
Clinical, Histological, and Molecular Features of Solitary Fibrous Tumor of Bone: A Single Institution Retrospective Review

 , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Histopathology Evaluation
2.2. Immunohistochemistry
2.3. DNA and RNA Isolation
2.4. Detection of NAB2-STAT6 Fusion Variants
2.5. TERT Promoter Mutation Analysis
2.6. Analysis of p53 Mutation
2.7. Statistics
3. Results
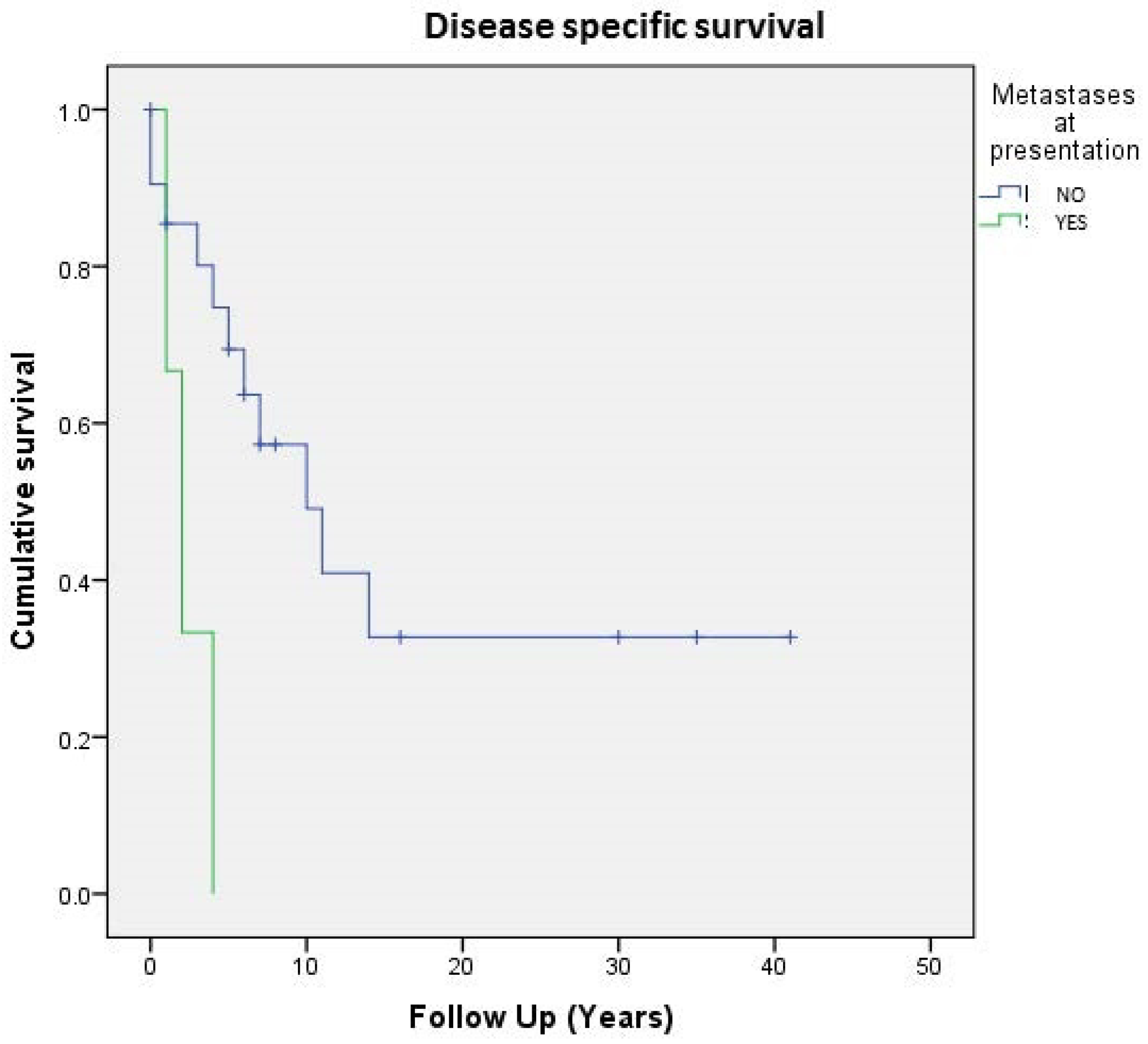
3.1. Clinicopathological Evaluation
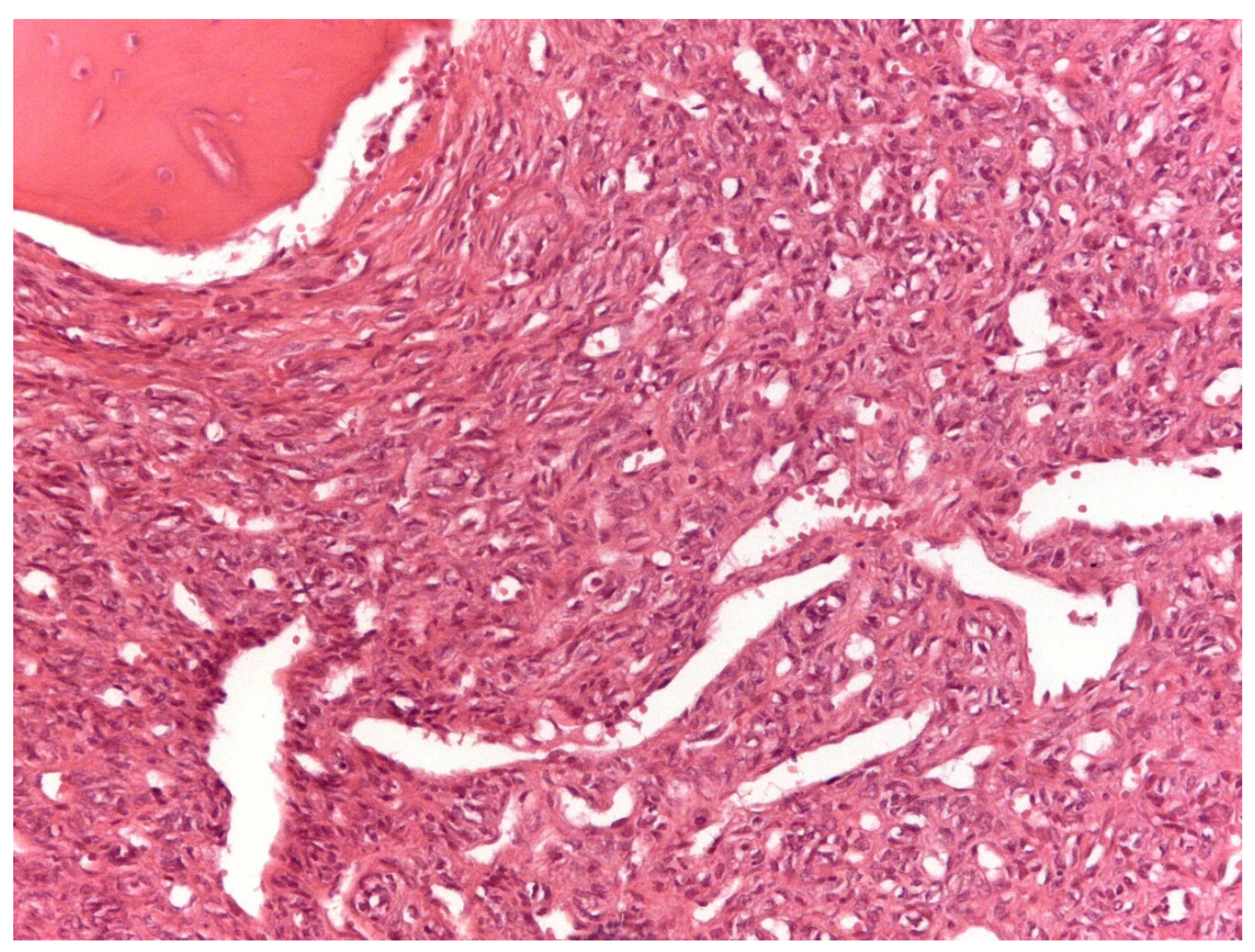
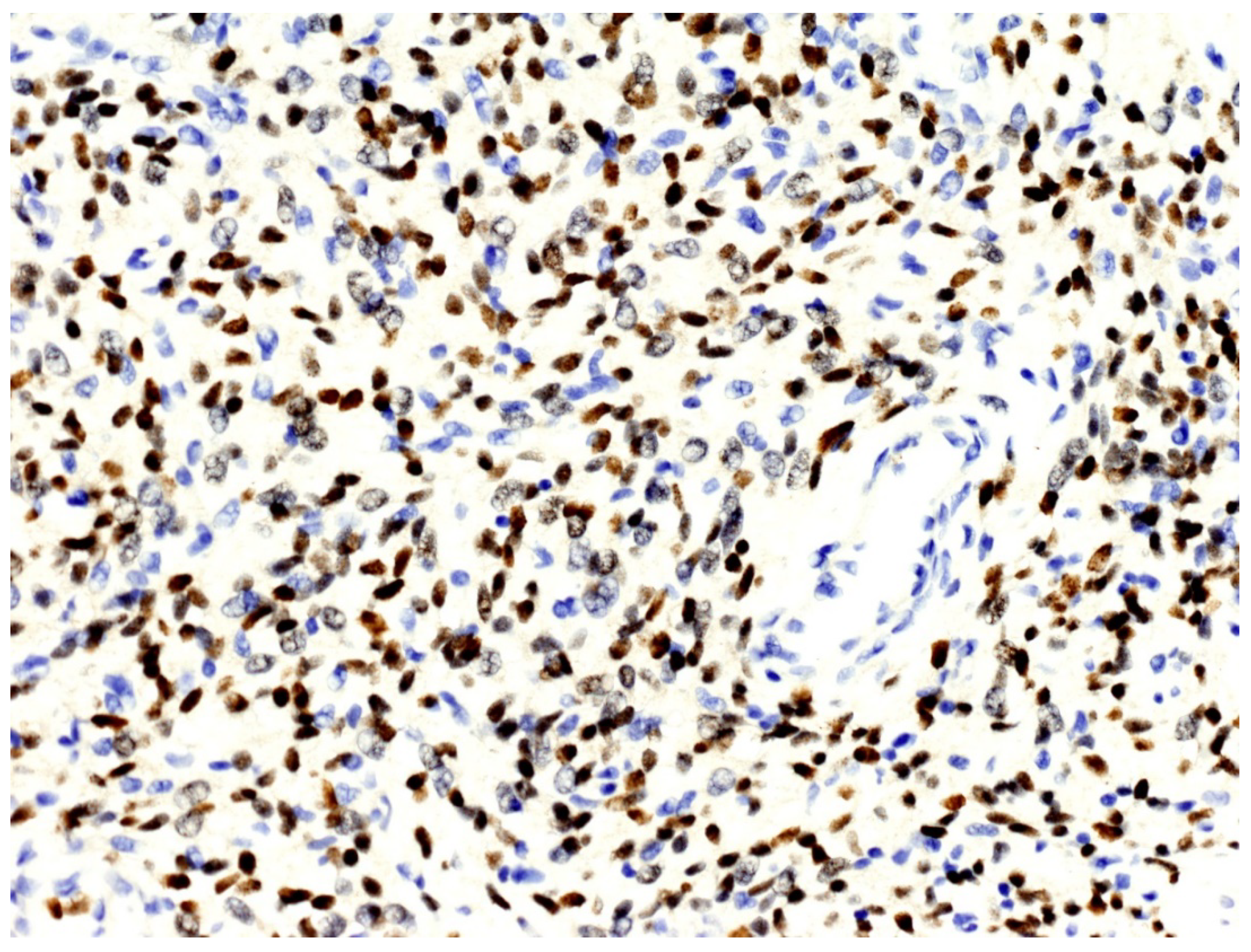
3.2. Histopathological and Immunohistochemical Features
3.3. NAB2–STAT6 Fusion Variants
3.4. TERT Promoter Mutations: C228T and C250T
3.5. p53 Mutations
3.6. Correlations between Clinicopathological, Immunohistochemical, and Molecular Data
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Demicco, E.G.; Fritchie, K.J.; Han, A. Solitary fibrous tumor. In WHO Classification of Tumours 5th Edition: Soft Tissue and Bone Tumours; WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; Volume 5, pp. 104–108. [Google Scholar]
- Klemperer, P.; Rabin, C.B. Primary neoplasms of the pleura. Arch. Pathol. 1931, 11, 385–412. [Google Scholar]
- Picci, P.; Manfrini, M.; Donati, D.M.; Gambarotti, M.; Righi, A.; Vanel, D.; Dei Tos, A.P. Diagnosis of Musculoskeletal Tumors and Tumor-Like Conditions. Clinical, Radiological and Histological Correlations—The Rizzoli Case Archive, 2nd ed.; Springer: Cham, Switzerland, 2020; pp. 3–11. [Google Scholar]
- Verbeke, S.L.; Fletcher, C.D.; Alberghini, M.; Daugaard, S.; Flanagan, A.M.; Parratt, T.; Kroon, H.M.; Hogendoorn, P.; Bovée, J.V. A Reappraisal of Hemangiopericytoma of Bone; Analysis of Cases Reclassified as Synovial Sarcoma and Solitary Fibrous Tumor of Bone. Am. J. Surg. Pathol. 2010, 34, 777–783. [Google Scholar] [CrossRef]
