
Inactivating Mutations of the IK Gene Weaken Ku80/Ku70-Mediated DNA Repair and Sensitize Endometrial Cancer to Chemotherapy

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
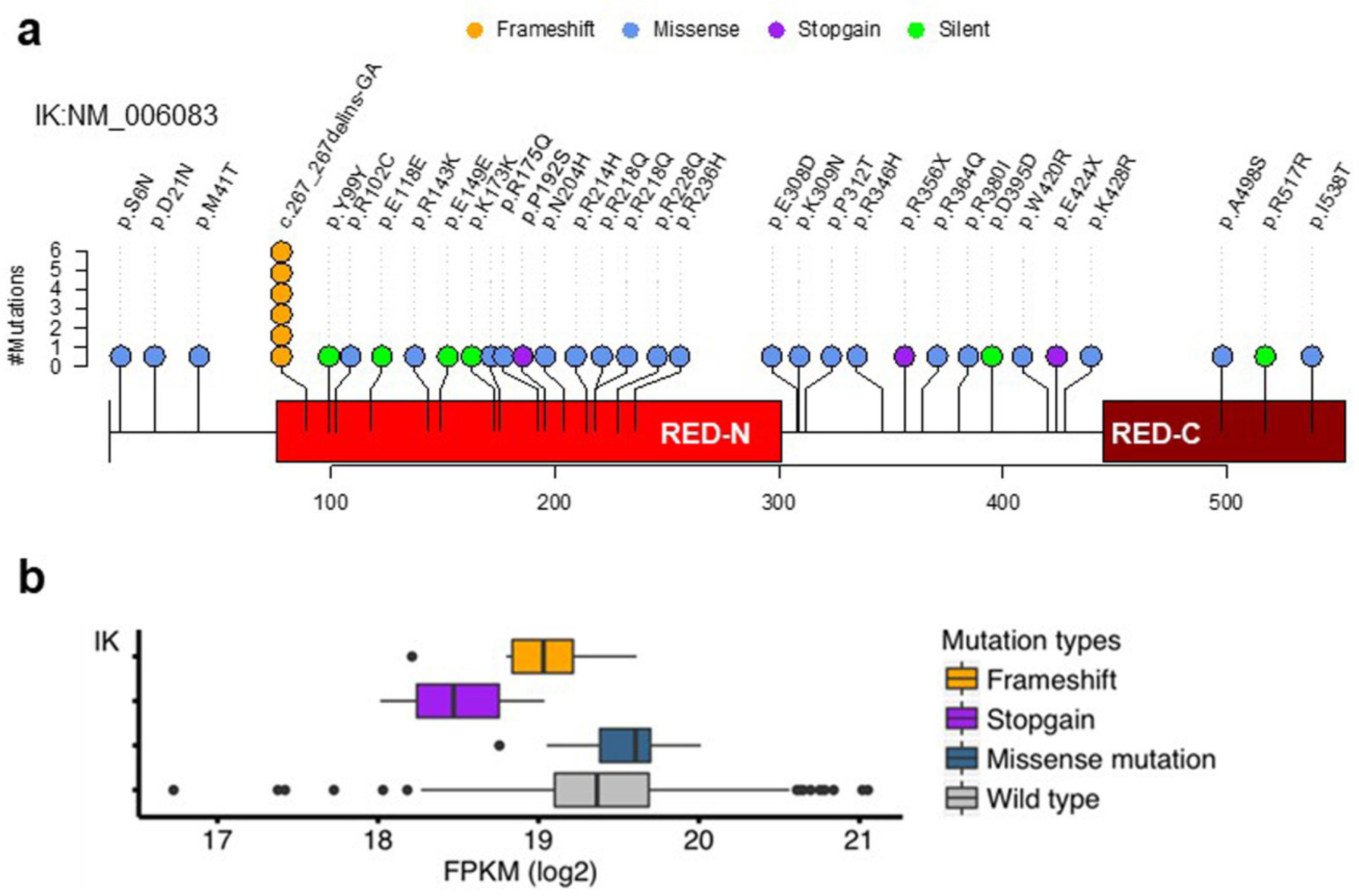
2.1. Somatic Mutations of the IK Gene in EEC
2.2. IK Attenuation Affects Cell Cycle and Sensitizes EC to Cisplatin In Vitro and In Vivo
2.3. IK Interacts with Ku80 and Modulates Ku80 Complexing with Ku70
2.4. IK Knockdown Leads to Inactivation of DNA Repair Signaling
2.5. Mutations of IK/Ku80 Are Associated with Higher Tumor Mutation Burden
3. Discussion
4. Materials and Methods
4.1. Somatic Mutation Calling for IK and DNA Repair Genes
4.2. Manual Inspection of an Indel of IK, exon5:c_267_267delines-GA
4.3. Cell Culture, Transfections, and Reagents
4.4. Western Blotting and IP Analysis
4.5. Cell Cycle Assays
4.6. Cell Viability Assays
4.7. Colony Formation Assays
4.8. Cell Death Assays
4.9. Cell Apoptosis Assays
4.10. Tumor Growth Assays
4.11. Silver Staining Assays
4.12. DNA Damage/γ-H2AX Assays
4.13. DNA Repair Assays
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silverberg, E.; Lubera, J. Cancer statistics, 1987. CA Cancer J. Clin. 1987, 37, 2–19. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suehiro, Y.; Umayahara, K.; Ogata, H.; Numa, F.; Yamashita, Y.; Oga, A.; Morioka, H.; Ito, T.; Kato, H.; Sasaki, K. Genetic aberrations detected by comparative genomic hybridization predict outcome in patients with endometrioid carcinoma. Genes Chromosomes Cancer 2000, 29, 75–82. [Google Scholar] [CrossRef]
- Talhouk, A.; Hoang, L.N.; McConechy, M.K.; Nakonechny, Q.; Leo, J.; Cheng, A.; Leung, S.; Yang, W.; Lum, A.; Kobel, M.; et al. Molecular classification of endometrial carcinoma on diagnostic specimens is highly concordant with final hysterectomy: Earlier prognostic information to guide treatment. Gynecol. Oncol. 2016, 143, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talhouk, A.; McConechy, M.K.; Leung, S.; Yang, W.; Lum, A.; Senz, J.; Boyd, N.; Pike, J.; Anglesio, M.; Kwon, J.S.; et al. Confirmation of ProMisE: A simple, genomics-based clinical classifier for endometrial cancer. Cancer 2017, 123, 802–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, J.; Pinto, C.; Pinto, D.; Pinheiro, M.; Silva, R.; Peixoto, A.; Rocha, P.; Veiga, I.; Santos, C.; Santos, R.; et al. POLE somatic mutations in advanced colorectal cancer. Cancer Med. 2017, 6, 2966–2971. [Google Scholar] [CrossRef] [PubMed]
- Guenther, M.; Veninga, V.; Kumbrink, J.; Haas, M.; Westphalen, C.B.; Kruger, S.; Heinemann, V.; Kirchner, T.; Boeck, S.; Jung, A.; et al. POLE gene hotspot mutations in advanced pancreatic cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 2161–2166. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Nakayama, K.; Nakamura, K.; Ono, R.; Yamashita, H.; Ishibashi, T.; Minamoto, T.; Iida, K.; Razia, S.; Ishikawa, N.; et al. High frequency of POLE mutations in synchronous endometrial and ovarian carcinoma. Hum. Pathol. 2019, 85, 92–100. [Google Scholar] [CrossRef]
- Li, Y.; Bian, Y.; Wang, K.; Wan, X.P. POLE mutations improve the prognosis of endometrial cancer via regulating cellular metabolism through AMF/AMFR signal transduction. BMC Med. Genet. 2019, 20, 202. [Google Scholar] [CrossRef]
