
Chasing the Target: New Phenomena of Resistance to Novel Selective RET Inhibitors in Lung Cancer. Updated Evidence and Future Perspectives

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
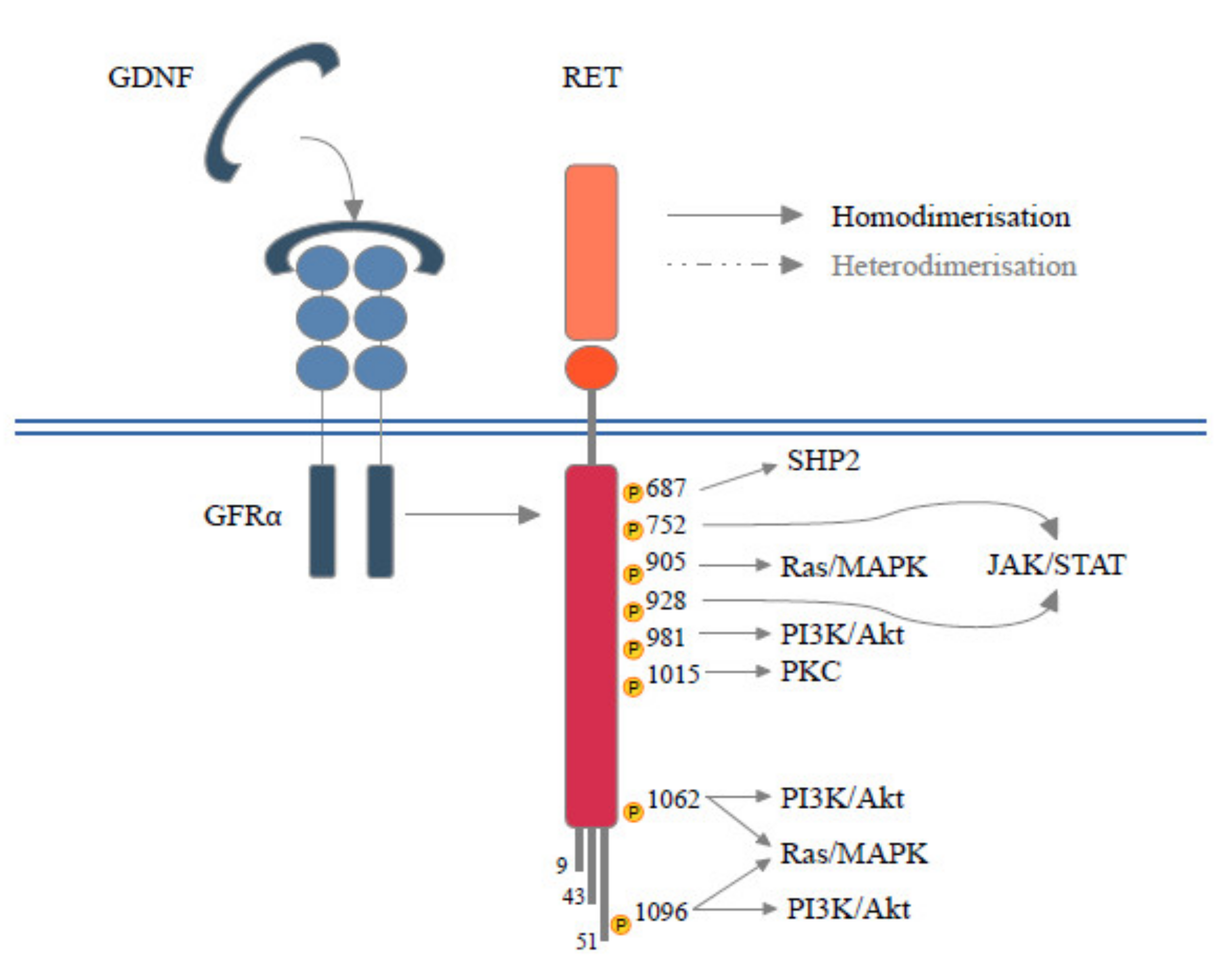
2. RET in Pills
2.1. Germline Mutations
2.2. Somatic Mutations and Cancer
2.3. RET in Lung Cancer
3. Activity of MKIs in RET-Positive NSCLC
3.1. Vandetanib
3.2. Cabozantinib
3.3. Lenvatinib
3.4. Other MKIs
4. New RET-Selective Inhibitors
5. Immune Checkpoint Inhibitors (ICI) and Chemotherapy (CT)
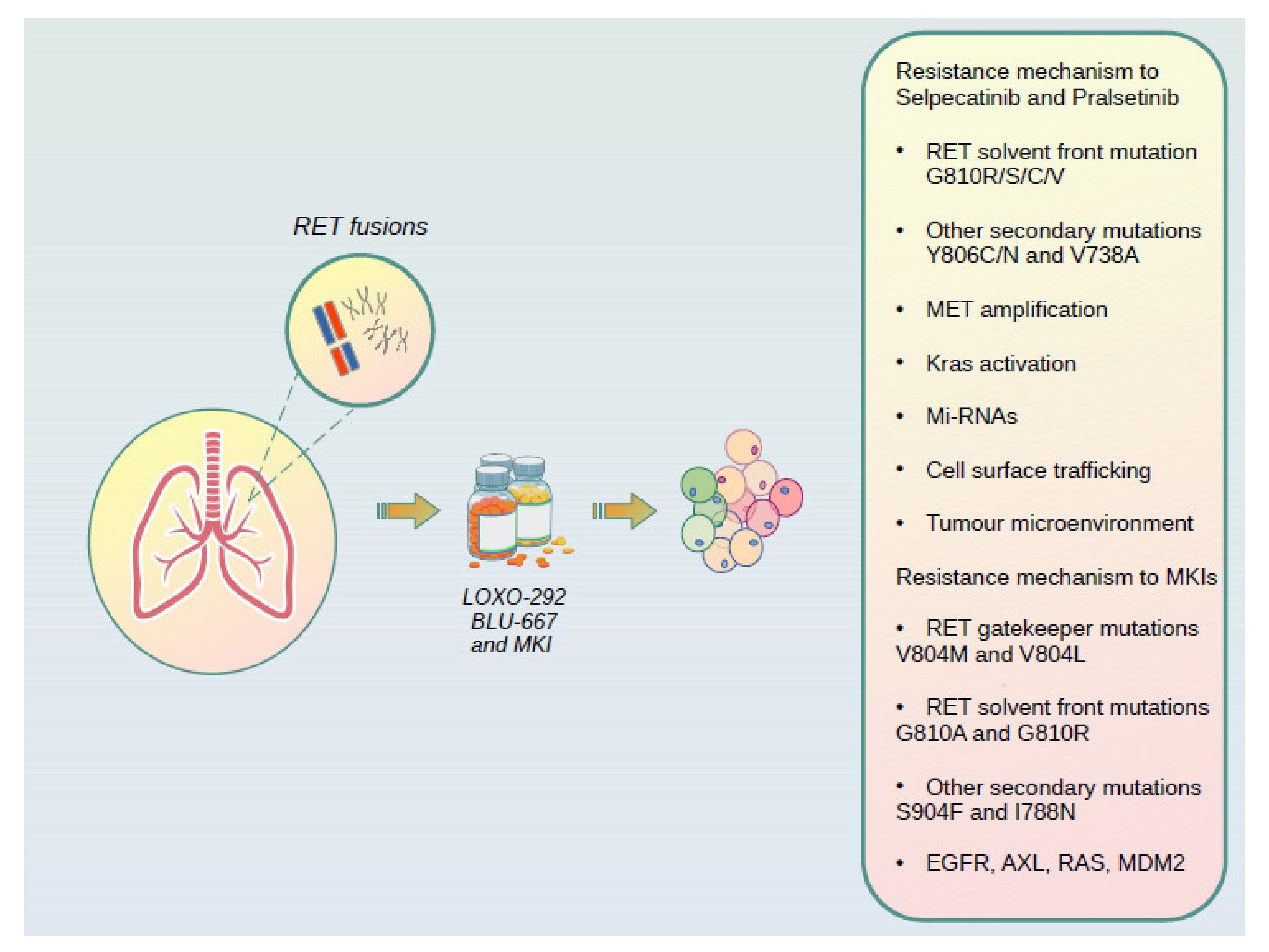
6. Resistance
7. Combination Strategies and Future Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takahashi, M.; Ritz, J.; Cooper, G.M. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 1985, 42, 581–588. [Google Scholar] [CrossRef]
- Andrew, S.D.; Delhanty, P.J.D.; Mulligan, L.M.; Robinson, B.G. Sp1 and Sp3 transactivate the RET proto-oncogene promoter. Gene 2000, 256, 283–291. [Google Scholar] [CrossRef]
- Lang, D.; Epstein, J.A. Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer. Hum. Mol. Genet. 2003, 12, 937–945. [Google Scholar] [CrossRef] [Green Version]
- Leon, T.Y.Y.; Ngan, E.S.W.; Poon, H.-C.; So, M.-T.; Lui, V.C.H.; Tam, P.K.H.; Garcia-Barcelo, M.M. Transcriptional regulation of RET by Nkx2-1, Phox2b, Sox10, and Pax3. J. Pediatr. Surg. 2009, 44, 1904–1912. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, C.F. Structure and Physiology of the RET Receptor Tyrosine Kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a009134. [Google Scholar] [CrossRef] [PubMed]
- Arighi, E.; Borrello, M.G.; Sariola, H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005, 16, 441–467. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ho, J.X.; Ruble, J.R.; Rose, J.; Rüker, F.; Ellenburg, M.; Murphy, R.; Click, J.; Soistman, E.; Wilkerson, L.; et al. Structural studies of several clinically important oncology drugs in complex with human serum albumin. Biochim. Biophys. Acta BBA Gen. Subj. 2013, 1830, 5356–5374. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shang, G.; Chen, Y.J.; Brautigam, C.A.; Liou, J.; Zhang, X.; Bai, X.C. Cryo-EM analyses reveal the common mechanism and diversification in the activation of RET by different ligands. Elife 2019, 8, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Cooke, M.P.; Ching, K.A.; Hakak, Y.; Walker, J.R.; Wiltshire, T.; Orth, A.P.; Vega, R.G.; Sapinoso, L.M.; Moqrich, A.; et al. Large-Scale Analysis of the Human and Mouse Transcriptomes. Proc. Natl. Acad. Sci. USA 2002, 99, 4465–4470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avantaggiato, V.; Dathan, N.A.; Grieco, M.; Fabien, N.; Lazzaro, D.; Fusco, A.; Simeone, A.; Santoro, M. Develop-mental Expression of the RET Protooncogene. Cell Growth Differ. 1994, 5, 305–311. [Google Scholar] [PubMed]
- Schuchardt, A.; Dagati, V.D.; Larsson-Blomberg, L.; Costantini, F.; Pachnis, V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nat. Cell Biol. 1994, 367, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Durbec, P.L.; Larsson-Blomberg, L.B.; Schuchardt, A.; Costantini, F.; Pachnis, V. Common origin and developmental dependence on c-ret of subsets of enteric and sympathetic neuroblasts. Development 1996, 122, 349–358. [Google Scholar]
- Meng, X.; Lindahl, M.; Hyvönen, M.E.; Parvinen, M.; De Rooij, D.G.; Hess, M.W.; Raatikainen-Ahokas, A.; Sainio, K.; Rauvala, H.; Lakso, M.; et al. Regulation of Cell Fate Decision of Undifferentiated Spermatogonia by GDNF. Science 2000, 287, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Mederer, T.; Schmitteckert, S.; Volz, J.; Martínez, C.; Röth, R.; Thumberger, T.; Eckstein, V.; Scheuerer, J.; Thöni, C.; Lasitschka, F.; et al. A complementary study approach unravels novel players in the pathoetiology of Hirschsprung disease. PLoS Genet. 2020, 16, e1009106. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, N.; Renkema, K.Y.; Bongers, E.M.H.F.; Giles, R.H.; Knoers, N.V.A.M. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat. Rev. Nephrol. 2015, 11, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Pereira, D.; Arroz-Madeira, S.; Rodrigues-Campos, M.; Barbosa, I.A.M.; Domingues, R.G.; Bento, T.; Almeida, A.R.M.; Ribeiro, H.; Potocnik, A.J.; Enomoto, H.; et al. The neurotrophic factor receptor RET drives haematopoietic stem cell survival and function. Nat. Cell Biol. 2014, 514, 98–101. [Google Scholar] [CrossRef]
- Marx, S.J. Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nat. Rev. Cancer 2005, 5, 367–375. [Google Scholar] [CrossRef]
- Kouvaraki, M.A.; Shapiro, S.E.; Perrier, N.D.; Cote, G.J.; Gagel, R.F.; Hoff, A.O.; Sherman, S.I.; Lee, J.E.; Evans, D.B. RET Proto-Oncogene: A Review and Update of Genotype–Phenotype Correlations in Hereditary Medullary Thyroid Cancer and Associated Endocrine Tumors. Thyroid 2005, 15, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohllk, N.; Cote, G.J.; Evans, D.B.; Goepfert, H.; Ordòñez, N.G.; Gagel, R.F. Application of Genetic Screening Information to the Management of Medullary Thyroid Carcinoma and Multiple Endocrine Neoplasia Type 2. Endocrinol. Metab. Clin. N. Am. 1996, 25, 1–25. [Google Scholar] [CrossRef]