- Dei Tos, A.P.; Righi, A.; Gambarotti, M.; Vanel, D.; Ferrari, C.; Benini, S.; Ferrari, S.; Picci, P. Reappraisal of primary spindle/pleomorphic sarcoma of bone. Mod. Pathol. 2014, 27, 15A. [Google Scholar]
- Fletcher, C.D. Haemangiopericytoma- A dying breed? Reappraisal of an ‘entity’ and its variants: A hypothesis. Curr. Diagn. Pathol. 1994, 1, 19–23. [Google Scholar] [CrossRef]
- Fletcher, C.D. The evolving classification of soft tissue tumours: An update based on the new WHO classification. Histopathology 2006, 48, 3–12. [Google Scholar] [CrossRef]
- Coca-Pelaz, A.; Llorente-Pendás, J.L.; Vivanco-Allende, B.; Suarez-Nieto, C. Solitary fibrous tumor of the petrous bone: A successful treatment option. Acta Oto-Laryngol. 2011, 131, 1349–1352. [Google Scholar] [CrossRef]
- Ge, X.; Liao, J.; Choo, R.J.; Yan, J.; Zhang, J. Solitary fibrous tumor of the ilium. Medicine 2017, 96, e9355. [Google Scholar] [CrossRef]
- Suarez-Zamora, D.A.; Rodriguez-Urrego, P.A.; Soto-Montoya, C.; Rivero-Rapalino, O.; Palau-Lazaro, M.A. Malignant Solitary Fibrous Tumor of the Humerus: A Case Report of an Extremely Rare Primary Bone Tumor. Int. J. Surg. Pathol. 2018, 26, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Crim, J.; Evenski, A.; Layfield, L.J. Solitary fibrous tumor of bone developing lung metastases on long-term follow-up. Skelet. Radiol. 2020, 49, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Sbaraglia, M.; Righi, A.; Gambarotti, M.; Vanel, D.; Picci, P.; Tos, A.P.D. Soft Tissue Tumors Rarely Presenting Primary in Bone; Diagnostic Pitfalls. Surg. Pathol. Clin. 2017, 10, 705–730. [Google Scholar] [CrossRef] [PubMed]
- England, D.M.; Hochholzer, L.; McCarthy, M.J. Localized Benign and Malignant Fibrous Tumors of the Pleura. Am. J. Surg. Pathol. 1989, 13, 640–658. [Google Scholar] [CrossRef] [PubMed]
