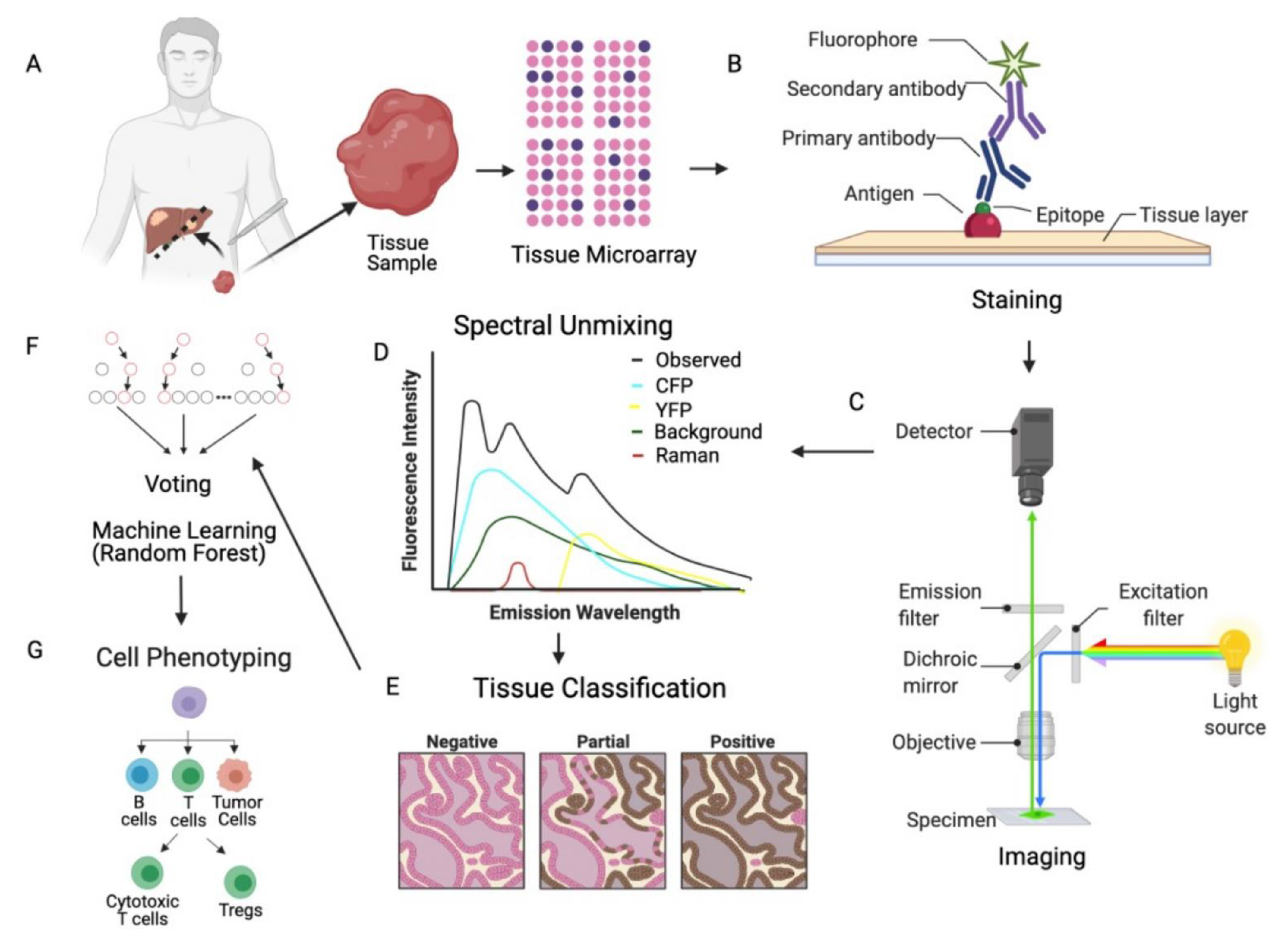
2. Data Preprocessing and Quality Control of mIF Data
2.1. mIF Assay and Data Generation
2.2. Quality Control of Generated Data
2.2.1. Conflicting Information between Markers (CD8 and FOXP3)
2.2.2. Batch Effects
3. Analysis of Summary Data
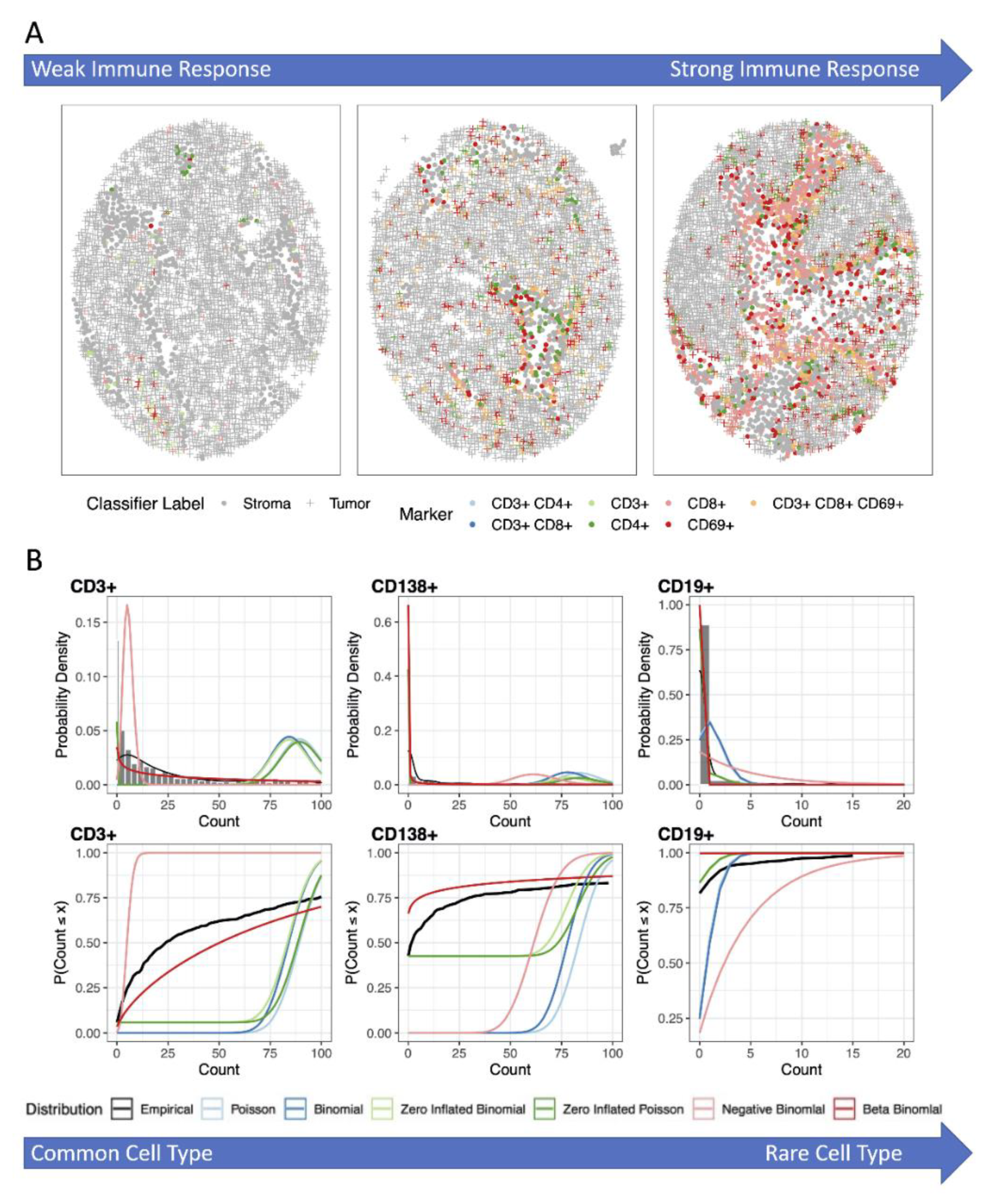
3.1. Analysis of the Number, Percentage or Density of Cells Positive for Immune Marker
3.2. Analysis Using Zero-Inflated and Over-Dispersed Distributions
3.3. Repeated Measurements
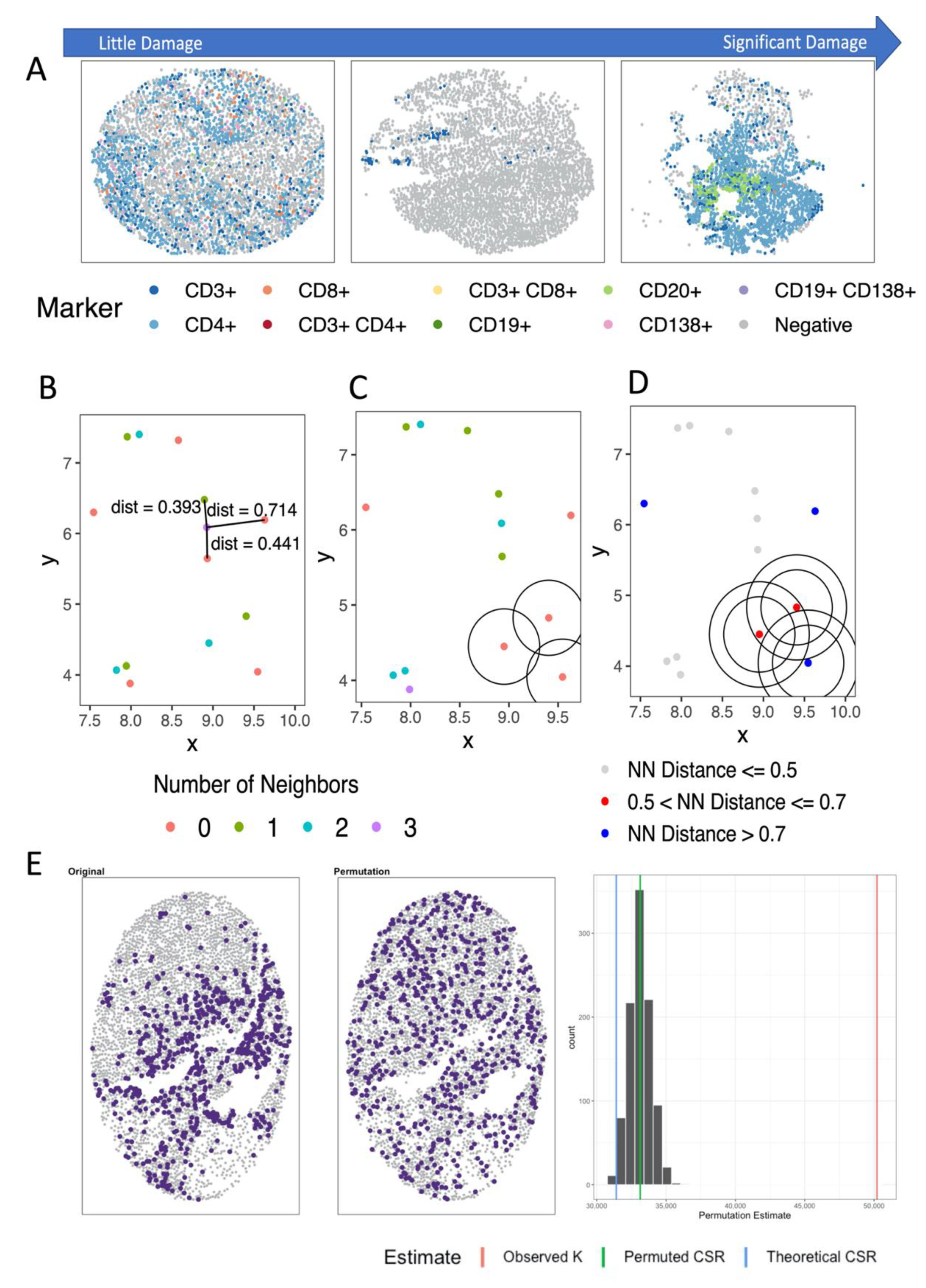
4. Clustering and Cooccurrence in Spatial Analysis of mIF
4.1. Pixel or Region-Based Methods
4.2. Distance- and Nearest Neighbor-Based Methods
4.3. Spatial Point Process Based Methods
4.3.1. Analyzing Number of Neighbors
4.3.2. Analyzing Distance to Neighbor
4.3.3. Considerations
5. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Drake, C.G.; Lipson, E.J.; Brahmer, J.R. Breathing new life into immunotherapy: Review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 2014, 11, 24–37. [Google Scholar] [CrossRef]
- Menon, S.; Shin, S.; Dy, G. Advances in Cancer Immunotherapy in Solid Tumors. Cancers 2016, 8, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emens, L.A.; Ascierto, P.A.; Darcy, P.K.; Demaria, S.; Eggermont, A.M.M.; Redmond, S.L.; Seliger, B.; Marincola, F.M. Cancer immunotherapy: Opportunities and challenges in the rapidly evolving clinical landscape. Eur. J. Cancer 2017, 81, 116–129. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Suantès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Gooden, M.J.; de Bock, G.H.; Leffers, N.; Daeman, T.; Nijman, H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 2011, 105, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind., P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Galon, J.; Fridman, W.H.; Pages, F. The adaptive immunologic microenvironment in colorectal cancer: A novel perspective. Cancer Res. 2007, 67, 1883–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angell, H.K.; Bruni, D.; Barrett, J.C.; Herbst, R.; Galon, J. The Immunoscore: Colon Cancer and Beyond. Clin. Cancer Res. 2020, 26, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Lugli, A.; Bifulco, C.; Pagès, F.; Masucci, G.; Marincola, F.M.; Ascierto, P.A. World-Wide Immunoscore Task Force: Meeting report from the "Melanoma Bridge", Napoli, November 30th–December 3rd, 2016. J. Transl. Med. 2017, 15, 212. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Pagès, F.; Marincola, F.M.; Thurin, M.; Trinchieri, G.; Fox, B.A.; Gajewski, T.F.; Ascierto, P.A. The immune score as a new possible approach for the classification of cancer. J. Transl. Med. 2012, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Pagès, F.; Marincola, F.M.; Angell, H.; Thurin, M.; Lugli, A.; Zlobec, I.; Berger, A.; Bifulco, C.; Botti, G.; et al. Cancer classification using the Immunoscore: A worldwide task force. J. Transl. Med. 2012, 10, 205. [Google Scholar] [CrossRef]