- Stelloo, E.; Bosse, T.; Nout, R.A.; MacKay, H.J.; Church, D.N.; Nijman, H.W.; Leary, A.; Edmondson, R.J.; Powell, M.E.; Crosbie, E.J.; et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod. Pathol. 2015, 28, 836–844. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Patel, L.; Mills, G.B.; Lu, K.H.; Sood, A.K.; Ding, L.; Kucherlapati, R.; Mardis, E.R.; Levine, D.A.; Shmulevich, I.; et al. Clinical significance of CTNNB1 mutation and Wnt pathway activation in endometrioid endometrial carcinoma. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.R.; Reske, J.J.; Holladay, J.; Wilber, G.E.; Rhodes, M.; Koeman, J.; Adams, M.; Johnson, B.; Su, R.W.; Joshi, N.R.; et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat. Commun. 2019, 10, 3554. [Google Scholar] [CrossRef]
- Assier, E.; Bouzinba-Segard, H.; Stolzenberg, M.C.; Stephens, R.; Bardos, J.; Freemont, P.; Charron, D.; Trowsdale, J.; Rich, T. Isolation, sequencing and expression of RED, a novel human gene encoding an acidic-basic dipeptide repeat. Gene 1999, 230, 145–154. [Google Scholar] [CrossRef]
- Krief, P.; Boucheix, C.; Billard, C.; Mishal, Z.; Van Agthoven, A.; Fiers, W.; Azzarone, B. Modulation of expression of class II histocompatibility antigens by secretion of a cellular inhibitor in K562 leukemic cells. Eur. J. Immunol. 1987, 17, 1021–1025. [Google Scholar] [CrossRef]
- Zhou, Z.; Licklider, L.J.; Gygi, S.P.; Reed, R. Comprehensive proteomic analysis of the human spliceosome. Nature 2002, 419, 182–185. [Google Scholar] [CrossRef]
- Perutz, M.F.; Johnson, T.; Suzuki, M.; Finch, J.T. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 1994, 91, 5355–5358. [Google Scholar] [CrossRef] [Green Version]
- Pelsue, S.; Agris, P.F. Immunoreactivity between a monoclonal lupus autoantibody and the arginine/aspartic acid repeats within the U1-snRNP 70K autoantigen is conformationally restricted. J. Protein. Chem. 1994, 13, 401–408. [Google Scholar] [CrossRef]
- Cao, L.X.; Le Bousse-Kerdiles, M.C.; Clay, D.; Oshevski, S.; Jasmin, C.; Krief, P. Implication of a new molecule IK in CD34+ hematopoietic progenitor cell proliferation and differentiation. Blood 1997, 89, 3615–3623. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Han, S.; Jeong, A.L.; Park, J.S.; Yang, Y. Depletion of IK causes mitotic arrest through aberrant regulation of mitotic kinases and phosphatases. FEBS Lett. 2014, 588, 2844–2850. [Google Scholar] [CrossRef] [Green Version]
- Yeh, P.C.; Yeh, C.C.; Chen, Y.C.; Juang, Y.L. RED, a spindle pole-associated protein, is required for kinetochore localization of MAD1, mitotic progression, and activation of the spindle assembly checkpoint. J. Biol. Chem. 2012, 287, 11704–11716. [Google Scholar] [CrossRef] [Green Version]
- Neumann, B.; Walter, T.; Heriche, J.K.; Bulkescher, J.; Erfle, H.; Conrad, C.; Rogers, P.; Poser, I.; Held, M.; Liebel, U.; et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 2010, 464, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Filiaci, V.; Randall, M.E.; Mutch, D.; Steinhoff, M.M.; DiSilvestro, P.A.; Moxley, K.M.; Kim, Y.M.; Powell, M.A.; O’Malley, D.M.; et al. Adjuvant Chemotherapy plus Radiation for Locally Advanced Endometrial Cancer. N. Engl. J. Med. 2019, 380, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Xue, F.; Zhang, W. miR-506: A regulator of chemo-sensitivity through suppression of the RAD51-homologous recombination axis. Chin. J. Cancer 2015, 34, 485–487. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.; Davis, A.J.; Fattah, K.R.; So, S.; Sun, J.; Lee, K.J.; Harrison, L.; Yang, J.; Chen, D.J. Persistently bound Ku at DNA ends attenuates DNA end resection and homologous recombination. DNA Repair 2012, 11, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.J.; Yan, C.T. Regulation of DNA repair in the absence of classical non-homologous end joining. DNA Repair 2018, 68, 34–40. [Google Scholar] [CrossRef]
- Yang, G.; Komaki, Y.; Yoshida, I.; Ibuki, Y. Formaldehyde inhibits UV-induced phosphorylation of histone H2AX. Toxicol. In Vitro 2019, 61, 104687. [Google Scholar] [CrossRef]
- Feng, X.; Tang, R.; Zhang, R.; Wang, H.; Ji, Z.; Shao, Y.; Wang, S.; Zhong, T.; Gu, Y.; Meng, J. A comprehensive analysis of IDO1 expression with tumour-infiltrating immune cells and mutation burden in gynaecologic and breast cancers. J. Cell Mol. Med. 2020, 24, 5238–5248. [Google Scholar] [CrossRef] [Green Version]
- Temko, D.; Van Gool, I.C.; Rayner, E.; Glaire, M.; Makino, S.; Brown, M.; Chegwidden, L.; Palles, C.; Depreeuw, J.; Beggs, A.; et al. Somatic POLE exonuclease domain mutations are early events in sporadic endometrial and colorectal carcinogenesis, determining driver mutational landscape, clonal neoantigen burden and immune response. J. Pathol. 2018, 245, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shmulevich, I.; Sood, A.K.; Zhang, W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA 2011, 306, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Papadopoulos, N.; McKinley, A.J.; Farrington, S.M.; Curtis, L.J.; Wyllie, A.H.; Zheng, S.; Willson, J.K.; Markowitz, S.D.; Morin, P.; et al. APC mutations in colorectal tumors with mismatch repair deficiency. Proc. Natl. Acad. Sci. USA 1996, 93, 9049–9054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Jeong, A.L.; Park, J.S.; Han, S.; Jang, C.Y.; Kim, K.I.; Kim, Y.; Park, J.H.; Lim, J.S.; Lee, M.S.; et al. IK-guided PP2A suppresses Aurora B activity in the interphase of tumor cells. Cell Mol. Life Sci. 2016, 73, 3375–3386. [Google Scholar] [CrossRef] [PubMed]
- Richardson, G.S.; Dickersin, G.R.; Atkins, L.; MacLaughlin, D.T.; Raam, S.; Merk, L.P.; Bradley, F.M. KLE: A cell line with defective estrogen receptor derived from undifferentiated endometrial cancer. Gynecol. Oncol. 1984, 17, 213–230. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Jin, X.; Medina, C.B.; Leonhardt, S.A.; Kiessling, V.; Bennett, B.C.; Shu, S.; Tamm, L.K.; Yeager, M.; Ravichandran, K.S.; et al. A quantized mechanism for activation of pannexin channels. Nat. Commun. 2017, 8, 14324. [Google Scholar] [CrossRef]
- Kim, M.J.; Kwon, S.B.; Kim, M.S.; Jin, S.W.; Ryu, H.W.; Oh, S.R.; Yoon, D.Y. Trifolin induces apoptosis via extrinsic and intrinsic pathways in the NCI-H460 human non-small cell lung-cancer cell line. Phytomedicine 2016, 23, 998–1004. [Google Scholar] [CrossRef]
- Wu, Y.H.; Huang, Y.F.; Chen, C.C.; Huang, C.Y.; Chou, C.Y. Comparing PI3K/Akt Inhibitors Used in Ovarian Cancer Treatment. Front. Pharmacol. 2020, 11, 206. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef]