- Gold, J.S.; Antonescu, C.R.; Hajdu, C.; Ferrone, C.R.; Hussain, M.; Lewis, J.J.; Brennan, M.F.; Coit, D.G. Clinicopathologic correlates of solitary fibrous tumors. Cancer 2002, 94, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Gholami, S.; Cassidy, M.R.; Kirane, A.; Kuk, D.; Zanchelli, B.; Antonescu, C.R.; Singer, S.; Brennan, M. Size and Location are the Most Important Risk Factors for Malignant Behavior in Resected Solitary Fibrous Tumors. Ann. Surg. Oncol. 2017, 24, 3865–3871. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Choi, Y.-L.; Kim, Y.J.; Park, H.K.; Kima, J.M.; Choia, Y.-L.; Parka, H.K. Comparison and evaluation of risk factors for meningeal, pleural, and extrapleural solitary fibrous tumors: A clinicopathological study of 92 cases confirmed by STAT6 immunohistochemical staining. Pathol. Res. Pract. 2017, 213, 619–625. [Google Scholar] [CrossRef]
- Doyle, L.A.; Fletcher, C.D.M. Predicting behavior of solitary fibrous tumor: Are we getting closer to more accurate risk assessment? Ann. Surg. Oncol. 2013, 20, 4055–4056. [Google Scholar] [CrossRef] [Green Version]
- Demicco, E.G.; Park, M.S.; Araujo, D.M.; Fox, P.S.; Bassett, R.L.; E Pollock, R.; Lazar, A.J.; Wang, W.-L. Solitary fibrous tumor: A clinicopathological study of 110 cases and proposed risk assessment model. Mod. Pathol. 2012, 25, 1298–1306. [Google Scholar] [CrossRef] [Green Version]
- Pasquali, S.; Gronchi, A.; Strauss, D.; Bonvalot, S.; Jeys, L.; Stacchiotti, S.; Hayes, A.; Honore, C.; Collini, P.; Renne, S.L.; et al. Resectable extra-pleural and extra-meningeal solitary fibrous tumours: A multi-centre prognostic study. Eur. J. Surg. Oncol. (EJSO) 2016, 42, 1064–1070. [Google Scholar] [CrossRef]
- Salas, S.; Resseguier, N.; Blay, J.Y.; Le Cesne, A.; Italiano, A.; Chevreau, C.; Rosset, P.; Isambert, N.; Soulie, P.; Cupissol, D.; et al. Prediction of local and metastatic recurrence in solitary fibrous tumor: Construction of a risk calculator in a multicenter cohort from the French Sarcoma Group (FSG) database. Ann. Oncol. 2017, 28, 1779–1787. [Google Scholar] [CrossRef]