- Yeong, J.; Tan, T.; Chow, Z.L.; Cheng, Q.; Lee, B.; Seet, A.; Lim, J.X.; Lim, J.C.T.; Ong, C.C.H.; Thike, A.A.; et al. Multiplex immunohistochemistry/immunofluorescence (mIHC/IF) for PD-L1 testing in triple-negative breast cancer: A translational assay compared with conventional IHC. J. Clin. Pathol. 2020, 73, 557–562. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- de Kanter, J.K.; Lijnzaad, P.; Candelli, T.; Margaritis, T.; Holstege, F.C.P. CHETAH: A selective, hierarchical cell type identification method for single-cell RNA sequencing. Nucleic Acids Res. 2019, 47, e95. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.M.; Fridley, B.L.; Conejo-Garcia, J.R.; Wang, X.; Yu, X. Wide and deep learning for automatic cell type identification. Comput. Struct. Biotechnol. J. 2021, 19, 1052–1062. [Google Scholar] [CrossRef]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Zirlado, S.B.; Wheeler, T.B.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Abbas-Aghababazadeh, F.; Chen, Y.A.; Fridley, B.L. Statistical and Bioinformatics Analysis of Data from Bulk and Single-Cell RNA Sequencing Experiments. Methods Mol. Biol. 2021, 2194, 143–175. [Google Scholar] [PubMed]
- Young, Y.K.; Bolt, A.M.; Ahn, R.; Mann, K.K. Analyzing the Tumor Microenvironment by Flow Cytometry. Methods Mol. Biol. 2016, 1458, 95–110. [Google Scholar]
- Carvajal-Hausdorf, D.E.; Patsenker, J.; Stanton, K.P.; Villarroel-Espindola, F.; Esch, A.; Montgomery, R.R.; Psyrri, A.; Kalogeros, K.T.; Kotoula, V.; Foutzilas, G.; et al. Multiplexed (18-Plex) Measurement of Signaling Targets and Cytotoxic T Cells in Trastuzumab-Treated Patients using Imaging Mass Cytometry. Clin. Cancer Res. 2019, 25, 3054–3062. [Google Scholar] [CrossRef] [Green Version]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schuffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef]
- Martinez-Morilla, S.; Villarroel-Espindola, F.; Wong, P.F.; Toki, M.I.; Aung, T.N.; Pelekanou, V.; Bourke-Martin, B.; Schalper, K.A.; Kluger, H.M.; Rimm, D.L. Biomarker Discovery in Patients with Immunotherapy-Treated Melanoma with Imaging Mass Cytometry. Clin. Cancer Res. 2021, 27, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Baharlou, H.; Canete, N.P.; Cunningham, A.L.; Harman, A.N.; Patrick, E. Mass Cytometry Imaging for the Study of Human Diseases-Applications and Data Analysis Strategies. Front Immunol. 2019, 10, 2657. [Google Scholar] [CrossRef] [PubMed]
- Angelo, M.; Bendall, S.C.; Finck, R.; Hale, M.B.; Hitzman, C.; Borowsky, A.D.; Levenson, R.M.; Lowe, J.B.; Liu, S.D.; Zhao, S.; et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 2014, 20, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.T.; Loewke, N.O.; Mandella, M.J.; Levenson, R.M.; Crawford, J.M.; Contag, C.H. Point-of-care pathology with miniature microscopes. Anal. Cell Pathol. 2011, 34, 81–98. [Google Scholar] [CrossRef]
- Sheffield, B.S. Immunohistochemistry as a Practical Tool in Molecular Pathology. Arch. Pathol. Lab. Med. 2016, 140, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L. Histotechnology a Self Instructional Text, 5th ed.; American Society of Clinical Oncology: Alexandria, VA, USA, 2020. [Google Scholar]
- Prophet, E.B.; Armed, P. Forces Institute of, Laboratory Methods in Histotechnology; American Registry of Pathology: Washington, DC, USA, 1992. [Google Scholar]
