
Conformation-Specific Inhibitory Anti-MMP-7 Monoclonal Antibody Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Chemotherapeutic Cell Kill

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibody Generation and Purification
2.2. Antibody Digestion with Papain for Fab Generation
2.3. MMP-7 Elisa Binding Assay
2.4. MMP-7 Enzymatic Kinetic Assay
2.5. Antibody Sequencing
2.6. Protein Crystallography of GSM-192 Fab
2.7. Computational Modeling and Docking
2.8. Constructing and Analyzing, MMP Ortholog-Based Multiple Sequence Alignment (MSA)
2.9. Structural Alignment and Analysis
2.10. Cell Culture
2.11. MMP-7 Lentiviral Silencing and FACS Analysis
2.12. MTT Cell Proliferation Assay
2.13. FACS Annexin-V Apoptosis Detection Assay
2.14. Western Blots
2.15. Wound Healing Assay (Scratch Assay)
2.16. Trans-Well Migration Assay
2.17. Immunohistochemistry
2.18. Co-Treatment with Chemotherapeutics
2.19. In Vitro Dose Response Matrix and Synergistic Effect
2.20. Statistics
3. Results
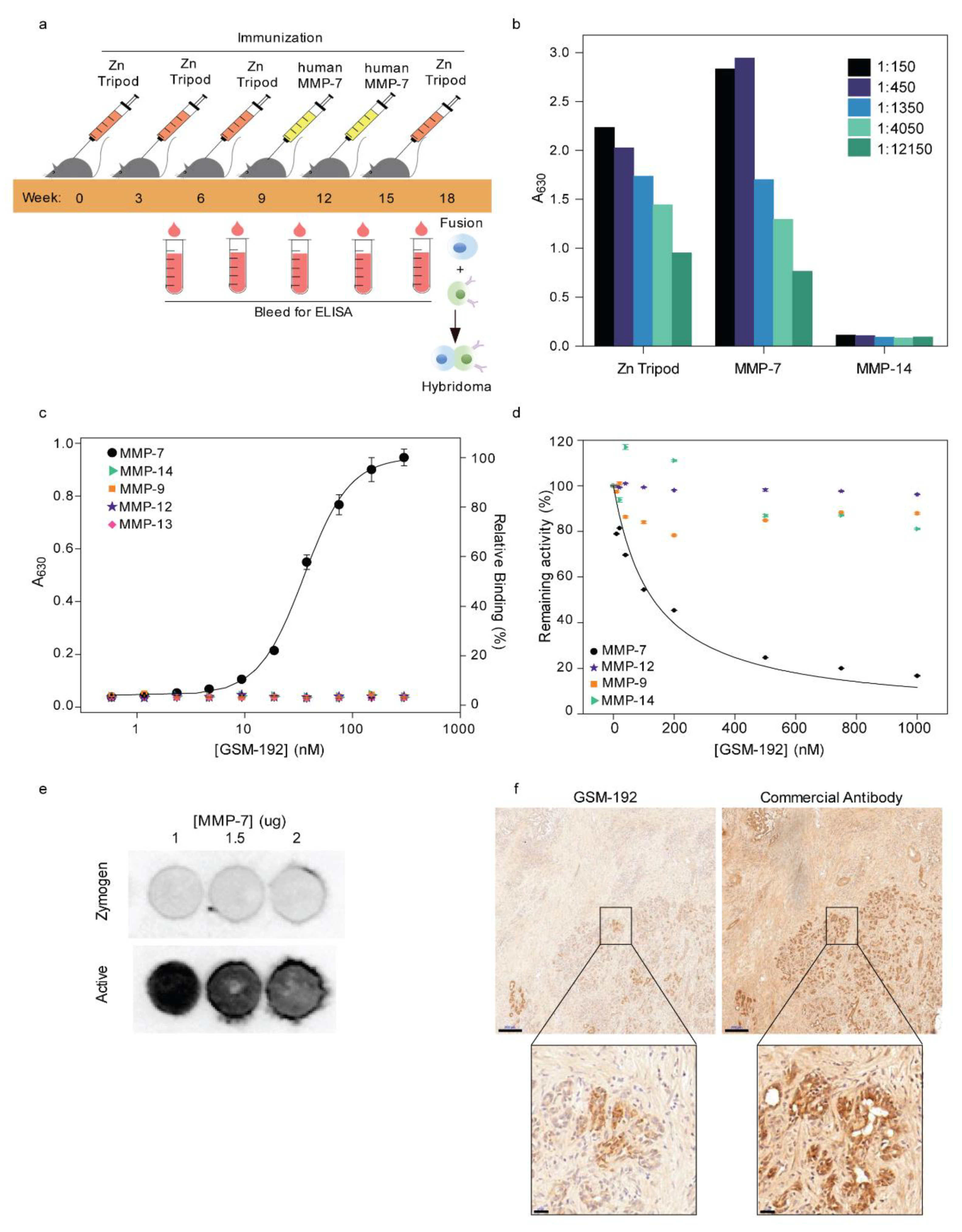
3.1. Alternating Antigen Immunization Strategy Yields High Affinity Monoclonal Antibody (mAb) Selectively Targeting Active MMP-7
3.2. GSM-192 Selectively Binds to the Active form of Human MMP-7 and Inhibits its Catalytic Activity
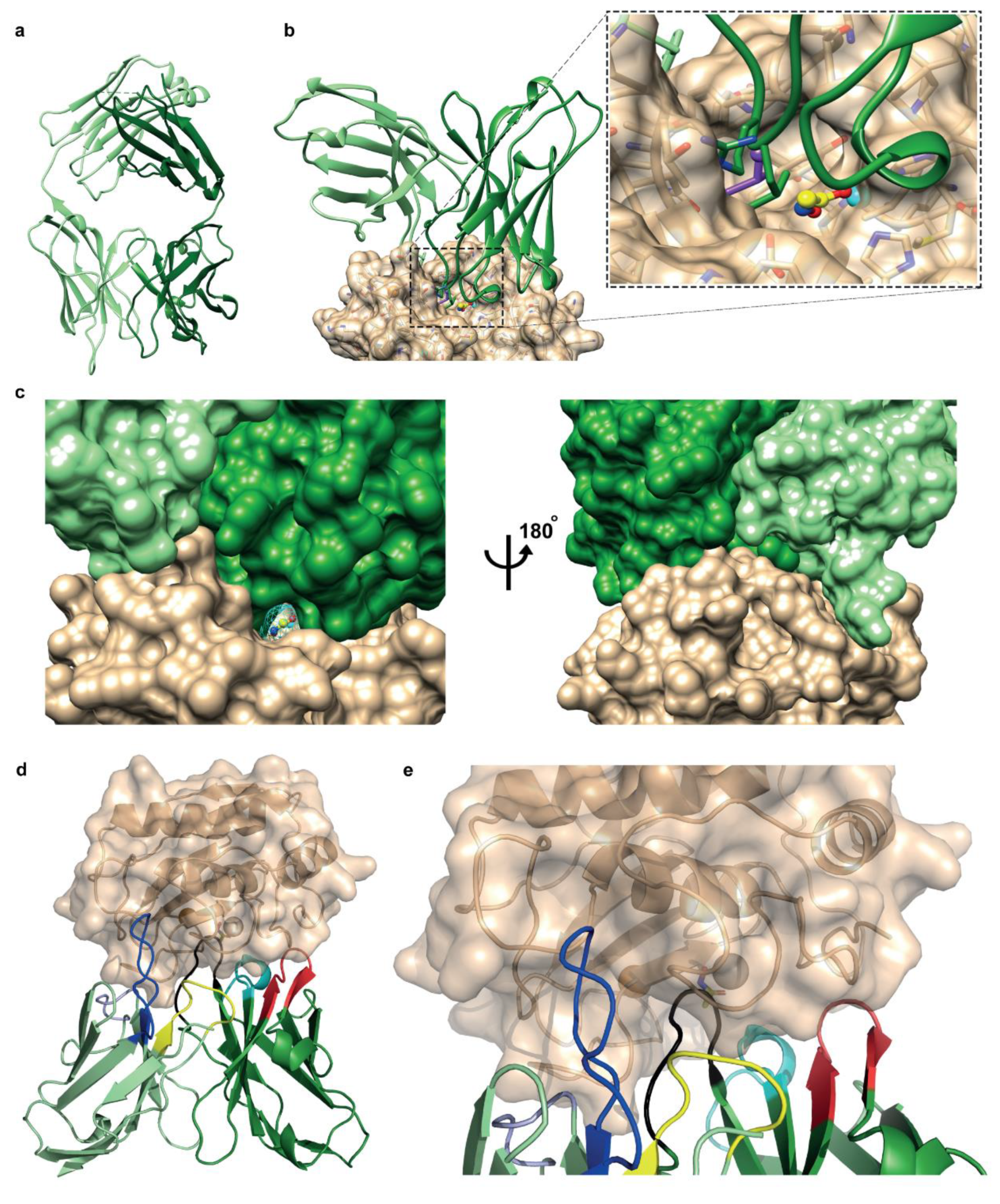
3.3. Fab Fragment Crystal Structure and Docking Model Show Antibody’s Affinity to Bind Sites Close to the Conserved Active Site
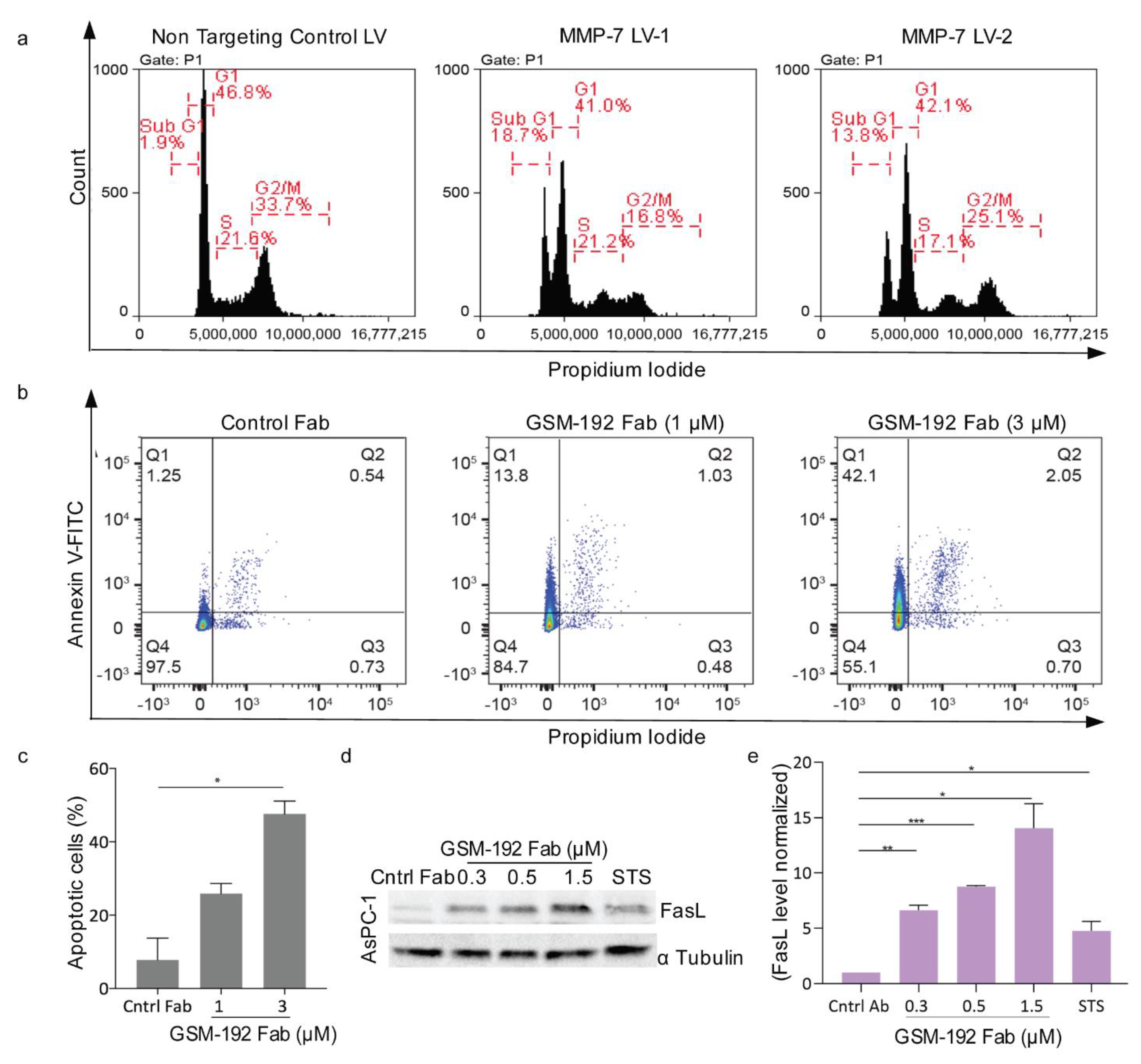
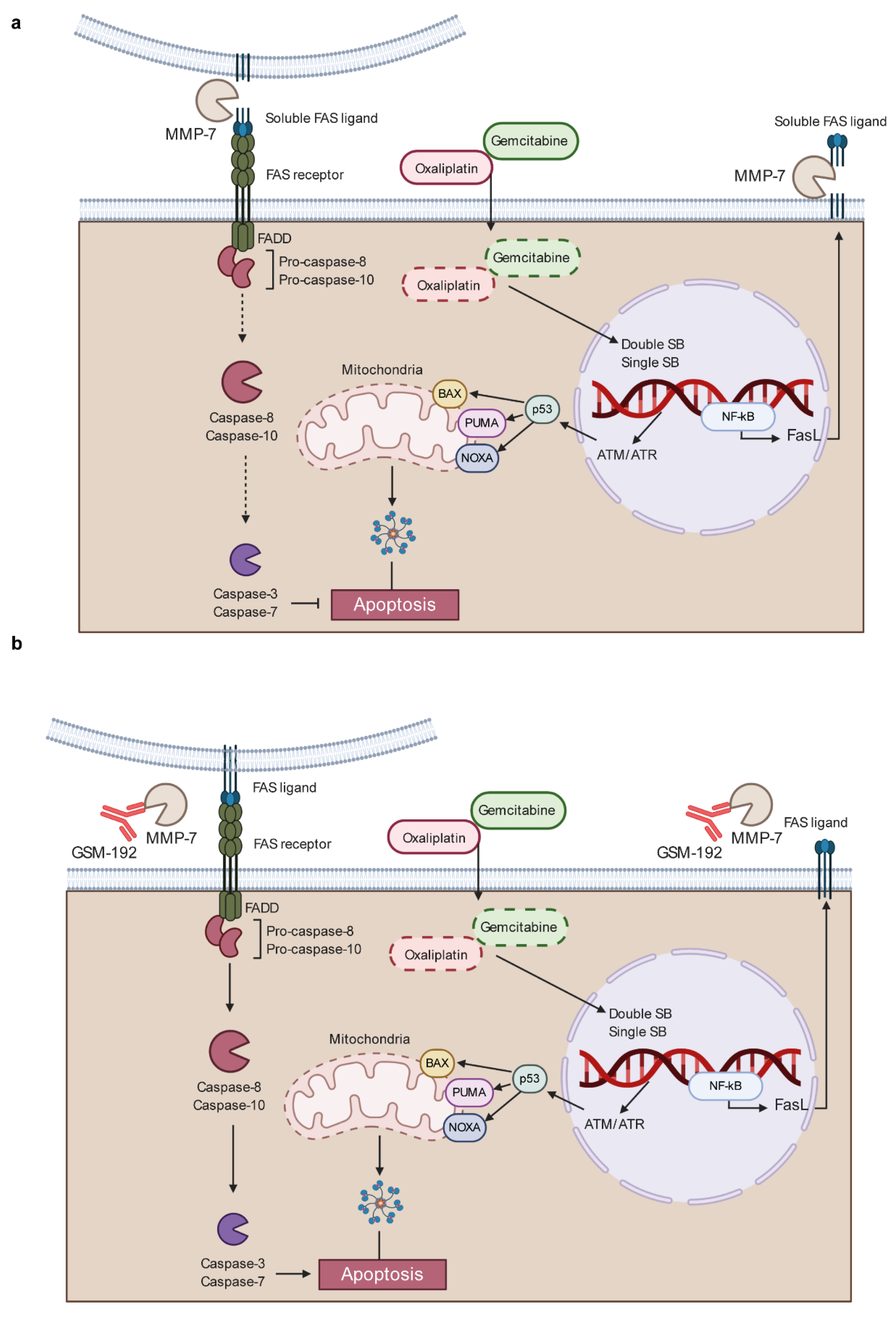
3.4. Anti MMP-7 Fab Inhibits Fas Ligand Shedding Leading to Pancreatic Tumor Cell Death via Apoptosis
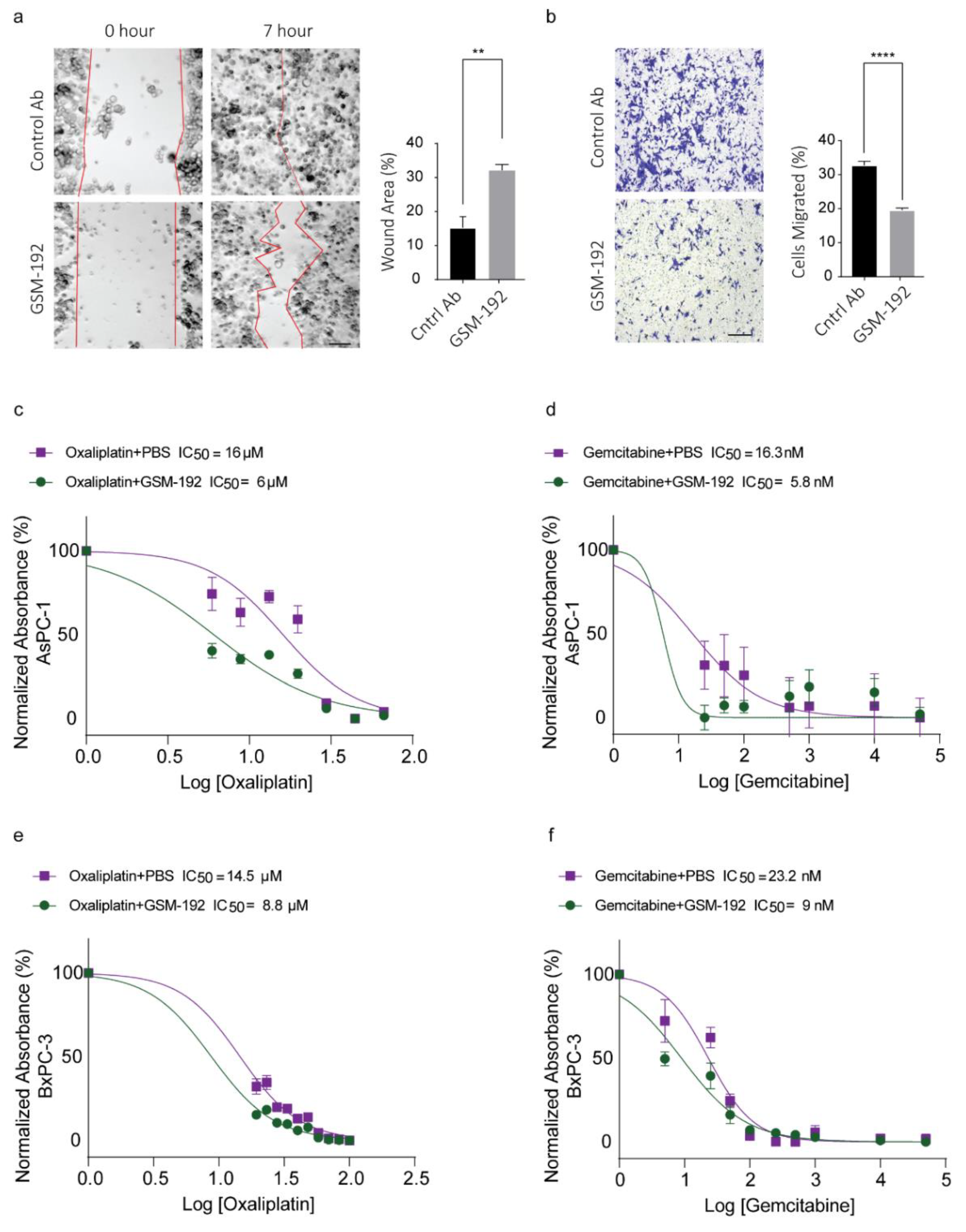
3.5. GSM-192 Fab Reduces Cell Motility In Vitro
3.6. Co-Treatment with GSM-192 Decreases IC50 of Gemcitabine (GEM) and Oxaliplatin (OX)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Drews, J. Drug discovery: A historical perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overall, C.M.; Kleifeld, O. Tumour microenvironment—Opinion: Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.; Udi, Y.; Solomonov, I.; Sagi, I. Next generation matrix metalloproteinase inhibitors—Novel strategies bring new prospects. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 1927–1939. [Google Scholar] [CrossRef]