- Machado, I.; Morales, G.N.; Cruz, J.; Lavernia, J.; Giner, F.; Navarro, S.; Ferrandez, A.; Llombart-Bosch, A. Solitary fibrous tumor: A case series identifying pathological adverse factors—implications for risk stratification and classification. Virchows Arch. 2019, 476, 597–607. [Google Scholar] [CrossRef]
- Demicco, E.G.; Wagner, M.J.; Maki, R.G.; Gupta, V.; Iofin, I.; Lazar, A.J.; Wang, W.-L. Risk assessment in solitary fibrous tumors: Validation and refinement of a risk stratification model. Mod. Pathol. 2017, 30, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Crago, A.M.; Rosenberg, M.; O’Connor, R.; Walker, S.R.; Ambrogio, L.; Auclair, D.; McKenna, A.; Heinrich, M.C.; Frank, D.A.; et al. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat. Genet. 2013, 45, 131–132. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.R.; Wu, Y.-M.; Kalyana-Sundaram, S.; Cao, X.; Lonigro, R.J.; Sung, Y.-S.; Chen, C.-L.; Zhang, L.; Wang, R.; Su, F.; et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat. Genet. 2013, 45, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Barthelmeß, S.; Geddert, H.; Boltze, C.; Moskalev, E.A.; Bieg, M.; Sirbu, H.; Brors, B.; Wiemann, S.; Hartmann, A.; Agaimy, A.; et al. Solitary Fibrous Tumors/Hemangiopericytomas with Different Variants of the NAB2-STAT6 Gene Fusion Are Characterized by Specific Histomorphology and Distinct Clinicopathological Features. Am. J. Pathol. 2014, 184, 1209–1218. [Google Scholar] [CrossRef]
- Bahrami, A.; Lee, S.; Schaefer, I.-M.; Boland, J.M.; Patton, K.T.; Pounds, S.; Fletcher, C.D. TERT promoter mutations and prognosis in solitary fibrous tumor. Mod. Pathol. 2016, 29, 1511–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, G.; Sambri, A.; Pedrini, E.; Pazzaglia, L.; Sangiorgi, L.; Ruengwanichayakun, P.; Donati, D.; Benassi, M.S.; Righi, A. Histological and molecular features of solitary fibrous tumor of the extremities: Clinical correlation. Virchows Arch. 2020, 476, 445–454. [Google Scholar] [CrossRef]