- Gnjatic, S.; Bronte, V.; Brunet, L.R.; Butler, M.O.; Disis, M.L.; Galon, J.; Hakansson, L.G.; Hanks, B.A.; Karanikas, V.; Khleif, S.N.; et al. Identifying baseline immune-related biomarkers to predict clinical outcome of immunotherapy. J. Immunother. Cancer 2017, 5, 44. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Hedvat, C.V. Digital microscopy: Past, present, and future. Arch. Pathol. Lab. Med. 2010, 134, 1666–1670. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Akturk, G.; Angelo, M.; Engle, E.L.; Gnjatic, S.; Greenbaum, S.; Greenwald, N.F.; Hedvat, C.V.; Hollmann, T.J.; Juco, J.; et al. The Society for Immunotherapy of Cancer statement on best practices for multiplex immunohistochemistry (IHC) and immunofluorescence (IF) staining and validation. J. Immunother. Cancer 2020, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Tsakiroglou, A.M.; Fergie, M.; Oguejiofor, K.; Linton, K.; Thomson, D.; Stern, P.L.; Astley, S.; Byers, R.; West, C.M.L. Spatial proximity between T and PD-L1 expressing cells as a prognostic biomarker for oropharyngeal squamous cell carcinoma. Br. J. Cancer 2020, 122, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.K.; Wang, M.; Sun, Y.; Di Costanzo, N.; Mitchell, C.; Achuthan, A.; Hamilton, J.A.; Busuttil, R.A.; Boussioutas, A. Macrophage spatial heterogeneity in gastric cancer defined by multiplex immunohistochemistry. Nat. Commun. 2019, 10, 3928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vayrynen, S.A.; Zhang, J.; Yuan, C.; Vayrynen, J.P.; Dias Costa, A.; Williams, H.; Morales-Oyarvide, V.; Lau, M.C.; Rubinson, D.A.; Dunne, R.F.; et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 1069–1081. [Google Scholar] [CrossRef]
- Belanger, C.F.; Hennekens, C.H.; Rosner, B.; Speizer, F.E. The nurses’ health study. Am. J. Nurs. 1978, 78, 1039–1040. [Google Scholar] [CrossRef]
- Birmann, B.M.; Barnard, M.E.; Bertrand, K.A.; Bao, Y.; Crous-Bou, M.; Wolpin, B.M.; De Vivo, I.; Tworoger, S.S. Nurses’ Health Study Contributions on the Epidemiology of Less Common Cancers: Endometrial, Ovarian, Pancreatic, and Hematologic. Am. J. Public Health 2016, 106, 1608–1615. [Google Scholar] [CrossRef]
- Boutot, M.E.; Purdue-Smithe, A.; Whitcomb, B.W.; Szegda, K.L.; Manson, J.E.; Hankinson, S.E.; Rosner, B.A.; Bertone-Johnson, E.R. Dietary Protein Intake and Early Menopause in the Nurses’ Health Study II. Am. J. Epidemiol. 2018, 187, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Schildkraut, J.M.; Alberg, A.J.; Bandera, E.V.; Barnholtz-Sloan, J.; Bondy, M.; Cote, M.L.; Funkhouser, E.; Peters, E.; Schwartz, A.G.; Terry, P.; et al. A multi-center population-based case-control study of ovarian cancer in African-American women: The African American Cancer Epidemiology Study (AACES). BMC Cancer 2014, 14, 688. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Mandal, G.; Payne, K.K.; Anadon, C.M.; Gatenbee, C.D.; Chaurio, R.A.; Costich, T.L.; Moran, C.; Harro, C.M.; Rigolizzo, K.E.; et al. IgA transcytosis and antigen recognition govern ovarian cancer immunity. Nature 2021, 591, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Hajiran, A.; Chakiryan, N.; Aydin, A.M.; Zemp, L.; Nguyen, J.; Laborde, J.M.; Chahoud, J.; Spiess, P.E.; Zaman, S.; Falasiri, S.; et al. Reconnaissance of tumor immune microenvironment spatial heterogeneity in metastatic renal cell carcinoma and correlation with immunotherapy response. Clin. Exp. Immunol. 2021, 204, 96–106. [Google Scholar] [CrossRef]
- Kamal, Y.; Dwan, D.; Hoehn, H.J.; Sanz-Pamplona, R.; Alonso, M.H.; Moreno, V.; Cheng, C.; Schell, M.J.; Kim, Y.; Felder, S.I. Tumor immune infiltration estimated from gene expression profiles predicts colorectal cancer relapse. OncoImmunology 2021, 10, 1862529. [Google Scholar] [CrossRef] [PubMed]