- Wells, S.A. Advances in the management of MEN2: From improved surgical and medical treatment to novel kinase inhibitors. Endocr. Relat. Cancer 2018, 25, T1–T13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloos, R.T.; Eng, C.; Evans, D.B.; Francis, G.L.; Gagel, R.F.; Gharib, H.; Moley, J.F.; Pacini, F.; Ringel, M.D.; Schlumberger, M.; et al. Medullary Thyroid Cancer: Management Guidelines of the American Thyroid Association. Thyroid Off. J. Am. Thyroid Assoc. 2009, 19, 565–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Saenko, V.; Yamashita, S.; Mitsutake, N. Radiation-Induced Thyroid Cancers: Overview of Molecular Signatures. Cancers 2019, 11, 1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikiforov, Y.E.; Rowland, J.M.; Bove, K.E.; Monforte-Munoz, H.; Fagin, J.A. Distinct Pattern of Ret Oncogene Rear-rangements in Morphological Variants of Radiation-Induced and Sporadic Thyroid Papillary Carcinomas in Children. Cancer Res. 1997, 57, 1690–1694. [Google Scholar] [PubMed]
- Santoro, M.; Moccia, M.; Federico, G.; Carlomagno, F. RET Gene Fusions in Malignancies of the Thyroid and Other Tissues. Genes 2020, 11, 424. [Google Scholar] [CrossRef] [Green Version]
- Boulay, A.; Breuleux, M.; Stephan, C.; Fux, C.; Brisken, C.; Fiche, M.; Wartmann, M.; Stumm, M.; Lane, H.A.; Hynes, N.E. The Ret Receptor Tyrosine Kinase Pathway Functionally Interacts with the ERα Pathway in Breast Cancer. Cancer Res. 2008, 68, 3743–3751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morandi, A.; Plaza-Menacho, I.; Isacke, C.M. RET in breast cancer: Functional and therapeutic implications. Trends Mol. Med. 2011, 17, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Watanabe, S.; Rothenberg, S.M.; Kherani, J.; French, P.P.; Olek, E. Abstract 5236: Complete response to selpercatinib (LOXO-292), a highly selective RET inhibitor, in a patient withRETfusion-positive breast cancer. Cancer Res. 2020, 80, 5236. [Google Scholar] [CrossRef]
- Le Rolle, A.-F.; Klempner, S.J.; Garrett, C.R.; Seery, T.; Sanford, E.M.; Balasubramanian, S.; Ross, J.S.; Stephens, P.J.; Miller, V.A.; Ali, S.M.; et al. Identification and characterization of RET fusions in advanced colorectal cancer. Oncotarget 2015, 6, 28929–28937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, C.; Sanz-Pamplona, R.; Salazar, R. RET-fusions: A novel paradigm in colorectal cancer. Ann. Oncol. 2018, 29, 1340–1343. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.; Lee, J.; Morano, F.; Fucà, G.; Nikolinakos, P.; Drilon, A.; Hechtman, J.; Christiansen, J.; et al. RET fusions in a small subset of advanced colorectal cancers at risk of being neglected. Ann. Oncol. 2018, 29, 1394–1401. [Google Scholar] [CrossRef]
- Kohno, T.; Tabata, J.; Nakaoku, T. REToma: A cancer subtype with a shared driver oncogene. Carcinogenesis 2019, 41, 123–129. [Google Scholar] [CrossRef]
- Kohno, T.; Ichikawa, H.; Totoki, Y.; Yasuda, K.; Hiramoto, M.; Nammo, T.; Sakamoto, H.; Tsuta, K.; Furuta, K.; Shimada, Y.; et al. KIF5B-RET fusions in lung adenocarcinoma. Nat. Med. 2012, 18, 375–377. [Google Scholar] [CrossRef]
- Ju, Y.S.; Lee, W.-C.; Shin, J.-Y.; Lee, S.; Bleazard, T.; Won, J.-K.; Kim, Y.T.; Kim, J.-I.; Kang, J.-H.; Seo, J.-S. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2011, 22, 436–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Z.; Ye, B.; Wang, K.; Zhou, P.; Zhao, S.; Li, W.; Tian, P. Unique Genetic Characteristics and Clinical Prognosis of Female Patients with Lung Cancer Harboring RET Fusion Gene. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Mizukami, T.; Shiraishi, K.; Shimada, Y.; Ogiwara, H.; Tsuta, K.; Ichikawa, H.; Sakamoto, H.; Kato, M.; Shibata, T.; Nakano, T.; et al. Molecular Mechanisms Underlying Oncogenic RET Fusion in Lung Adenocarcinoma. J. Thorac. Oncol. 2014, 9, 622–630. [Google Scholar] [CrossRef] [Green Version]
- Lipson, D.; Capelletti, M.; Yelensky, R.; Otto, G.; Parker, A.; Jarosz, M.; Curran, J.A.; Balasubramanian, S.; Bloom, T.; Brennan, K.W.; et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med. 2012, 18, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Subbiah, V.; Marchlik, E.; Elkin, S.K.; Carter, J.L.; Kurzrock, R. RET Aberrations in Diverse Cancers: Next-Generation Sequencing of 4,871 Patients. Clin. Cancer Res. 2017, 23, 1988–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, T.-H.; Wu, S.-G.; Hsieh, M.-S.; Jin-Yuan, S.; Yang, J.C.-H.; Shih, J.-Y. Clinical and prognostic implications of RET rearrangements in metastatic lung adenocarcinoma patients with malignant pleural effusion. Lung Cancer 2015, 88, 208–214. [Google Scholar] [CrossRef]
- Suzuki, M.; Makinoshima, H.; Matsumoto, S.; Suzuki, A.; Mimaki, S.; Matsushima, K.; Yoh, K.; Goto, K.; Suzuki, Y.; Ishii, G.; et al. Identification of a lung adenocarcinoma cell line with CCDC 6- RET fusion gene and the effect of RET inhibitors in vitro and in vivo. Cancer Sci. 2013, 104, 896–903. [Google Scholar] [CrossRef]
- Melillo, R.M.; Castellone, M.D.; Guarino, V.; De Falco, V.; Cirafici, A.M.; Salvatore, G.; Caiazzo, F.; Basolo, F.; Giannini, R.; Kruhoffer, M.; et al. The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J. Clin. Investig. 2016, 126, 1603. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Ishigame, T.; Tsuta, K.; Kumamoto, K.; Imai, T.; Kohno, T. A mouse model of KIF5B-RET fusion-dependent lung tumorigenesis. Carcinogenesis 2014, 35, 2452–2456. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Shen, J.; Xiang, J.; Li, H.; Li, B.; Zhang, T.; Zhang, L.; Mao, X.; Jian, H.; Shu, Y. Characterization of acquired receptor tyrosine–kinase fusions as mechanisms of resistance to EGFR tyrosine–kinase inhibitors. Cancer Manag. Res. 2019, 11, 6343–6351. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Rekhtman, N.; Arcila, M.; Wang, L.; Ni, A.; Albano, M.; Van Voorthuysen, M.; Somwar, R.; Smith, R.S.; Montecalvo, J.; et al. Cabozantinib in patients with advanced RET -rearranged non-small-cell lung cancer: An open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. 2016, 17, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Lira, M.E.; Choi, Y.