- Park, H.K.; Yu, D.B.; Sung, M.; Oh, E.; Kim, M.; Song, J.-Y.; Lee, M.-S.; Jung, K.; Noh, K.-W.; An, S.; et al. Molecular changes in solitary fibrous tumor progression. J. Mol. Med. 2019, 97, 1413–1425. [Google Scholar] [CrossRef] [Green Version]
- Tai, H.-C.; Chuang, I.-C.; Chen, T.-C.; Li, C.-F.; Huang, S.-C.; Kao, Y.-C.; Lin, P.-C.; Tsai, J.-W.; Lan, J.; Yu, S.-C.; et al. NAB2–STAT6 fusion types account for clinicopathological variations in solitary fibrous tumors. Mod. Pathol. 2015, 28, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Akaike, K.; Kurisaki-Arakawa, A.; Hara, K.; Suehara, Y.; Takagi, T.; Mitani, K.; Kaneko, K.; Yao, T.; Saito, T. Distinct clinicopathological features of NAB2-STAT6 fusion gene variants in solitary fibrous tumor with emphasis on the acquisition of highly malignant potential. Hum. Pathol. 2015, 46, 347–356. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient | Age | Sex | Anatomical Site | Tumor Size (cm) | Metastases | Surgical Procedure | Surgical Margins | DeMicco Score | Local Recurrence | Follow-Up (Months) | Status |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 75 | F | Proximal tibia | 15 | Yes (3) | Thigh amputation | Wide | High | No | 8 | DOD |
| 2 | 61 | M | Distal femur | 12.5 | Yes (5) | Thigh amputation | Wide | High | No | 15 | DOD |
| 3 | 66 | M | Distal fibula | 14 | Yes (0) | Leg Amputation | Radical | Intermediate | No | 58 | DOD |
| 4 | 71 | F | Scapula | 8 | Yes (0) | Resection | Wide | Intermediate | No | 28 | DOD |
| 5 | 84 | M | Iliac wing | 20 | No | Inoperable | NA | Intermediate Inter | Yes (2) | 2 | DOD |
| 6 | 52 | F | Ileum pubic branch | 7 | No | Inoperable | NA | Low | Yes (24) | 56 | DOD |