- Sela-Passwell, N.; Kikkeri, R.; Dym, O.; Rozenberg, H.; Margalit, R.; Arad-Yellin, R.; Eisenstein, M.; Brenner, O.; Shoham, T.; Danon, T.; et al. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat. Med. 2011, 18, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Crawford, H.C.; Scoggins, C.R.; Washington, M.K.; Matrisian, L.M.; Leach, S.D. Matrix metalloproteinase-7 is expressed by pancreatic cancer precursors and regulates acinar-to-ductal metaplasia in exocrine pancreas. J. Clin. Investig. 2002, 109, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A.; Wang, S.C.; Morris IV, J.P.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.M.; Firpo, M.A.; Mulvihill, S.J. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer cell 2011, 19, 441–455. [Google Scholar] [CrossRef] [Green Version]
- Wagenaar-Miller, R.A.; Hanley, G.; Shattuck-Brandt, R.; DuBois, R.N.; Bell, R.L.; Matrisian, L.M.; Morgan, D.W. Cooperative effects of matrix metalloproteinase and cyclooxygenase-2 inhibition on intestinal adenoma reduction. Br. J. Cancer 2003, 88, 1445–1452. [Google Scholar] [CrossRef] [Green Version]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Burke, B. The role of matrix metalloproteinase 7 in innate immunity. Immunobiology 2004, 209, 51–56. [Google Scholar] [CrossRef]
- Vandenbroucke, R.; Vanlaere, I.; Van Hauwermeiren, F.; Van Wonterghem, E.; Wilson, C.; Libert, C. Pro-inflammatory effects of matrix metalloproteinase 7 in acute inflammation. Mucosal. Immunol. 2014, 7, 579–588. [Google Scholar] [CrossRef]
- Shiomi, T.; Okada, Y. MT1-MMP and MMP-7 in invasion and metastasis of human cancers. Cancer Metastasis. Rev. 2003, 22, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Dufour, A.; Overall, C.M. Missing the target: Matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol. Sci. 2013, 34, 233–242. [Google Scholar] [CrossRef]