-L.; Lim, S.M.; Deng, S.; Huang, D.; Ozeck, M.; Han, J.; Jeong, J.Y.; Shim, H.S.; Cho, B.C.; et al. A Single-Tube Multiplexed Assay for Detecting ALK, ROS1, and RET Fusions in Lung Cancer. J. Mol. Diagn. 2014, 16, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Gautschi, O.; Milia, J.; Filleron, T.; Wolf, J.; Carbone, D.P.; Owen, D.; Camidge, R.; Narayanan, V.; Doebele, R.C.; Besse, B.; et al. Targeting RET in Patients With RET-Rearranged Lung Cancers: Results From the Global, Multicenter RET Registry. J. Clin. Oncol. 2017, 35, 1403–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velcheti, V.; Thawani, R.; Khunger, M.; Mukhopadhyay, S.; Chute, D.J.; Schrock, A.B.; Ali, S.M. FRMD4A / RET: A Novel RET Oncogenic Fusion Variant in Non–Small Cell Lung Carcinoma. J. Thorac. Oncol. 2017, 12, e15–e16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, Y.; Liu, C.; Wang, W.; Li, M.; Lv, D.; Sun, G.; Chen, H.; Dong, X.; Miao, Z.; et al. Identification of a novel KIF13A-RET fusion in lung adenocarcinoma by next-generation sequencing. Lung Cancer 2018, 118, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Nakaoku, T.; Tsuta, K.; Ichikawa, H.; Shiraishi, K.; Sakamoto, H.; Enari, M.; Furuta, K.; Shimada, Y.; Ogiwara, H.; Watanabe, S.-I.; et al. Druggable Oncogene Fusions in Invasive Mucinous Lung Adenocarcinoma. Clin. Cancer Res. 2014, 20, 3087–3093. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-H.; Lee, J.-K.; Ahn, M.-J.; Kim, D.-W.; Sun, J.-M.; Keam, B.; Kim, T.M.; Heo, D.S.; Ahn, J.S.; Choi, Y.-L.; et al. Vandetanib in pretreated patients with advanced non-small cell lung cancer-harboring RET rearrangement: A phase II clinical trial. Ann. Oncol. 2017, 28, 292–297. [Google Scholar] [CrossRef]
- Wang, R.; Hu, H.; Pan, Y.; Li, Y.; Ye, T.; Li, C.; Luo, X.; Wang, L.; Li, H.; Zhang, Y.; et al. RET Fusions Define a Unique Molecular and Clinicopathologic Subtype of Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2012, 30, 4352–4359. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Liebers, M.; Zhelyazkova, B.; Cao, Y.; Panditi, D.; Lynch, K.D.; Chen, J.; Robinson, H.E.; Shim, H.S.; Chmielecki, J.; et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 2014, 20, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.; Zheng, Y.; Lv, J. TBC1D32-RET: A Novel RET Oncogenic Fusion in Lung Adenocarcinoma. J. Thorac. Oncol. 2019, 14, e7–e9. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Wang, L.; Hasanovic, A.; Suehara, Y.; Lipson, D.; Stephens, P.; Ross, J.; Miller, V.; Ginsberg, M.; Zakowski, M.F.; et al. Response to Cabozantinib in Patients with RET Fusion-Positive Lung Adenocarcinomas. Cancer Discov. 2013, 3, 630–635. [Google Scholar] [CrossRef] [Green Version]
- Velcheti, V.; Madison, R.; Ali, S.M.; Schrock, A.B. WAC/RET: A Novel RET Oncogenic Fusion Variant in Non–Small Cell Lung Carcinoma. J. Thorac. Oncol. 2018, 13, e122–e123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, P.P.; Murray-Rust, J.; Kjær, S.; Scott, R.P.; Hanrahan, S.; Santoro, M.; Ibáñez, C.F.; McDonald, N.Q. Structure and Chemical Inhibition of the RET Tyrosine Kinase Domain. J. Biol. Chem. 2006, 281, 33577–33587. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, M.G.; Di Noia, V.; D’Argento, E.; Vita, E.; Damiano, P.; Cannella, A.; Ribelli, M.; Pilotto, S.; Milella, M.; Tortora, G.; et al. Oncogene-Addicted Non-Small-Cell Lung Cancer: Treatment Opportunities and Future Perspectives. Cancers 2020, 12, 1196. [Google Scholar] [CrossRef]
- Huang, Q.; Schneeberger, V.E.; Luetteke, N.; Jin, C.; Afzal, R.; Budzevich, M.M.; Makanji, R.J.; Martinez, G.V.; Shen, T.; Zhao, L.; et al. Preclinical Modeling of KIF5B–RET Fusion Lung Adenocarcinoma. Mol. Cancer Ther. 2016, 15, 2521–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehara, H.; et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Kennedy, E.; Sequist, L.V.; Brastianos, P.K.; Goodwin, K.E.; Stevens, S.; Wanat, A.C.; Stober, L.L.; Digumarthy, S.R.; Engelman, J.A.; et al. Clinical Activity of Alectinib in Advanced RET -Rearranged Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2016, 11, 2027–2032. [Google Scholar] [CrossRef] [PubMed]
- Yoh, K.; Seto, T.; Satouchi, M.; Nishio, M.; Yamamoto, N.; Murakami, H.; Nogami, N.; Matsumoto, S.; Kohno, T.; Tsuta, K.; et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): An open-label, multicentre phase 2 trial. Lancet Respir. Med. 2017, 5, 42–50. [Google Scholar] [CrossRef]
- Hida, T.; Velcheti, V.; Reckamp, K.L.; Nokihara, H.; Sachdev, P.; Kubota, T.; Nakada, T.; Dutcus, C.E.; Ren, M.; Tamura, T. A phase 2 study of lenvatinib in patients with RET fusion-positive lung adenocarcinoma. Lung Cancer 2019, 138, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Fu, S.; Patel, M.R.; Fakih, M.; Wang, D.; Olszanski, A.J.; Morgensztern, D.; Liu, S.V.; Cho, B.C.; Bazhenova, L.; et al. A Phase I/Ib Trial of the VEGFR-Sparing Multikinase RET Inhibitor RXDX-105. Cancer Discov. 2018, 9, 384–395. [Google Scholar] [CrossRef] [Green Version]
- Velcheti, V.; Hida, T.; Reckamp, K.L.; Yang, J.C.; Nokihara, H.; Sachdev, P.; Feit, K.; Kubota, T.; Nakada, T.; Dutcus, C.E.; et al. Phase 2 study of lenvatinib (LN) in patients (Pts) with RET fusion-positive adenocarcinoma of the lung. Ann. Oncol. 2016, 27, vi417. [Google Scholar] [CrossRef] [Green Version]
- Platt, A.; Morten, J.; Ji, Q.; Elvin, P.; Womack, C.; Su, X.; Donald, E.; Gray, N.; Read, J.; Bigley, G.; et al. A retrospective analysis of RET translocation, gene copy number gain and expression in NSCLC patients treated with vandetanib in four randomized Phase III studies. BMC Cancer 2015, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Horiike, A.; Takeuchi, K.; Uenami, T.; Kawano, Y.; Tanimoto, A.; Kaburaki, K.; Tambo, Y.; Kudo, K.; Yanagitani, N.; Ohyanagi, F.; et al. Sorafenib treatment for patients with RET fusion-positive non-small cell lung cancer. Lung Cancer 2016, 93, 43–46. [Google Scholar] [CrossRef]