| 7 | 54 | F | Femoral shaft | 11 | Yes (108) | Resection | Wide | Intermediate | Yes (149) | 168 | DOD |
| 8 | 58 | M | Sacrum | 7.5 | Yes (75) | Curettage | Marginal | Intermediate | Yes (97) | 122 | DOD |
| 9 | 44 | F | Distal femur | 8.5 | Yes (66) | Resection | Wide | Low | No | 87 | DOD |
| 10 | 39 | F | Proximal tibia | 5 | No | Resection | Wide | Low | No | 421 | NED |
| 11 | 47 | M | Scapula | 6 | No | Scapulectomy | Wide | Intermediate Intermediate | No | 96 | DOC |
| 12 | 44 | M | Sacrum | 7 | No | Inoperable | NA | Low | No | 73 | DOD |
| 13 | 50 | F | Proximal humerus | 16 | Yes (30) | ISTA | Radical | Intermediate | No | 361 | NED |
| 14 | 58 | M | Ischium pubic branch | 8 | No | Resection | Wide | Low | No | 0 | DOC |
| 15 | 61 | F | Iliac wing | 8 | Yes (84) | Resection | Intralesional | Low | No | 137 | DOD |
| 16 | 27 | F | Distal femur | 17 | No | IIAA | Intralesional | Intermediate | No | 495 | NED |
| 17 | 32 | F | Sacrum | 15 | No | Inoperable | NA | High | No | 41 | DOD |
| 18 | 54 | M | Sacrum | 10 | No | Resection | Wide | Low | No | 67 | NED |
| 19 | 49 | M | Proximal humerus | 19 | No | ISTA | Radical | High | No | 92 | DOC |
| 20 | 31 | F | 4th lumbar vertebra | 8 | Yes (38) | Vertebrectomy | Wide | Low | No | 66 | DOD |
| 21 | 21 | F | Proximal humerus | 9.5 | Yes (72) | Resection | Wide | Intermediate | Yes (72) | 72 | AWD |
| 22 | 7 | F | Humeral shaft | 11 | No | Inoperable | NA | Intermediate | No | 196 | NED |
| 23 | 60 | F | Proximal femur | 7.3 | Yes (0) | Resection | Wide | Intermediate | No | 21 | DOD |
| 24 | 69 | M | Femoral shaft | 10.5 | No | Resection | Wide | High | No | 18 | NED |
| Patient | Mitotic Count (X10 HPF) | Necrosis (≥10%) | STAT6 | CD34 | TERT Mutation | NAB2-STAT6 Fusion | P53 Variants (Variant Type) | Grade of Malignancy |
|---|---|---|---|---|---|---|---|---|
| 1 | ≥4 | Yes | Pos | Pos | C250C/C228C | NA | yes (nonsense) | HIGH |