- Strand, S.; Vollmer, P.; van den Abeelen, L.; Gottfried, D.; Alla, V.; Heid, H.; Kuball, J.; Theobald, M.; Galle, P.R.; Strand, D. Cleavage of CD95 by matrix metalloproteinase-7 induces apoptosis resistance in tumour cells. Oncogene 2004, 23, 3732–3736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.K.; Ishii, G.; Chiba, H.; Ochiai, A. The VEGF angiogenic switch of fibroblasts is regulated by MMP-7 from cancer cells. Oncogene 2007, 26, 7194–7203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, B.C.; Sang, Q.A. Angiostatin-converting enzyme activities of human matrilysin (MMP-7) and gelatinase B/type IV collagenase (MMP-9). J. Biol. Chem 1997, 272, 28823–28825. [Google Scholar] [CrossRef] [Green Version]
- McGuire, J.K.; Li, Q.; Parks, W.C. Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am. J. Pathol. 2003, 162, 1831–1843. [Google Scholar] [CrossRef] [Green Version]
- McCawley, L.J.; Matrisian, L.M. Matrix metalloproteinases: They’re not just for matrix anymore! Curr. Opin. Cell Biol. 2001, 13, 534–540. [Google Scholar] [CrossRef]
- Lynch, C.C.; Vargo-Gogola, T.; Martin, M.D.; Fingleton, B.; Crawford, H.C.; Matrisian, L.M. Matrix metalloproteinase 7 mediates mammary epithelial cell tumorigenesis through the ErbB4 receptor. Cancer Res. 2007, 67, 6760–6767. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Liu, S.; Parajuli, K.R.; Zhang, W.; Zhang, K.; Mo, Z.; Liu, J.; Chen, Z.; Yang, S.; Wang, A.R.; et al. Interleukin-17 promotes prostate cancer via MMP7-induced epithelial-to-mesenchymal transition. Oncogene 2016, 36, 687–699. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Jung, A.; Dag, S.; Hlubek, F.; Kirchner, T. beta-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am. J. Pathol 1999, 155, 1033–1038. [Google Scholar] [CrossRef]