- Wu, H.; Shih, J.-Y.; Yang, J.C.-H. Rapid Response to Sunitinib in a Patient with Lung Adenocarcinoma Harboring KIF5B-RET Fusion Gene. J. Thorac. Oncol. 2015, 10, e95–e96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velcheti, V.; Ahluwalia, M. Intracranial and Systemic Response to Alectinib in a Patient with RET-KIF5B Oncogenic Fusion. J. Thorac. Oncol. 2017, 12, e98–e99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, M.F.S.A.; Alessi, J.V.M.; Oliveira, L.J.C.; Gongora, A.B.L.; Sacardo, K.P.; Zucchetti, B.M.; Shimada, A.K.; de Galiza Barbosa, F.; Feher, O.; Katz, A. Alectinib activity in chemotherapy-refractory metastatic RET-rearranged non-small cell lung carcinomas: A case series. Lung Cancer 2020, 139, 9–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosaki, K.; Takeuchi, S.; Takahara, S.; Kawakami, T.; Yoh, K.; Kono, Y.; Horiike, A.; Seto, T.; Goto, K.; Yoshimura, K.; et al. Safety of alectinib in non-small cell lung cancer patients with RET fusion gene (ALL-RET): Results from the dose-finding portion of a phase 1/2 study. Ann. Oncol. 2017, 28, x127. [Google Scholar] [CrossRef]
- Yanagitani, N.; Takeuchi, S.; Murayama, T.; Yoshimura, K.; Imai, Y.; Takahara, S.; Kawakami, T.; Seto, T.; Hattori, Y.; Ohashi, K.; et al. OA02.01 Alectinib in Previously Treated RET-Rearranged Advanced Non-Small-Cell Lung Cancer: A Phase 1/2 Trial (ALL-RET). J. Thorac. Oncol. 2019, 14, S207. [Google Scholar] [CrossRef]
- Li, G.G.; Somwar, R.; Joseph, J.; Smith, R.S.; Hayashi, T.; Martin, L.; Franovic, A.; Schairer, A.; Martin, E.; Riely, G.J.; et al. Antitumor Activity of RXDX-105 in Multiple Cancer Types with RET Rearrangements or Mutations. Clin. Cancer Res. 2017, 23, 2981–2990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.; Loong, H.H.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef]
- Gainor, J.F.; Lee, D.H.; Curigliano, G.; Doebele, R.C.; Kim, D.-W.; Baik, C.S.; Tan, D.S.-W.; Lopes, G.; Gadgeel, S.M.; Cassier, P.A.; et al. Clinical activity and tolerability of BLU-667, a highly potent and selective RET inhibitor, in patients (pts) with advanced RET-fusion+ non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2019, 37, 9008. [Google Scholar] [CrossRef]
- Brandhuber, B.; Haas, J.; Tuch, B.; Ebata, K.; Bouhana, K.; McFaddin, E.; Williams, L.; Winski, S.; Brown, E.; Burkhard, M.; et al. The development of a potent, KDR/VEGFR2-sparing RET kinase inhibitor for treating patients with RET-dependent cancers. Eur. J. Cancer 2016, 69, S144. [Google Scholar] [CrossRef]
- Drilon, A.; Lin, J.J.; Filleron, T.; Ni, A.; Milia, J.; Bergagnini, I.; Hatzoglou, V.; Velcheti, V.; Offin, M.; Li, B.; et al. Frequency of Brain Metastases and Multikinase Inhibitor Outcomes in Patients With RET–Rearranged Lung Cancers. J. Thorac. Oncol. 2018, 13, 1595–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbiah, V.; Velcheti, V.; Tuch, B.; Ebata, K.; Busaidy, N.; Cabanillas, M.; Wirth, L.; Stock, S.; Smith, S.; Lauriault, V.; et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann. Oncol. 2018, 29, 1869–1876. [Google Scholar] [CrossRef]
- Solomon, B.J.; Zhou, C.C.; Drilon, A.; Park, K.; Wolf, J.; Elamin, Y.; Davis, H.M.; Soldatenkova, V.; Sashegyi, A.; Lin, A.B.; et al. Phase III study of selpercatinib versus chemotherapy ± pembrolizumab in untreated RET positive non-small-cell lung cancer. Future Oncol. 2021, 17, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Rahal, R.; Maynard, M.; Hu, W.; Brubaker, J.; Cao, Q.; Kim, J.L.; Sheets, M.P.; Wilson, D.P.; Wilson, K.J.; DiPietro, L.; et al. Abstract B151: BLU-667 Is a Potent and Highly Selective RET Inhibitor Being Developed for RET—Driven Cancers. In Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, Philadelphia, PA, USA, 26–30 October 2017; American Association for Cancer Research (AACR): Philadelphia, PA, USA, 2018; Volume 17, p. B151. [Google Scholar]
- Subbiah, V.; Gainor, J.F.; Rahal, R.; Brubaker, J.D.; Kim, J.L.; Maynard, M.; Hu, W.; Cao, Q.; Sheets, M.P.; Wilson, D.; et al. Precision Targeted Therapy with BLU-667 for RET-Driven Cancers. Cancer Discov. 2018, 8, 836–849. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Curigliano, G.; Kim, D.-W.; Lee, D.H.; Besse, B.; Baik, C.S.; Doebele, R.C.; Cassier, P.A.; Lopes, G.; Tan, D.S.-W.; et al. Registrational dataset from the phase I/II ARROW trial of pralsetinib (BLU-667) in patients (pts) with advanced RET fusion+ non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2020, 38, 9515. [Google Scholar] [CrossRef]
- Besse, B.; Felip, E.; Clifford, C.; Louie-Gao, M.; Green, J.; Turner, C.D.; Popat, S. AcceleRET Lung: A phase III study of first-line pralsetinib in patients (pts) with RET-fusion+ advanced/metastatic non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2020, 38, TPS9633. [Google Scholar] [CrossRef]
- Offin, M.; Guo, R.; Wu, S.L.; Sabari, J.; Land, J.D.; Ni, A.; Montecalvo, J.; Halpenny, D.F.; Buie, L.W.; Pak, T.; et al. Immunophenotype and Response to Immunotherapy of RET-Rearranged Lung Cancers. JCO Precis. Oncol. 2019, 3, 1–8. [Google Scholar] [CrossRef]