- Akoya Biosciences. Opal Mulitplex IHC Assay Development Guide; Akoya Biosciences: Marlborough, MA, USA, 2019. [Google Scholar]
- Lim, J.C.T.; Yeong, J.P.S.; Lim, C.J.; Ong, C.C.H.; Wong, S.C.; Chew, V.S.P.; Ahmed, S.S.; Tan, P.H.; Iqbal, J. An automated staining protocol for seven-colour immunofluorescence of human tissue sections for diagnostic and prognostic use. Pathology 2018, 50, 333–341. [Google Scholar] [CrossRef]
- Garini, Y.; Young, I.T.; McNamara, G. Spectral imaging: Principles and applications. Cytometry A 2006, 69, 735–747. [Google Scholar] [CrossRef]
- Abel, E.J.; Bauman, T.M.; Weiker, M.; Shi, F.; Downs, T.M.; Jarrard, D.F.; Huang, W. Analysis and validation of tissue biomarkers for renal cell carcinoma using automated high-throughput evaluation of protein expression. Hum. Pathol. 2014, 45, 1092–1099. [Google Scholar] [CrossRef] [Green Version]
- Ghaznavi, F.; Evans, A.; Madabhushi, A.; Feldman, M. Digital imaging in pathology: Whole-slide imaging and beyond. Annu. Rev. Pathol. 2013, 8, 331–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, E.R.; Uraoka, N.; Jiang, M.; Cook, P.; Gibbons, D.; Forget, M.-A.; Bernatchez, C.; Haymaker, C.; Wistuba, I.I.; Rodriguez-Canales, J. Validation of multiplex immunofluorescence panels using multispectral microscopy for immune-profiling of formalin-fixed and paraffin-embedded human tumor tissues. Sci. Rep. 2017, 7, 13380. [Google Scholar] [CrossRef] [Green Version]
- Acs, B.; Pelekanou, V.; Bai, Y.; Martinez-Morilla, S.; Toki, M.; Leung, S.C.Y.; Nielsen, T.O.; Rimm, D.L. Ki67 reproducibility using digital image analysis: An inter-platform and inter-operator study. Lab. Investig. 2019, 99, 107–117. [Google Scholar] [CrossRef]
- Horai, Y.; Mizukawa, M.; Nishina, H.; Nishikawa, S.; Ono, Y.; Takemoto, K.; Baba, N. Quantification of histopathological findings using a novel image analysis platform. J. Toxicol. Pathol. 2019, 32, 319–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakya, R.; Nguyen, T.H.; Waterhouse, N.; Khanna, R. Immune contexture analysis in immuno-oncology: Applications and challenges of multiplex fluorescent immunohistochemistry. Clin. Transl. Immunol. 2020, 9, e1183. [Google Scholar] [CrossRef] [PubMed]
- Stack, E.C.; Wang, C.; Roman, K.A.; Hoyt, C.C. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 2014, 70, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef] [Green Version]
- Gorris, M.A.J.; Halilovic, A.; Rabold, K.; van Duffelen, A.; Wickramasinghe, I.N.; Verweij, D.; Wortel, I.M.N.; Textor, J.C.; de Vries, I.J.M.; Figdor, C.G. Eight-Color Multiplex Immunohistochemistry for Simultaneous Detection of Multiple Immune Checkpoint Molecules within the Tumor Microenvironment. J. Immunol. 2018, 200, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Mezheyeuski, A.; Bergsland, C.H.; Backman, M.; Djureinovic, D.; Sjoblom, T.; Bruun, J.; Micke, P. Multispectral imaging for quantitative and compartment-specific immune infiltrates reveals distinct immune profiles that classify lung cancer patients. J. Pathol. 2018, 244, 421–431. [Google Scholar] [CrossRef]
- Mori, H.; Bolen, J.; Schuetter, L.; Massion, P.; Hoyt, C.C.; VandenBerg, S.; Esserman, L.; Borowsky, A.D.; Campbell, M.J. Characterizing the Tumor Immune Microenvironment with Tyramide-Based Multiplex Immunofluorescence. J. Mammary Gland. Biol. Neoplasia 2020, 25, 417–432. [Google Scholar] [CrossRef]