- Almendro, V.; Ametller, E.; García-Recio, S.; Collazo, O.; Casas, I.; Augé, J.M.; Maurel, J.; Gascón, P. The role of MMP7 and its cross-talk with the FAS/FASL system during the acquisition of chemoresistance to oxaliplatin. PLoS ONE 2009, 4, e4728. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yu, H.; Lei, H.; Xie, C.; Zhong, Y. Matrix metalloproteinase 7 is a useful marker for 5-fluorouracil-based adjuvant chemotherapy in stage II and stage III colorectal cancer patients. Med. Oncol. 2014, 31, 824. [Google Scholar] [CrossRef] [PubMed]
- Knight, C.G.; Willenbrock, F.; Murphy, G. A novel coumarin-labelled peptide for sensitive continuous assays of the matrix metalloproteinases. FEBS Lett. 1992, 296, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Terwilliger, T.C. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 965–972. [Google Scholar] [CrossRef] [PubMed]
- French, S.; Wilson, K. On the treatment of negative intensity observations. Acta Crystallogr. Sect. A 1978, 34, 517–525. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.C.; Berendzen, J. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 849–861. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Suhre, K.; Sanejouand, Y.H. ElNemo: A normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004, 32, W610–W614. [Google Scholar] [CrossRef] [PubMed]
- Katchalski-Katzir, E.; Shariv, I.; Eisenstein, M.; Friesem, A.A.; Aflalo, C.; Vakser, I.A. Molecular surface recognition: Determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. USA 1992, 89, 2195–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heifetz, A.; Katchalski-Katzir, E.; Eisenstein, M. Electrostatics in protein-protein docking. Protein Sci. 2002, 11, 571–587. [Google Scholar] [CrossRef]
- Berchanski, A.; Shapira, B.; Eisenstein, M. Hydrophobic complementarity in protein-protein docking. Proteins 2004, 56, 130–142. [Google Scholar] [CrossRef]
- Kowalsman, N.; Eisenstein, M. Combining interface core and whole interface descriptors in postscan processing of protein-protein docking models. Proteins 2009, 77, 297–318. [Google Scholar] [CrossRef]