- Guisier, F.; Dubos-Arvis, C.; Viñas, F.; Doubre, H.; Ricordel, C.; Ropert, S.; Janicot, H.; Bernardi, M.; Fournel, P.; Lamy, R.; et al. Efficacy and Safety of Anti–PD-1 Immunotherapy in Patients with Advanced NSCLC with BRAF, HER2, or MET Mutations or RET Translocation: GFPC 01-2018. J. Thorac. Oncol. 2020, 15, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.; Mezquita, L.; Thai, A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Hegde, A.; Andreev-Drakhlin, A.Y.; Roszik, J.; Huang, L.; Liu, S.; Hess, K.; Cabanillas, M.; Hu, M.I.; Busaidy, N.L.; Sherman, S.I.; et al. Responsiveness to immune checkpoint inhibitors versus other systemic therapies in RET-aberrant malignancies. ESMO Open 2020, 5, e000799. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Cao, W.; Ren, Z.; Tang, Y.; Zhang, C.; Yang, R.; Chen, Y.; Liu, Z.; Peng, C.; Wang, L.; et al. GDNF secreted by nerves enhances PD-L1 expression via JAK2-STAT1 signaling activation in HNSCC. OncoImmunology 2017, 6, e1353860. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Yang, X.; Shi, J.; Chen, X.; Jiang, Y.; Zhao, H. Efficacy of Cabozantinib and Nivolumab in Treating Hepatocellular Carcinoma with RET Amplification, High Tumor Mutational Burden, and PD-L1 Expression. Oncologist 2020, 25, 470–474. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.J.; Shaw, A.T. Resisting Resistance: Targeted Therapies in Lung Cancer. Trends Cancer 2016, 2, 350–364. [Google Scholar] [CrossRef] [Green Version]
- Pailler, E.; Faugeroux, V.; Oulhen, M.; Mezquita, L.; Laporte, M.; Honoré, A.; Lecluse, Y.; Queffelec, P.; Ngo-Camus, M.; Nicotra, C.; et al. Acquired Resistance Mutations to ALK Inhibitors Identified by Single Circulating Tumor Cell Sequencing in ALK-Rearranged Non–Small-Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6671–6682. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A.; Ou, S.H.I.; Cho, B.C.; Kim, D.W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (Tpx-0005) is a Next-Generation ros1/Trk/Alk Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent-Front Mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A.; Hu, Z.I.; Lai, G.G.Y.; Tan, D.S.W. Targeting RET-driven cancers: Lessons from evolving preclinical and clinical landscapes. Nat. Rev. Clin. Oncol. 2017, 15, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, F.; Guida, T.; Anaganti, S.; Vecchio, G.; Fusco, A.; Ryan, A.J.; Billaud, M.; Santoro, M. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 2004, 23, 6056–6063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, I.; Stevens, S.E.; Lin, J.J.; Nagy, R.; Ferris, L.; Shaw, A.T.; Gainor, J.F. Emergence of a RET V804M Gatekeeper Mutation During Treatment With Vandetanib in RET-Rearranged NSCLC. J. Thorac. Oncol. 2018, 13, e226–e227. [Google Scholar] [CrossRef] [Green Version]
- Wirth, L.J.; Kohno, T.; Udagawa, H.; Matsumoto, S.; Ishii, G.; Ebata, K.; Tuch, B.B.; Zhu, E.Y.; Nguyen, M.; Smith, S.; et al. Emergence and Targeting of Acquired and Hereditary Resistance to Multikinase RET Inhibition in Patients With RET-Altered Cancer. JCO Precis. Oncol. 2019, 3, 1–7. [Google Scholar] [CrossRef]
- Nakaoku, T.; Kohno, T.; Araki, M.; Niho, S.; Chauhan, R.; Knowles, P.P.; Tsuchihara, K.; Matsumoto, S.; Shimada, Y.; Mimaki, S.; et al. A secondary RET mutation in the activation loop conferring resistance to vandetanib. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Cosci, B.; Vivaldi, A.; Romei, C.; Gemignani, F.; Landi, S.; Ciampi, R.; Tacito, A.; Molinaro, E.; Agate, L.; Bottici, V.; et al. In silico and in vitro analysis of rare germline allelic variants of RET oncogene associated with medullary thyroid cancer. Endocr. Relat. Cancer 2011, 18, 603–612. [Google Scholar] [CrossRef]
- Plenker, D.; Riedel, M.; Brägelmann, J.; Dammert, M.A.; Chauhan, R.; Knowles, P.P.; Lorenz, C.; Keul, M.; Bührmann, M.; Pagel, O.; et al. Drugging the catalytically inactive state of RET kinase in RET-rearranged tumors. Sci. Transl. Med. 2017, 9, eaah6144. [Google Scholar] [CrossRef] [Green Version]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [Green Version]
- Nelson-Taylor, S.K.; Le, A.T.; Yoo, M.; Schubert, L.; Mishall, K.M.; Doak, A.; Varella-Garcia, M.; Tan, A.-C.; Doebele, R.C. Resistance to RET-Inhibition in RET-Rearranged NSCLC Is Mediated By Reactivation of RAS/MAPK Signaling. Mol. Cancer Ther. 2017, 16, 1623–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaishnavi, A.; Schubert, L.; Rix, U.; Marek, L.A.; Le, A.T.; Keysar, S.B.; Glogowska, M.J.; Smith, M.A.; Kako, S.; Sumi, N.J.; et al. EGFR Mediates Responses to Small-Molecule Drugs Targeting Oncogenic Fusion Kinases. Cancer Res. 2017, 77, 3551–3563. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Sung, J.H.; Moon, S.U.; Kim, H.-S.; Kim, J.W.; Lee, J.S. EGF Induced RET Inhibitor Resistance inCCDC6-RETLung Cancer Cells. Yonsei Med. J. 2017, 58, 9–18. [Google Scholar] [CrossRef]
- Bhinge, K.; Yang, L.; Terra, S.; Nasir, A.; Muppa, P.; Aubry, M.C.; Yi, J.; Janaki, N.; Kovtun, I.V.; Murphy, S.J.; et al. EGFR mediates activation of RET in lung adenocarcinoma with neuroendocrine differentiation characterized by ASCL1 expression. Oncotarget 2017, 8, 27155–27165. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.W.; Jang, K.W.; Sohn, J.; Kim, S.-M.; Pyo, K.-H.; Kim, H.; Yun, M.R.; Na Kang, H.; Kim, H.R.; Lim, S.M.; et al. Antitumor Activity and Acquired Resistance Mechanism of Dovitinib (TKI258) in RET-Rearranged Lung Adenocarcinoma. Mol. Cancer Ther. 2015, 14, 2238–2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somwar, R.; Smith, R.; Hayashi, T.; Ishizawa, K.; Charen, A.S.; Khodos, I.; Mattar, M.; He, J.; Balasubramanian, S.; Stephens, P.; et al. MDM2 amplification (Amp) to mediate cabozantinib resistance in patients (Pts) with advanced RET-rearranged lung cancers. J. Clin. Oncol. 2016, 34, 9068. [Google Scholar] [CrossRef]