- Amancio, D.R.; Comin, C.H.; Casanova, D.; Travieso, G.; Bruno, O.M.; Rodrigues, F.A.; Costa Lda, F. A systematic comparison of supervised classifiers. PLoS ONE 2014, 9, e94137. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Thurin, M.; Cesano, A.; Marincola, F. Biomarkers for Immunotherapy of Cancer. Methods in Molecular Biology; Thurin, M., Cesano, A., Marincola, F., Eds.; Springer: New York, NY, USA, 2020. [Google Scholar]
- Viratham Pulsawatdi, A.; Craig, S.G.; Bingham, V.; McCombe, K.; Humphries, M.P.; Senevirathne, S.; Richman, S.D.; Quirke, P.; Campo, L.; Domingo, E.; et al. A robust multiplex immunofluorescence and digital pathology workflow for the characterisation of the tumour immune microenvironment. Mol. Oncol. 2020, 14, 2384–2402. [Google Scholar] [CrossRef]
- Blessin, N.C.; Simon, R.; Kluth, M.; Fischer, K.; Hube-Magg, C.; Li, W.; Makrypidi-Fraune, G.; Wellge, B.; Mandelkow, T.; Debatin, N.F.; et al. Patterns of TIGIT Expression in Lymphatic Tissue, Inflammation, and Cancer. Dis. Markers 2019, 2019, 5160565. [Google Scholar] [CrossRef] [PubMed]
- Govek, K.W.; Troisi, E.C.; Miao, Z.; Aubin, R.G.; Woodhouse, S.; Camara, P.G. Single-cell transcriptomic analysis of mIHC images via antigen mapping. Sci. Adv. 2021, 7, eabc5464. [Google Scholar] [CrossRef]
- Tworoger, S.S.; Hankinson, S.E. Use of biomarkers in epidemiologic studies: Minimizing the influence of measurement error in the study design and analysis. Cancer Causes Control. 2006, 17, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Parra, E.R.; Jiang, M.; Solis, L.; Mino, B.; Laberiano, C.; Hernandez, S.; Gite, S.; Verma, A.; Tetzlaff, M.; Haymaker, C.; et al. Procedural Requirements and Recommendations for Multiplex Immunofluorescence Tyramide Signal Amplification Assays to Support Translational Oncology Studies. Cancers 2020, 12, 255. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-W.; Ren, Y.J.; Marella, M.; Wang, M.; Hartke, J.; Couto, S.S. Multiplex immunofluorescence staining and image analysis assay for diffuse large B cell lymphoma. J. Immunol. Methods 2020, 478, 112714. [Google Scholar] [CrossRef]
- Diem, K.; Magaret, A.; Klock, A.; Jin, L.; Zhu, J.; Corey, L. Image analysis for accurately counting CD4+ and CD8+ T cells in human tissue. J. Virol. Methods. 2015, 222, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, K.; Kim, I.; Chun, B.; Pucilowska, J.; Redmond, W.L.; Urba, W.J.; Martel, M.; Wu, Y.; Campbell, M.; Sun, Z.; et al. Multiplex immunofluorescence to measure dynamic changes in tumor-infiltrating lymphocytes and PD-L1 in early-stage breast cancer. Breast Cancer Res. 2021, 23, 2. [Google Scholar] [CrossRef]
- McCullagh, P.; Nelder, J.A. Generalized Linear Models; Chapman & Hall/CRC: Boca Raton, FL, USA, 1999; p. 511. [Google Scholar]
- Agresti, A. Categorical Data Analysis, 2nd ed.; Wiley Series in Probability and Statistics; ohn Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002; p. 710. [Google Scholar]
- Genser, B.; Cooper, P.J.; Yazdanbakhsh, M.; Barreto, M.L.; Rodrigues, L.C. A guide to modern statistical analysis of immunological data. BMC Immunol. 2007, 8, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, F.L.; Schafer, D.W. The Statistical Sleuth: A Course in Methods of Data Analysis, 3rd ed.; Brooks/Cole, Cengage Learning: Boston, MA, USA, 2013. [Google Scholar]