- Kowalsman, N.; Eisenstein, M. Inherent limitations in protein-protein docking procedures. Bioinformatics 2007, 23, 421–426. [Google Scholar] [CrossRef]
- Ben-Shimon, A.; Eisenstein, M. Computational mapping of anchoring spots on protein surfaces. J. Mol. Biol. 2010, 402, 259–277. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gish, W.; States, D.J. Identification of protein coding regions by database similarity search. Nat. Genet. 1993, 3, 266–272. [Google Scholar] [CrossRef]
- Yee, J.K.; Friedmann, T.; Burns, J.C. Generation of high-titer pseudotyped retroviral vectors with very broad host range. Methods Cell Biol. 1994, 43 Pt A, 99–112. [Google Scholar]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutelingsperger, C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef] [PubMed]
- Ratnikov, B.I.; Cieplak, P.; Gramatikoff, K.; Pierce, J.; Eroshkin, A.; Igarashi, Y.; Kazanov, M.; Sun, Q.; Godzik, A.; Osterman, A. Basis for substrate recognition and distinction by matrix metalloproteinases. Proc. Natl. Acad. Sci. USA 2014, 111, E4148–E4155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, M.; Amar, D.; Aharoni, A. Employing directed evolution for the functional analysis of multi-specific proteins. Bioorg. Med. Chem. 2013, 21, 3511–3516. [Google Scholar] [CrossRef]
- Rausell, A.; Juan, D.; Pazos, F.; Valencia, A. Protein interactions and ligand binding: From protein subfamilies to functional specificity. Proc. Natl. Acad. Sci. USA 2010, 107, 1995–2000. [Google Scholar] [CrossRef] [Green Version]
- Crabbe, T.; Willenbrock, F.; Eaton, D.; Hynds, P.; Carne, A.F.; Murphy, G.; Docherty, A.J. Biochemical characterization of matrilysin. Activation conforms to the stepwise mechanisms proposed for other matrix metalloproteinases. Biochemistry 1992, 31, 8500–8507. [Google Scholar] [CrossRef]