- Solomon, B.J.; Tan, L.; Lin, J.J.; Wong, S.Q.; Hollizeck, S.; Ebata, K.; Tuch, B.B.; Yoda, S.; Gainor, J.F.; Sequist, L.V.; et al. RET Solvent Front Mutations Mediate Acquired Resistance to Selective RET Inhibition in RET-Driven Malignancies. J. Thorac. Oncol. 2020, 15, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Shen, T.; Terzyan, S.; Liu, X.; Hu, X.; Patel, K.; Hu, M.; Cabanillas, M.; Behrang, A.; Meric-Bernstam, F.; et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann. Oncol. 2021, 32, 261–268. [Google Scholar] [CrossRef]
- Lin, J.J.; Liu, S.V.; McCoach, C.E.; Zhu, V.W.; Tan, A.C.; Yoda, S.; Peterson, J.; Do, A.; Prutisto-Chang, K.; Dagogo-Jack, I.; et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann. Oncol. 2020, 31, 1725–1733. [Google Scholar] [CrossRef]
- Zhu, V.W.; Madison, R.; Schrock, A.B.; Ou, S.-H.I. Emergence of High Level of MET Amplification as Off-Target Resistance to Selpercatinib Treatment in KIF5B-RET NSCLC. J. Thorac. Oncol. 2020, 15, e124–e127. [Google Scholar] [CrossRef]
- Liu, X.; Shen, T.; Mooers, B.H.M.; Hilberg, F.; Wu, J. Drug resistance profiles of mutations in the RET kinase domain. Br. J. Pharmacol. 2018, 175, 3504–3515. [Google Scholar] [CrossRef] [Green Version]
- Carlomagno, F.; Guida, T.; Anaganti, S.; Provitera, L.; Kjaer, S.; McDonald, N.Q.; Ryan, A.J.; Santoro, M. Identification of tyrosine 806 as a molecular determinant of RET kinase sensitivity to ZD6474. Endocr. Relat. Cancer 2009, 16, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulligan, L.M. GDNF and the RET Receptor in Cancer: New Insights and Therapeutic Potential. Front. Physiol. 2019, 9, 1873. [Google Scholar] [CrossRef]
- Nakasatomi, M.; Takahashi, S.; Sakairi, T.; Ikeuchi, H.; Kaneko, Y.; Hiromura, K.; Nojima, Y.; Maeshima, A. Enhancement of HGF-induced tubulogenesis by endothelial cell-derived GDNF. PLoS ONE 2019, 14, e0212991. [Google Scholar] [CrossRef] [PubMed]
- Garnis, C.; Campbell, J.; Davies, J.J.; Macaulay, C.; Lam, S.; Lam, W.L. Involvement of multiple developmental genes on chromosome 1p in lung tumorigenesis. Hum. Mol. Genet. 2004, 14, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Joo, L.J.S.; Weiss, J.; Gill, A.J.; Clifton-Bligh, R.; Brahmbhatt, H.; MacDiarmid, J.A.; Gild, M.L.; Robinson, B.G.; Zhao, J.T.; Sidhu, S.B. RET Kinase-Regulated MicroRNA-153-3p Improves Therapeutic Efficacy in Medullary Thyroid Carcinoma. Thyroid. 2019, 29, 830–844. [Google Scholar] [CrossRef]
- Zhang, W.; Dong, Y.-Z.; Du, X.; Peng, X.-N.; Shen, Q.-M. MiRNA-153-3p promotes gefitinib-sensitivity in non-small cell lung cancer by inhibiting ATG5 expression and autophagy. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2444–2452. [Google Scholar]
- Chen, W.-J.; Zhang, E.-N.; Zhong, Z.-K.; Jiang, M.-Z.; Yang, X.-F.; Zhou, D.-M.; Wang, X.-W. MicroRNA-153 Expres-sion and Prognosis in Non-Small Cell Lung Cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 8671–8675. [Google Scholar]
- Zhang, M.; Liu, L.; Lin, X. Article A Translocation Pathway for Vesicle-Mediated Article A Translo-cation Pathway for Vesicle-Mediated Unconventional Protein Secretion. Cell 2020, 1–16. [Google Scholar] [CrossRef]
- Xiao, G.-Y.; Schmid, S. Abstract 118: FCHSD2 controls oncogenic ERK1/2 signaling by regulating endocytic trafficking in non-small-cell lung cancer. Immunology 2020, 80, 118. [Google Scholar] [CrossRef]
- Batrouni, A.G.; Baskin, J.M. A MAP for PI3K Activation on Endosomes. Nat. Cell Biol. 2020, 22, 1292–1294. [Google Scholar] [CrossRef]
- Crupi, M.J.F.; Maritan, S.M.; Reyes-Alvarez, E.; Lian, E.Y.; Hyndman, B.D.; Rekab, A.N.; Moodley, S.; Antonescu, C.N.; Mulligan, L.M. GGA3-mediated recycling of the RET receptor tyrosine kinase contributes to cell migration and invasion. Oncogene 2020, 39, 1361–1377. [Google Scholar] [CrossRef] [PubMed]
- Cascone, T.; Subbiah, V.; Hess, K.R.; Nelson, S.; Nilsson, M.B.; Subbiah, I.M.; Ali, S.M.; Carbone, D.P.; Salgia, R.; Owonikoko, T.K.; et al. Significant systemic and CNS activity of RET inhibitor vandetanib combined with mTOR inhibitor everolimus in patients with advanced NSCLC with RET fusion. J. Clin. Oncol. 2016, 34, 9069. [Google Scholar] [CrossRef]
- Subbiah, V.; Berry, J.; Roxas, M.; Guha-Thakurta, N.; Subbiah, I.M.; Ali, S.M.; McMahon, C.; Miller, V.; Cascone, T.; Pai, S.; et al. Systemic and CNS activity of the RET inhibitor vandetanib combined with the mTOR inhibitor everolimus in KIF5B-RET re-arranged non-small cell lung cancer with brain metastases. Lung Cancer 2015, 89, 76–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, E.Y.; Johnson, M.L.; Clifford, S.E.; Somwar, R.; Kherani, J.F.; Son, J.; Bertram, A.A.; Davare, M.A.; Gladstone, E.G.; Ivanova, E.V.; et al. Overcoming MET-Dependent Resistance to Selective RET Inhibition in Patients with RET Fusion–Positive Lung Cancer by Combining Selpercatinib with Crizotinib. Clin. Cancer Res. 2021, 27, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Koga, K.; Hattori, Y.; Komori, M.; Narishima, R.; Yamasaki, M.; Hakoshima, M.; Fukui, T.; Maitani, Y. Combination of RET siRNA and irinotecan inhibited the growth of medullary thyroid carcinoma TT cells and xenografts via apoptosis. Cancer Sci. 2010, 101, 941–947. [Google Scholar] [CrossRef]
- Bertazza, L.; Barollo, S.; Radu, C.M.; Cavedon, E.; Simioni, P.; Faggian, D.; Plebani, M.; Pelizzo, M.R.; Rubin, B.; Boscaro, M.; et al. Synergistic antitumour activity of RAF265 and ZSTK474 on human TT medullary thyroid cancer cells. J. Cell. Mol. Med. 2015, 19, 2244–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, N.; Jiang, T.; Rosen, D.M.; Nelkin, B.D.; Ball, D.W. Synergistic Action of a RAF Inhibitor and a Dual PI3K/mTOR Inhibitor in Thyroid Cancer. Clin. Cancer Res. 2011, 17, 6482–6489. [Google Scholar] [CrossRef] [Green Version]