| 2 | ≥4 | Yes | Pos | Pos | C250C/C228C | EX6-EX17 | yes (CNV deletion) | HIGH |
| 3 | <4 | Yes | Pos | Pos | NA | NA | NA | LOW |
| 4 | ≥4 | Yes | Pos | Pos | C250C/C228C | NA | NA | HIGH |
| 5 | <4 | No | Pos | Pos | NA | NA | NA | LOW |
| 6 | <4 | No | Pos | Pos | NA | NA | NA | LOW |
| 7 | ≥4 | Yes | Pos | Pos | C250C/C228C | EX2-EX2 | yes (CNV deletion) | HIGH |
| 8 | ≥4 | Yes | Pos | Pos | C250C/C228C | EX6-EX16 | NA | HIGH |
| 9 | <4 | No | Pos | Neg | NA | NA | NA | LOW |
| 10 | ≥4 | No | Pos | Pos | NA | NA | NA | HIGH |
| 11 | ≥4 | Yes | Pos | Pos | C250C/C228C | NA | yes (splice site) | HIGH |
| 12 | <4 | No | Pos | Pos | C250C/C228C | EX4-EX2 | NA | LOW |
| 13 | ≥4 | No | Pos | Pos | C250C/C228C | NA | yes (CNV deletion) | HIGH |
| 14 | <4 | No | Pos | Pos | C250C/C228T | OTHER | no | LOW |
| 15 | <4 | No | Pos | Pos | C250C/C228C | OTHER | yes (CNV deletion) | LOW |
| 16 | <4 | Yes | Pos | Pos | C250C/C228C | EX6-EX16/EX6-EX17 | yes (missense + CNV amplification) | LOW |
| 17 | ≥4 | Yes | Pos | Pos | NA | NA | NA | HIGH |
| 18 | <4 | No | Pos | Pos | C250C/C228C | EX4-EX2 | yes (CNV deletion) | LOW |
| 19 | ≥4 | Yes | Pos | Pos | C250C/C228C | EX6-EX17 | yes (CNV deletion) | HIGH |
| 20 | ≥4 | No | Pos | Pos | NA | NA | NA | HIGH |
| 21 | ≥4 | Yes | Pos | Pos | C250C/C228C | EX6-EX17 | yes (CNV duplication) | HIGH |
| 22 | ≥4 | No | Pos | Pos | NA | NA | NA | HIGH |
| 23 | ≥4 | Yes | Pos | Pos | C250C/C228C | EX6-EX17 | yes (CNV deletion) | HIGH |
| 24 | ≥4 | Yes | Pos | Pos | C250C/C228C | EX4-EX2 | NA | HIGH |
| Variables | Disease Specific Survival (24 pts) | Localized Disease * (16 pts) | |||||
|---|---|---|---|---|---|---|---|
| 5 Years-DSS | 10 Years-DSS | p-Value | 5 Years-DSS | 10 Years-DSS | p-Value | ||
| Histological Grade | |||||||
| Low | 62% | 31% | 0.52 | 100% | 67% | 0.84 | |
| High | 65% | 58% | 82% | 71% | |||
| Size | |||||||