- Hammond, M.E.H.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Immunohistochemical Testing of Estrogen and Progesterone Receptors in Breast Cancer. J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef] [Green Version]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847. [Google Scholar] [CrossRef] [Green Version]
- Ilie, M.; Hofman, V.; Dietel, M.; Soria, J.C.; Hofman, P. Assessment of the PD-L1 status by immunohistochemistry: Challenges and perspectives for therapeutic strategies in lung cancer patients. Virchows Arch. 2016, 468, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Hu-Lieskovan, S. What does PD-L1 positive or negative mean? J. Exp. Med. 2016, 213, 2835–2840. [Google Scholar] [CrossRef] [Green Version]
- Bouwmeester, W.; Zuithoff, N.P.; Mallett, S.; Geerlings, M.I.; Vergouwe, Y.; Steyerberg, E.W.; Altman, D.G.; Moons, K.G. Reporting and methods in clinical prediction research: A systematic review. PLoS Med. 2012, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mabikwa, O.V.; Greenwood, D.C.; Baxter, P.D.; Fleming, S.J. Assessing the reporting of categorised quantitative variables in observational epidemiological studies. BMC Health Serv. Res. 2017, 17, 201. [Google Scholar]
- Altman, D.G.; Lausen, B.; Sauerbrei, W.; Schumacher, M. Dangers of using “optimal” cutpoints in the evaluation of prognostic factors. J. Natl. Cancer Inst. 1994, 86, 829–835. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.; Thapa, R.; Creed, J.; Nguyen, J.; Segura, C.M.; Gerke, T.; Schildkraut, K.; Peres, L.; Fridley, B.L. Statistical framework for studying the spatial architecture of the tumor immune microenvironment. medRxiv 2021. [Google Scholar] [CrossRef]
- Hall, D.B. Zero-inflated Poisson and binomial regression with random effects: A case study. Biometrics 2000, 56, 1030–1039. [Google Scholar] [CrossRef]
- Lee, A.H.; Lee, A.H.; Wang, K.; Scott, J.A.; Yau, K.K.; McLachlan, G.J. Multi-level zero-inflated poisson regression modelling of correlated count data with excess zeros. Stat. Methods Med. Res. 2006, 15, 47–61. [Google Scholar] [CrossRef]
- Jiang, S.; Xiao, G.; Koh, A.Y.; Kim, J.; Li, Q.; Zhan, X. A Bayesian zero-inflated negative binomial regression model for the integrative analysis of microbiome data. Biostatistics 2019. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yi, N. Fast zero-inflated negative binomial mixed modeling approach for analyzing longitudinal metagenomics data. Bioinformatics 2020, 36, 2345–2351. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Gallins, P.; Zhou, Y.H. A Zero-inflated Beta-binomial Model for Microbiome Data Analysis. Stat. Int. Stat. Inst. 2018, 7, e185. [Google Scholar] [CrossRef] [PubMed]
- Yau, K.K.; Lee, A.H. Zero-inflated Poisson regression with random effects to evaluate an occupational injury prevention programme. Stat. Med. 2001, 20, 2907–2920. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Smyth, G.K. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 2007, 23, 2881–2887. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Smyth, G.K. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 2008, 9, 321–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- J-Express 2012, Visualization and Analysis of Microarray Data. pp. 161–177. Available online: https://mybiosoftware.com/j-express-2009-analysis-visualization-microarray-data.html (accessed on 20 May 2021).