- Caccia, D.; Miccichè, F.; Cassinelli, G.; Mondellini, P.; Casalini, P.; Bongarzone, I. Dasatinib reduces FAK phosphorylation increasing the effects of RPI-1 inhibition in a RET/PTC1-expressing cell line. Mol. Cancer 2010, 9, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, Y.W.; Shah, M.H.; Agarwal, K.; Mccarty, S.K.; Koo, B.S.; Brendel, V.J.; Wang, C.; Porter, K.; Jarjoura, D.; Saji, M.; et al. Sorafenib and Mek inhibition is synergistic in medullary thyroid carcinoma in vitro. Endocr. Relat. Cancer 2012, 19, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.S.; Sebti, S.M.; Newman, R.A.; Blaskovich, M.A.; Ye, L.; Gagel, R.F.; Moulder, S.; Wheler, J.J.; Naing, A.; Tannir, N.M.; et al. Phase I Trial of a Combination of the Multikinase Inhibitor Sorafenib and the Farnesyltransferase Inhibitor Tipifarnib in Advanced Malignancies. Clin. Cancer Res. 2009, 15, 7061–7068. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Naganna, N.; Sintim, H.O. Identification of nicotinamide aminonaphthyridine compounds as potent RET kinase inhibitors and antitumor activities against RET rearranged lung adenocarcinoma. Bioorg. Chem. 2019, 90, 103052. [Google Scholar] [CrossRef]
- Nguyen, M.; Miyakawa, S.; Kato, J.; Mori, T.; Arai, T.; Armanini, M.; A Gelmon, K.; Yerushalmi, R.; Leung, S.; Gao, D.; et al. Preclinical Efficacy and Safety Assessment of an Antibody–Drug Conjugate Targeting the c-RET Proto-Oncogene for Breast Carcinoma. Clin. Cancer Res. 2015, 21, 5552–5562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosco, E.E.; Christie, R.J.; Carrasco, R.; Sabol, D.; Zha, J.; Dacosta, K.; Brown, L.; Kennedy, M.; Meekin, J.; Phipps, S.; et al. Preclinical evaluation of a GFRA1 targeted antibody-drug conjugate in breast cancer. Oncotarget 2018, 9, 22960–22975. [Google Scholar] [CrossRef] [Green Version]
- Bagheri-Yarmand, R.; Williams, M.D.; Grubbs, E.G.; Gagel, R.F. ATF4 Targets RET for Degradation and is a Candidate Tumor Suppressor Gene in Medullary Thyroid Cancer. J. Clin. Endocrinol. Metab. 2016, 102, 933–941. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Xia, Y.; Yin, Y.; Luo, J.; Liu, M.; Zhang, H.; Zhang, C.; Zhao, Y.; Yang, L.; Kong, L. ATF4 destabilizes RET through nonclassical GRP78 inhibition to enhance chemosensitivity to bortezomib in human osteosarcoma. Theranostics 2019, 9, 6334–6353. [Google Scholar] [CrossRef]
- Drilon, A.; Rogers, E.; Zhai, D.; Deng, W.; Zhang, X.; Lee, D.; Ung, J.; Whitten, J.; Zhuang, H.; Liu, J.; et al. TPX-0046 is a novel and potent RET/SRC inhibitor for RET-driven cancers. Ann. Oncol. 2019, 30, v190–v191. [Google Scholar] [CrossRef]
- Schoffski, P.; Aftimos, P.G.; Massard, C.; Italiano, A.; Jungels, C.; Andreas, K.; Keegan, M.; Ho, P.T.C. A phase I study of BOS172738 in patients with advanced solid tumors with RET gene alterations including non-small cell lung cancer and medullary thyroid cancer. J. Clin. Oncol. 2019, 37, TPS3162. [Google Scholar] [CrossRef]
- Frett, B.; Carlomagno, F.; Moccia, M.L.; Brescia, A.; Federico, G.; De Falco, V.; Admire, B.; Chen, Z.; Qi, W.; Santoro, M.; et al. Fragment-Based Discovery of a Dual pan-RET/VEGFR2 Kinase Inhibitor Optimized for Single-Agent Polypharmacology. Angew. Chem. Int. Ed. 2015, 54, 8717–8721. [Google Scholar] [CrossRef] [PubMed]
- Moccia, M.; Frett, B.; Zhang, L.; Lakkaniga, N.R.; Briggs, D.C.; Chauhan, R.; Brescia, A.; Federico, G.; Yan, W.; Santoro, M.; et al. Bioisosteric Discovery of NPA101.3, a Second-Generation RET/VEGFR2 Inhibitor Optimized for Single-Agent Polypharmacology. J. Med. Chem. 2020, 63, 4506–4516. [Google Scholar] [CrossRef] [PubMed]
- Larocque, E.; Naganna, N.; Ma, X.; Opoku-Temeng, C.; Carter-Cooper, B.; Chopra, G.; Lapidus, R.G.; Sintim, H.O. Aminoisoquinoline benzamides, FLT3 and Src-family kinase inhibitors, potently inhibit proliferation of acute myeloid leukemia cell lines. Future Med. Chem. 2017, 9, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, K.; Zhang, G.; Yang, Q.-Y.; Li, Y.-S.; Huang, S.-Z.; Wang, Y.-L.; Yang, W.; Jiang, X.-J.; Yan, H.-X.; et al. Structural optimization and structure-activity relationship studies of N-phenyl-7,8-dihydro-6H-pyrimido[5,4-b][1,4]oxazin-4-amine derivatives as a new class of inhibitors of RET and its drug resistance mutants. Eur. J. Med. Chem. 2018, 143, 1148–1164. [Google Scholar] [CrossRef] [PubMed]
- Lakkaniga, N.R.; Gunaganti, N.; Zhang, L.; Belachew, B.; Frett, B.; Leung, Y.-K.; Li, H. Pyrrolo[2,3-d]pyrimidine derivatives as inhibitors of RET: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2020, 206, 112691. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.-C.; Chen, W.; Feng, Z.-L.; Liu, Z.-P. Recent developments of RET protein kinase inhibitors with diverse scaffolds as hinge binders. Future Med. Chem. 2021, 13, 45–62. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Fusions | Histotype | Gender | Reference |
|---|---|---|---|
| CDC123-RET | ADC | F | [42] |
| CCDC6-RET | ADC, NE | M > F | [34] |
| CLIP1-RET | ADC | NA | [43] |
| CUX1-RET | ADC | M | [44] |
| EPHA5-RET | ADC | NA | [45] |
| ERC1-RET | ADC | NA | [43] |
| FRMD4A-RET | ADC | F | [46] |
| FYCO1-RET | ADC | F | [34] |
| ITGA8-RET | ADC | M | [34] |
| ITIH2-RET | AS | F | [34] |
| KIF13A-RET | ADC | F | [47] |
| KIF5B-RET | ADC, NE, NSCLC, AS | F > M | [34] |
| KIAA1468-RET | IMA | M | [48] |
| MIR3924-RET | SCC | M | [34] |
| MYO5C-RET | ADC | NA | [49] |
| NCOA4-RET | ADC | F | [50] |
| PICALM-RET | ADC | NA | [45] |
| RASSF4-RET | ADC | NA | [51] |
| RUFY2-RET | ADC | NA | [52] |
| SLC25A36-RET | ADC | F | [34] |
| SLC39A8-RET | ADC | F | [34] |