| (A) 0–4.99 cm | 100% | 100% | 0.44 | 100% | 100% | 0.54 | |
| (B) 5–9.99 cm | 70% | 36% | 87% | 45% | |||
| (C)10–14.99 cm | 62% | 62% | 80% | 80% | |||
| (D) >15 | 50% | 50% | 50% | 50% | |||
| Age | |||||||
| <55 years | 86% | 27% | 0.06 | 100% | 77% | 0.15 | |
| ≥55 years | 61% | 27% | 100% | 60% | |||
| Mitosis | |||||||
| (A) <1 | 60% | 30% | 0.54 | 60% | 30% | 0.33 | |
| (B) 1–3 | 66% | 33% | 100% | 50% | |||
| (C) ≥4 | 65% | 58% | 76% | 68% | |||
| Necrosis | |||||||
| <10% | 80% | 47% | 0.66 | 100% | 62% | 0.95 | |
| ≥10% | 51% | 51% | 78% | 78% | |||
| Gene Fusion | |||||||
| Exon6 | 80% | 40% | 0.68 | ||||
| Other | 100% | 67% | |||||
| Demicco Score Risk | |||||||
| High | 54% | 54% | 0.43 | ||||
| Intermediate | 72% | 46% | |||||
| Low | 64% | 28% | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianchi, G.; Lana, D.; Gambarotti, M.; Ferrari, C.; Sbaraglia, M.; Pedrini, E.; Pazzaglia, L.; Sangiorgi, L.; Bartolotti, I.; Dei Tos, A.P.; et al. Clinical, Histological, and Molecular Features of Solitary Fibrous Tumor of Bone: A Single Institution Retrospective Review. Cancers 2021, 13, 2470. https://doi.org/10.3390/cancers13102470
Bianchi G, Lana D, Gambarotti M, Ferrari C, Sbaraglia M, Pedrini E, Pazzaglia L, Sangiorgi L, Bartolotti I, Dei Tos AP, et al. Clinical, Histological, and Molecular Features of Solitary Fibrous Tumor of Bone: A Single Institution Retrospective Review. Cancers. 2021; 13(10):2470. https://doi.org/10.3390/cancers13102470
Chicago/Turabian StyleBianchi, Giuseppe, Debora Lana, Marco Gambarotti, Cristina Ferrari, Marta Sbaraglia, Elena Pedrini, Laura Pazzaglia, Luca Sangiorgi, Isabella Bartolotti, Angelo Paolo Dei Tos, and et al. 2021. "Clinical, Histological, and Molecular Features of Solitary Fibrous Tumor of Bone: A Single Institution Retrospective Review" Cancers 13, no. 10: 2470. https://doi.org/10.3390/cancers13102470