- Jeong, H.H.; Kim, S.Y.; Rousseaux, M.W.C.; Zoghbi, H.Y.; Liu, Z. Beta-binomial modeling of CRISPR pooled screen data identifies target genes with greater sensitivity and fewer false negatives. Genome Res. 2019, 29, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, J.H. The Validation of a Beta-Binomial Model for Overdispersed Binomial Data. Commun. Stat. Simul. Comput. 2017, 46, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.D.; Witten, D.; Willis, A.D. Modeling Microbial Abundances and Dysbiosis with Beta-Binomial Regression. Ann. Appl. Stat. 2020, 14, 94–115. [Google Scholar] [CrossRef] [Green Version]
- Najera-Zuloaga, J.; Lee, D.J.; Arostegui, I. Comparison of beta-binomial regression model approaches to analyze health-related quality of life data. Stat. Methods Med. Res. 2018, 27, 2989–3009. [Google Scholar] [CrossRef]
- Jakaitiene, A.; Avino, M.; Guarracino, M.R. Beta-Binomial Model for the Detection of Rare Mutations in Pooled Next-Generation Sequencing Experiments. J. Comput. Biol. 2017, 24, 357–367. [Google Scholar] [CrossRef]
- Congdon, P. Bayesian Statistical Modelling. Wiley Series in Probability and Statistics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007; p. 552. [Google Scholar]
- McCulloch, C.E.; Searle, S.R. Generalized, Linear, and Mixed Models. Wiley Series in Probability and Statistics Texts, References, and Pocketbooks Section; John Wiley & Sons, Inc.: New York, NY, USA, 2001; p. 325. [Google Scholar]
- Dempster, A.P.; Laird, N.M.; Rubin, D.B. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Ser. B 1977, 39, 1–38. [Google Scholar]
- Ypma, T.J. Historical Development of the Newton-Raphson Method. Soc. Ind. Appl. Math. Rev. 1995, 37, 531–551. [Google Scholar] [CrossRef] [Green Version]
- Geman, S.; Geman, D. Stochastic relaxation, Gibbs distributions, and the Bayesian restoration of images. IEEE Trans. Pattern Anal. Mach. Intell. 1984, 6, 721–741. [Google Scholar] [CrossRef]
- Gilks, W.R.; Richardson, S.; Spiegelhalter, D.J. Markov Chain Monte Carlo in Practice; Chapman & Hall/CRC: Boca Raton, FL, USA, 1996; p. 486. [Google Scholar]
- Sainani, K. The importance of accounting for correlated observations. Pm R. 2010, 2, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Schober, P.; Vetter, T.R. Repeated Measures Designs and Analysis of Longitudinal Data: If at First You Do Not Succeed-Try, Try Again. Anesth Analg. 2018, 127, 569–575. [Google Scholar] [CrossRef]
- Magurran, A.E. Biological Diversity. Curr. Biol. 2005, 15, R116–R118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, H.S. Measurement of overlap in comparative ecological studies. Am. Nat. 1966, 100, 419–424. [Google Scholar] [CrossRef]
- Wolda, H. Similarity indices, sample size and diversity. Oecologia 1981, 50, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Duncan, O.D.; Duncan, B. A Methodological Analysis of Segregation Indexes. Am. Sociol. Rev. 1955, 20, 210–217. [Google Scholar] [CrossRef]
- Yao, J.; Wong, D.; Bailey, N.; Minton, J. Spatial Segregation Measures: A Methodological Review: Spatial Segregation Measures. Tijdschr. Econ. Soc. Geogr. 2018, 110, 235–250. [Google Scholar] [CrossRef] [Green Version]
- Ripley, B.D. The second-order analysis of stationary point processes. J. Appl. Probab. 1976, 13, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Baddeley, A.; Rubak, E.; Turner, R. Spatial Point Patterns: Methodology and Applications with R; CRC: Boca Raton, FL, USA, 2015; 810p. [Google Scholar]
- Besag, J. Comments on Ripley’s paper. J. R. Stat. Soc. Ser. A 1977, 39, 193–195. [Google Scholar]
- Marcon, E.; Puech, F.; Traissac, S. Characterizing the Relative Spatial Structure of Point Patterns. Int. J. Ecol. 2012, 2012, 619281. [Google Scholar] [CrossRef] [Green Version]
- Bull, J.A.; Macklin, P.S.; Quaiser, T.; Braun, F.; Waters, S.L.; Pugh, C.W.; Byrne, H.M. Combining multiple spatial statistics enhances the description of immune cell localisation within tumours. Sci. Rep. 2020, 10, 18624. [Google Scholar] [CrossRef]
- Agnew, D.; Green, J.; Brown, T.M.; Simpson, M.J.; Binder, B.J. Distinguishing between mechanisms of cell aggregation using pair-correlation functions. J. Theor. Biol. 2014, 352, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Rose, C.J.; Naidoo, K.; Clay, V.; Linton, K.; Radford, J.A.; Byers, R.J. A statistical framework for analyzing hypothesized interactions between cells imaged using multispectral microscopy and multiple immunohistochemical markers. J. Pathol. Inf. 2013, 4 (Suppl. 4). [Google Scholar] [CrossRef]
- Baddeley, A.J.; Gill, R.D. The Empty Space Hazard of a Spatial Pattern; University of Western Australia. Department of Mathematics: Perth, Australia, 1994. [Google Scholar]
- Baddeley, A.; Gill, R.D. Kaplan-Meier Estimators of Distance Distributions for Spatial Point Processes. Ann. Stat. 1997, 25, 263–292. [Google Scholar] [CrossRef]
- Baddeley, A.J.; Kerscher, M.; Schladitz, K.; Scott, B.T. Estimating the J function without edge correction. Stat. Neerl. 2000, 54, 315–328. [Google Scholar] [CrossRef]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y. Spatial Heterogeneity in the Tumor Microenvironment. Cold Spring Harb. Perspect. Med. 2016, 6, a026583. [Google Scholar] [CrossRef] [Green Version]
- Maley, C.C.; Koelble, K.; Natrajan, R.; Aktipis, A.; Yuan, Y. An ecological measure of immune-cancer colocalization as a prognostic factor for breast cancer. Breast Cancer Res. BCR 2015, 17, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rempala, G.A.; Seweryn, M. Methods for diversity and overlap analysis in T-cell receptor populations. J. Math. Biol. 2013, 67, 1339–1368. [Google Scholar] [CrossRef] [Green Version]