- Imai, K.; Yokohama, Y.; Nakanishi, I.; Ohuchi, E.; Fujii, Y.; Nakai, N.; Okada, Y. Matrix metalloproteinase 7 (matrilysin) from human rectal carcinoma cells: Activation of the precursor, interaction with other matrix metalloproteinases and enzymic properties. J. Biol. Chem. 1995, 270, 6691–6697. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Jiang, W.; Wang, S.; Wang, L.; Xie, K. Role of Wnt/β-catenin signaling in drug resistance of pancreatic cancer. Curr. Pharm. Des. 2012, 18, 2464–2471. [Google Scholar] [CrossRef]
- Siena, L.; Pace, E.; Ferraro, M.; Di Sano, C.; Melis, M.; Profita, M.; Spatafora, M.; Gjomarkaj, M. Gemcitabine sensitizes lung cancer cells to Fas/FasL system-mediated killing. Immunology 2014, 141, 242–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, V.M.; Sheppard, B.C.; Sears, R.C.; Alani, A.W. Hypoxia: Friend or Foe for drug delivery in Pancreatic Cancer. Cancer Lett. 2020, 492, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Chari, S.T.; Kelly, K.; Hollingsworth, M.A.; Thayer, S.P.; Ahlquist, D.A.; Andersen, D.K.; Batra, S.K.; Brentnall, T.A.; Canto, M.; Cleeter, D.F.; et al. Early detection of sporadic pancreatic cancer: Summative review. Pancreas 2015, 44, 693–712. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.C.; Parekh, J.R.; Porembka, M.R.; Nathan, H.; D’Angelica, M.I.; DeMatteo, R.P.; Fong, Y.; Kingham, T.P.; Jarnagin, W.R.; Allen, P.J. A Pilot Study Evaluating Serum MMP7 as a Preoperative Prognostic Marker for Pancreatic Ductal Adenocarcinoma Patients. J. Gastrointest. Surg 2016, 20, 899–904. [Google Scholar] [CrossRef] [Green Version]
- Park, H.D.; Kang, E.S.; Kim, J.W.; Lee, K.T.; Lee, K.H.; Park, Y.S.; Park, J.O.; Lee, J.; Heo, J.S.; Choi, S.H.; et al. Serum CA19-9, cathepsin D, and matrix metalloproteinase-7 as a diagnostic panel for pancreatic ductal adenocarcinoma. Proteomics 2012, 12, 3590–3597. [Google Scholar] [CrossRef]