- Grundy, G.J.; Rulten, S.L.; Arribas-Bosacoma, R.; Davidson, K.; Kozik, Z.; Oliver, A.W.; Pearl, L.H.; Caldecott, K.W. The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nat. Commun. 2016, 7, 11242. [Google Scholar] [CrossRef]
- Gu, J.; Lu, H.; Tippin, B.; Shimazaki, N.; Goodman, M.F.; Lieber, M.R. XRCC4:DNA ligase IV can ligate incompatible DNA ends and can ligate across gaps. EMBO J. 2007, 26, 1010–1023. [Google Scholar] [CrossRef] [Green Version]
- Ahnesorg, P.; Smith, P.; Jackson, S.P. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 2006, 124, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Buck, D.; Malivert, L.; de Chasseval, R.; Barraud, A.; Fondaneche, M.C.; Sanal, O.; Plebani, A.; Stephan, J.L.; Hufnagel, M.; le Deist, F.; et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 2006, 124, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbs, T.A.; Tainer, J.A.; Lees-Miller, S.P. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair 2010, 9, 1307–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Difilippantonio, M.J.; Zhu, J.; Chen, H.T.; Meffre, E.; Nussenzweig, M.C.; Max, E.E.; Ried, T.; Nussenzweig, A. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 2000, 404, 510–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Tamburini, B.A.; Tyler, J.K. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol. Cell Biol. 2005, 25, 4903–4913. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xu, X. DNA double-strand break repair: A tale of pathway choices. Acta. Biochim. Biophys. Sin. 2016, 48, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of double-strand breaks by end joining. Cold Spring Harb. Perspect. Biol. 2013, 5, a012757. [Google Scholar] [CrossRef]
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell 2012, 47, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Sakogawa, K.; Aoki, Y.; Misumi, K.; Hamai, Y.; Emi, M.; Hihara, J.; Shi, L.; Kono, K.; Horikoshi, Y.; Sun, J.; et al. Involvement of homologous recombination in the synergism between cisplatin and poly (ADP-ribose) polymerase inhibition. Cancer Sci. 2013, 104, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, R.; Russo, N.; Liu, M.; Basrur, V.; Bellile, E.; Palanisamy, N.; Scanlon, C.S.; van Tubergen, E.; Inglehart, R.C.; Metwally, T.; et al. TRIP13 promotes error-prone nonhomologous end joining and induces chemoresistance in head and neck cancer. Nat. Commun. 2014, 5, 4527. [Google Scholar] [CrossRef] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Church, D.N.; Briggs, S.E.; Palles, C.; Domingo, E.; Kearsey, S.J.; Grimes, J.M.; Gorman, M.; Martin, L.; Howarth, K.M.; Hodgson, S.V.; et al. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 2013, 22, 2820–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome. Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, D.E.; Harris, C.C.; Chen, K.; Koboldt, D.C.; Abbott, T.E.; Dooling, D.J.; Ley, T.J.; Mardis, E.R.; Wilson, R.K.; Ding, L. SomaticSniper: Identification of somatic point mutations in whole genome sequencing data. Bioinformatics 2012, 28, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Skidmore, Z.L.; Wagner, A.H.; Lesurf, R.; Campbell, K.M.; Kunisaki, J.; Griffith, O.L.; Griffith, M. GenVisR: Genomic Visualizations in R. Bioinformatics 2016, 32, 3012–3014. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- Costello, M.; Pugh, T.J.; Fennell, T.J.; Stewart, C.; Lichtenstein, L.; Meldrim, J.C.; Fostel, J.L.; Friedrich, D.C.; Perrin, D.; Dionne, D.; et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013, 41, e67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Takahara, A.; Hagio, T.; Nishiko, R.; Kanayama, J.; Gotoh, O.; Mori, S. Sequencing artifacts derived from a library preparation method using enzymatic fragmentation. PLoS ONE 2020, 15, e0227427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Peng, C.; Chen, J.; Chen, D.; Yang, B.; He, B.; Hu, W.; Zhang, Y.; Liu, H.; Dai, L.; et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol. Cancer 2019, 18, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landen, C.N., Jr.; Chavez-Reyes, A.; Bucana, C.; Schmandt, R.; Deavers, M.T.; Lopez-Berestein, G.; Sood, A.K. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005, 65, 6910–6918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisamatsu, T.; McGuire, M.; Wu, S.Y.; Rupaimoole, R.; Pradeep, S.; Bayraktar, E.; Noh, K.; Hu, W.; Hansen, J.M.; Lyons, Y.; et al. PRKRA/PACT Expression Promotes Chemoresistance of Mucinous Ovarian Cancer. Mol. Cancer Ther. 2019, 18, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleire, L.; Skeie, B.S.; Netland, I.A.; Forde, H.E.; Dodoo, E.; Selheim, F.; Leiss, L.; Heggdal, J.I.; Pedersen, P.H.; Wang, J.; et al. Drug repurposing: Sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene 2015, 34, 5951–5959. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S.; Mukherjee, J.; See, W.L.; Pieper, R.O. Mutant IDH1-driven cellular transformation increases RAD51-mediated homologous recombination and temozolomide resistance. Cancer Res. 2014, 74, 4836–4844. [Google Scholar] [CrossRef] [Green Version]
- Bigot, N.; Mouche, A.; Preti, M.; Loisel, S.; Renoud, M.L.; Le Guevel, R.; Sensebe, L.; Tarte, K.; Pedeux, R. Hypoxia Differentially Modulates the Genomic Stability of Clinical-Grade ADSCs and BM-MSCs in Long-Term Culture. Stem. Cells 2015, 33, 3608–3620. [Google Scholar] [CrossRef] [Green Version]
- Turner, K.M.; Sun, Y.; Ji, P.; Granberg, K.J.; Bernard, B.; Hu, L.; Cogdell, D.E.; Zhou, X.; Yli-Harja, O.; Nykter, M.; et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3421–3426. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, C.; Jin, G.; Forbes, E.; Mangala, L.S.; Wang, Y.; Rodriguez-Aguayo, C.; Amero, P.; Bayraktar, E.; Yan, Y.; Lopez-Berestein, G.; et al. Inactivating Mutations of the IK Gene Weaken Ku80/Ku70-Mediated DNA Repair and Sensitize Endometrial Cancer to Chemotherapy. Cancers 2021, 13, 2487. https://doi.org/10.3390/cancers13102487
Gao C, Jin G, Forbes E, Mangala LS, Wang Y, Rodriguez-Aguayo C, Amero P, Bayraktar E, Yan Y, Lopez-Berestein G, et al. Inactivating Mutations of the IK Gene Weaken Ku80/Ku70-Mediated DNA Repair and Sensitize Endometrial Cancer to Chemotherapy. Cancers. 2021; 13(10):2487. https://doi.org/10.3390/cancers13102487
Chicago/Turabian StyleGao, Chao, Guangxu Jin, Elizabeth Forbes, Lingegowda S. Mangala, Yingmei Wang, Cristian Rodriguez-Aguayo, Paola Amero, Emine Bayraktar, Ye Yan, Gabriel Lopez-Berestein, and et al. 2021. "Inactivating Mutations of the IK Gene Weaken Ku80/Ku70-Mediated DNA Repair and Sensitize Endometrial Cancer to Chemotherapy" Cancers 13, no. 10: 2487. https://doi.org/10.3390/cancers13102487