- Roh, K.-H.; Lillemeier, B.F.; Wang, F.; Davis, M.M. The coreceptor CD4 is expressed in distinct nanoclusters and does not colocalize with T-cell receptor and active protein tyrosine kinase p56lck. Proc. Natl. Acad. Sci. USA 2015, 112, E1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.M. Voronoi Diagram. In Encyclopedia of GIS; Shekhar, S., Xiong, H., Eds.; Springer: Boston, MA, USA, 2008; pp. 1232–1235. [Google Scholar]
- Enfield, K.S.S.; Martin, S.D.; Marshall, E.A.; Kung, S.H.Y.; Gallagher, P.; Milne, K.; Chen, Z.; Nelson, B.H.; Lam, S.; English, J.C.; et al. Hyperspectral cell sociology reveals spatial tumor-immune cell interactions associated with lung cancer recurrence. J. Immunother. Cancer 2019, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gartrell, R.D.; Marks, D.K.; Hart, T.D.; Li, G.; Davari, D.R.; Wu, A.; Blake, Z.; Lu, Y.; Askin, K.N.; Monod, A.; et al. Quantitative Analysis of Immune Infiltrates in Primary Melanoma. Cancer Immunol. Res. 2018, 6, 481–493. [Google Scholar] [CrossRef] [Green Version]
- Parra, E.R.; Zhai, J.; Tamegnon, A.; Zhou, N.; Pandurengan, R.K.; Barreto, C.; Jiang, M.; Rice, D.C.; Creasy, C.; Vaporciyan, A.A.; et al. Identification of distinct immune landscapes using an automated nine-color multiplex immunofluorescence staining panel and image analysis in paraffin tumor tissues. Sci. Rep. 2021, 11, 4530. [Google Scholar] [CrossRef]
- Fassler, D.J.; Abousamra, S.; Gupta, R.; Chen, C.; Zhao, M.; Paredes, D.; Batool, S.A.; Knudsen, B.S.; Escobar-Hoyos, L.; Shroyer, K.R.; et al. Deep learning-based image analysis methods for brightfield-acquired multiplex immunohistochemistry images. Diagn. Pathol. 2020, 15, 100. [Google Scholar] [CrossRef] [PubMed]
- Moore, M. Spatial Statistics: Methodological Aspects and Applications; Springer: New York, NY, USA, 2001; p. 282. [Google Scholar]
- Cressie, N.A.C. Statistics for Spatial Data. Revised Edition; Wiley Series in Probability and Mathematical Statistics; John Wiley & Sons, Inc.: New York, NY, USA, 1993; p. 900. [Google Scholar]
- Barua, S.; Fang, P.; Sharma, A.; Fujimoto, J.; Wistuba, I.; Rao, A.U.K.; Lin, S.H. Spatial interaction of tumor cells and regulatory T cells correlates with survival in non-small cell lung cancer. Lung Cancer 2018, 117, 73–79. [Google Scholar] [CrossRef]
- Binder, B.J.; Simpson, M.J. Quantifying spatial structure in experimental observations and agent-based simulations using pair-correlation functions. Phys. Rev. E 2013, 88, 022705. [Google Scholar] [CrossRef] [Green Version]
- Cressie, N.; Hoef, J.M.V. Spatial Statistical Analysis of Environmental and Ecological Data; Oxford University Press: Oxford, UK, 1993; pp. 404–409. [Google Scholar]
- Gabriel, E.; Baddeley, A.; Rubak, E.; Turner, R. Spatial Point Patterns: Methodology and Applications with R. Math. Geosci. 2017, 49, 815–817. [Google Scholar] [CrossRef]
- Ramsay, J.O.; Silverman, B.W. Applied Functional Data Analysis: Methods and Case Studies; Springer Series in Statistics; Springer: New York, NY, USA, 2002. [Google Scholar]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leek, J.T.; Storey, J.D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007, 3, 1724–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thrane, K.; Eriksson, H.; Maaskola, J.; Hansson, J.; Lundeberg, J. Spatially Resolved Transcriptomics Enables Dissection of Genetic Heterogeneity in Stage III Cutaneous Malignant Melanoma. Cancer Res. 2018, 78, 5970–5979. [Google Scholar] [CrossRef] [Green Version]
- Stahl, P.L.; Salmen, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstrahle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.; Larsson, L.; et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Analysis | Name | Empirical Formula | Theoretical Value under CSR | Comments |
|---|---|---|---|---|
| Pixel/Area Based | Morisita Horn Index [110,111] | = |
| |
| Duncan Segregation Index [113] |
| |||
| Nearest Neighbor | Euclidean Distance | |||
| Nearest Neighbor | ||||
| Spatial Point Processes | Ripley’s K [115] |
| ||
| Besag’s L [117] | ||||
| Marcon’s M [118] | ||||
| Pairwise Correlation Function [119,120] |
| |||
| Hypothesized Interaction Distribution [121] |
| |||
| Empty Space Function [122] |
| |||
| Nearest Neighbor Function [116] | ||||
| Hazard Empty Space Function [123] or Hazard Nearest Neighbor Function |
| |||
| J-function [124] |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, C.M.; Ospina, O.E.; Townsend, M.K.; Nguyen, J.; Moran Segura, C.; Schildkraut, J.M.; Tworoger, S.S.; Peres, L.C.; Fridley, B.L. Challenges and Opportunities in the Statistical Analysis of Multiplex Immunofluorescence Data. Cancers 2021, 13, 3031. https://doi.org/10.3390/cancers13123031
Wilson CM, Ospina OE, Townsend MK, Nguyen J, Moran Segura C, Schildkraut JM, Tworoger SS, Peres LC, Fridley BL. Challenges and Opportunities in the Statistical Analysis of Multiplex Immunofluorescence Data. Cancers. 2021; 13(12):3031. https://doi.org/10.3390/cancers13123031
Chicago/Turabian StyleWilson, Christopher M., Oscar E. Ospina, Mary K. Townsend, Jonathan Nguyen, Carlos Moran Segura, Joellen M. Schildkraut, Shelley S. Tworoger, Lauren C. Peres, and Brooke L. Fridley. 2021. "Challenges and Opportunities in the Statistical Analysis of Multiplex Immunofluorescence Data" Cancers 13, no. 12: 3031. https://doi.org/10.3390/cancers13123031