- Kuhlmann, K.F.; van Till, J.W.; Boermeester, M.A.; de Reuver, P.R.; Tzvetanova, I.D.; Offerhaus, G.J.; Ten Kate, F.J.; Busch, O.R.; van Gulik, T.M.; Gouma, D.J.; et al. Evaluation of matrix metalloproteinase 7 in plasma and pancreatic juice as a biomarker for pancreatic cancer. Cancer Epidemiol Biomark. Prev. 2007, 16, 886–891. [Google Scholar] [CrossRef] [Green Version]
- Mitsiades, N.; Yu, W.-h.; Poulaki, V.; Tsokos, M.; Stamenkovic, I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 2001, 61, 577–581. [Google Scholar]
- Szarvas, T.; Csizmarik, A.; Váradi, M.; Fazekas, T.; Hüttl, A.; Nyirády, P.; Hadaschik, B.; Grünwald, V.; Tschirdewahn, S.; Shariat, S. The prognostic value of serum MMP-7 levels in prostate cancer patients who received docetaxel, abiraterone, or enzalutamide therapy. Urol. Oncol. Semin. Orig. Investig. 2020. [Google Scholar] [CrossRef]
- Kumar, P.; Siripini, S.; Sreedhar, A.S. The matrix metalloproteinase 7 (MMP7) links Hsp90 chaperone with acquired drug resistance and tumor metastasis. Cancer Rep. 2020, e1261. [Google Scholar] [CrossRef]
- Li, H.; Xu, X.; Liu, Y.; Li, S.; Zhang, D.; Meng, X.; Lu, L.; Li, Y. MMP7 Induces T-DM1 Resistance and Leads to the Poor Prognosis of Gastric Adenocarcinoma via a DKK1-Dependent Manner. Anti Cancer Agents Med. Chem. 2018, 18, 2010–2016. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Gavoille, C.; Samalin, E.; Ychou, M.; Ducreux, M. The role of the FOLFIRINOX regimen for advanced pancreatic cancer. Curr. Oncol. Rep. 2013, 15, 182–189. [Google Scholar] [CrossRef]
- Maréchal, R.; Bachet, J.B.; Mackey, J.R.; Dalban, C.; Demetter, P.; Graham, K.; Couvelard, A.; Svrcek, M.; Bardier–Dupas, A.; Hammel, P. Levels of gemcitabine transport and metabolism proteins predict survival times of patients treated with gemcitabine for pancreatic adenocarcinoma. Gastroenterology 2012, 143, 664–674. e666. [Google Scholar] [CrossRef] [PubMed]