| TBC1D32-RET | ADC | F | [53] |
| TRIM24-RET | ADC | NA | [52] |
| TRIM33-RET | ADC | F | [54] |
| WAC-RET | ADC | F | [55] |
| ZBTB41-RET | ADC | M | [34] |
| Drug | Principal Kinase Targets | Type of Study | No. of pts with RET Positive—NSCLC Treated | ORR (%) | DCR (%) | DRR (%) | Grade ≥ 3 AEs (%) | Common Grade 3 or 4 TEAEs (%) |
|---|---|---|---|---|---|---|---|---|
| Vandetanib | VEGFR, EGFR, RET | Phase II trial [61] | 19 | 47% | 90% | 53% | 58% | Hypertension (58%) Rash acneiform (16%) Diarrhea (11%) Prolonged QTc (11%) |
| Phase II trial [49] | 17 | 18% | 65% | 22% | 28% | Hypertension (17%) Prolonged QTc (11%) AST/ALT elevation (6%) | ||
| Retrospective series [45] | 11 | 18% | 45% | NA | NA | NA | ||
| Retrospective series [65] | 3 | 0% | 33% | 33% | NA | NA | ||
| Cabozantinib | VEGFR2, MET, AXL, c-KIT, FLT3, TIE-2, RET | Phase II trial [43] | 26 | 28% | 100% | 73% | 47% | AST/ALT elevation (16%) Lipase elevation (15%) Decreased platelet count (8%) Hypophosphatemia (8%) |
| Retrospective series [45] | 21 | 33% | 57% | NA | NA | NA | ||
| Lenvatinib | VEGFR1-3, FGFR1-4, PDGFR-A, c-KIT, RET | Phase II trial [62,64] | 25 | 16% | 76% | 64% | 92% | Hypertension (56%) Hyponatremia (20%) Proteinuria (16%) Pneumonia (16%) Nausea (12%) |
| Retrospective series [45] | 2 | 50% | 50% | NA | NA | NA | ||
| Other MKIs | ||||||||
| Sorafenib | VEGFR1-3, PDGFRB, c-KIT, FLT3, BRAF, c-RAF | Phase II trial [66] | 3 | 0% | 33% | 33% | 33% | HFS (33%) Infection (33%) |
| Retrospective series [45] | 2 | 0% | 100% | NA | NA | NA | ||
| Sunitinib | VEGFR1-3, PDGFRB, c-KIT, FLT3, RET | Retrospective series [45] | 10 | 22% | 50% | NA | NA | NA |
| Case report [67] | 1 | - | - | - | - | Fatigue Thrombocytopenia | ||
| Ponatinib | BCR-ABL, FLT3, SRC, c-KIT, FGFR, VEGFR, PDGFR, RET | Retrospective series [45] | 2 | 0% | 100% | NA | NA | NA |
| Alectinib | ALK, LTK, CHEK2, FLT3, RET | Case series [60] | 4 | 50% | 75% | 25% | 25% | Hyperbilirubinemia (25%) Increased CPK (25%) |
| Retrospective series [69] | 4 | 50% | 50% | 0% | 0% | None | ||
| Phase II trial [71] | 25 | 4% | 52% | 0% | 4% | Neutropenia Pneumonitis Diarrhea Hyponatremia Increased CPK Hyperbilirubinemia (percentages NA) | ||
| Retrospective series [45] | 2 | 0% | 0% | NA | NA | NA | ||
| Case report [68] | 1 | - | - | - | - | None | ||
| Nintedanib | PDGFRA-B, VEGFR1-3, FGFR1-3 | Retrospective series [45] | 2 | 50% | 100% | NA | NA | NA |
| Regorafenib | VEGFR1-3, PDGFRB, c-KIT, FGFR, RET, c-RAF | Retrospective series [45] | 1 | 0% | 0% | NA | NA | NA |
| RXDX-105 | RET, BRAF | Phase I/Ib trial [63] | 40 | 15% (19% in previous untreated pts) | 52% | 5% | NA | Hypophosphatemia AST/ALT elevation Rash Diarrhea Fatigue (percentages NA) |
| New RET-selective inhibitors | ||||||||
| Selpercatinib | RET | Phase I/II trial [73] | 144 (105 evaluable for response) | 64% (85% in previous untreated pts) | 92% (95% in previous untreated pts) | 30% of the safety population (3/531 pts) | 28% | AST/ALT elevation (23%) Hypertension (14%) Hyponatremia (6%) Lymphopenia (6%) |
| Pralsetinib | RET | Phase I/II trial [74] * | 116 | 61% (73% in previous untreated pts) | 93% | 4% of the safety population (4/120) | 28% | AST elevation (22%), Hypertension (18%) ALT elevation (17%) Fatigue (15%) Neutrophilia (15%) |
| Mutation Status | Cabozantinib [112] | Vandetanib [112] | Lenvatinib [112] | Ponatinib [112] | Selpercatinib [109] | Pralsetinib [109] | |
|---|---|---|---|---|---|---|---|
| Gatekeeper | V804M | 4.26 | 5.83 | 5.42 | 0.0339 | 0.0559 | 0.0168 |
| V804L | 3.22 | 6.10 | 10.60 | 0.43 [60] | 0.0172 | 0.0018 | |
| Solvent front | G810A | 0.22 | 2.76 | 0.11 | 0.008 [60] | - | - |
| G810R | - | - | - | - | 2.744 | 2.650 | |
| G810S | 1.05 | 5.47 | 0.67 | - | 0.8802 | 0.3906 | |
| G810C | - | - | - | - | 1.227 | 0.6417 | |
| Other | S904F | - | 0.908 [98] | - | - | - | - |
| Y806C | - | 0.933 [113] | - | - | 0.1744 | 0.2958 | |
| Y806N | 4.76 | 5.86 | 1.93 | - | 0.1498 | 0.2925 | |
| V738A | 1.20 | 1.05 | 2.35 | - | 0.2388 | 0.1775 | |
| Drugs | In Vitro | In Vivo | References | ||
|---|---|---|---|---|---|
| RET IC50 | VEGFR2 IC50 | RET V804M IC50 | Xenograft Mouse Model | ||
| Pz-1 | <0.001 μM | <0.001 μM | <0.001 μM | 10 mg/kg/day per os inhibition of tumor growth | [140] |
| NPA-101.3 | 0.001 μM | 0.003 μM | 0.008 μM | 10 mg/kg/day per os inhibition of tumor growth | [141] |
| HSN356 | - | - | - | - | [142] |
| HSN608 | 3.16 nM | - | - | - | [133] |
| 17d | 0.01 μM | - | 0.015 μM * | 10–30 mg/kg/day inhibition of tumor growth | [143] |
| 59 | 0.0068 μM | - | 13.51 nM | - | [144] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fancelli, S.; Caliman, E.; Mazzoni, F.; Brugia, M.; Castiglione, F.; Voltolini, L.; Pillozzi, S.; Antonuzzo, L. Chasing the Target: New Phenomena of Resistance to Novel Selective RET Inhibitors in Lung Cancer. Updated Evidence and Future Perspectives. Cancers 2021, 13, 1091. https://doi.org/10.3390/cancers13051091
Fancelli S, Caliman E, Mazzoni F, Brugia M, Castiglione F, Voltolini L, Pillozzi S, Antonuzzo L. Chasing the Target: New Phenomena of Resistance to Novel Selective RET Inhibitors in Lung Cancer. Updated Evidence and Future Perspectives. Cancers. 2021; 13(5):1091. https://doi.org/10.3390/cancers13051091
Chicago/Turabian StyleFancelli, Sara, Enrico Caliman, Francesca Mazzoni, Marco Brugia, Francesca Castiglione, Luca Voltolini, Serena Pillozzi, and Lorenzo Antonuzzo. 2021. "Chasing the Target: New Phenomena of Resistance to Novel Selective RET Inhibitors in Lung Cancer. Updated Evidence and Future Perspectives" Cancers 13, no. 5: 1091. https://doi.org/10.3390/cancers13051091