- Kunzmann, V.; Ramanathan, R.K.; Goldstein, D.; Liu, H.; Ferrara, S.; Lu, B.; Renschler, M.F.; Von Hoff, D.D. Tumor reduction in primary and metastatic pancreatic cancer lesions with nab-paclitaxel and gemcitabine: An exploratory analysis from a phase 3 study. Pancreas 2017, 46, 203. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Balibrea, E.; Martínez-Cardús, A.; Ginés, A.; de Porras, V.R.; Moutinho, C.; Layos, L.; Manzano, J.L.; Bugés, C.; Bystrup, S.; Esteller, M. Tumor-related molecular mechanisms of oxaliplatin resistance. Mol. Cancer Ther. 2015, 14, 1767–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H.; Voss, M.; Jansen, M.; Roeder, C.; Henne-Bruns, D.; Kremer, B.; Kalthoff, H. Human pancreatic adenocarcinomas express Fas and Fas ligand yet are resistant to Fas-mediated apoptosis. Cancer Res. 1998, 58, 1741–1749. [Google Scholar] [PubMed]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Fields, G.B. The rebirth of matrix metalloproteinase inhibitors: Moving beyond the dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef] [Green Version]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohan, V.; Gaffney, J.P.; Solomonov, I.; Levin, M.; Klepfish, M.; Akbareian, S.; Grünwald, B.; Dym, O.; Eisenstein, M.; Yu, K.H.; et al. Conformation-Specific Inhibitory Anti-MMP-7 Monoclonal Antibody Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Chemotherapeutic Cell Kill. Cancers 2021, 13, 1679. https://doi.org/10.3390/cancers13071679
Mohan V, Gaffney JP, Solomonov I, Levin M, Klepfish M, Akbareian S, Grünwald B, Dym O, Eisenstein M, Yu KH, et al. Conformation-Specific Inhibitory Anti-MMP-7 Monoclonal Antibody Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Chemotherapeutic Cell Kill. Cancers. 2021; 13(7):1679. https://doi.org/10.3390/cancers13071679
Chicago/Turabian StyleMohan, Vishnu, Jean P. Gaffney, Inna Solomonov, Maxim Levin, Mordehay Klepfish, Sophia Akbareian, Barbara Grünwald, Orly Dym, Miriam Eisenstein, Kenneth H. Yu, and et al. 2021. "Conformation-Specific Inhibitory Anti-MMP-7 Monoclonal Antibody Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Chemotherapeutic Cell Kill" Cancers 13, no. 7: 1679. https://doi.org/10.3390/cancers13071679