
Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets

 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract

1. Introduction
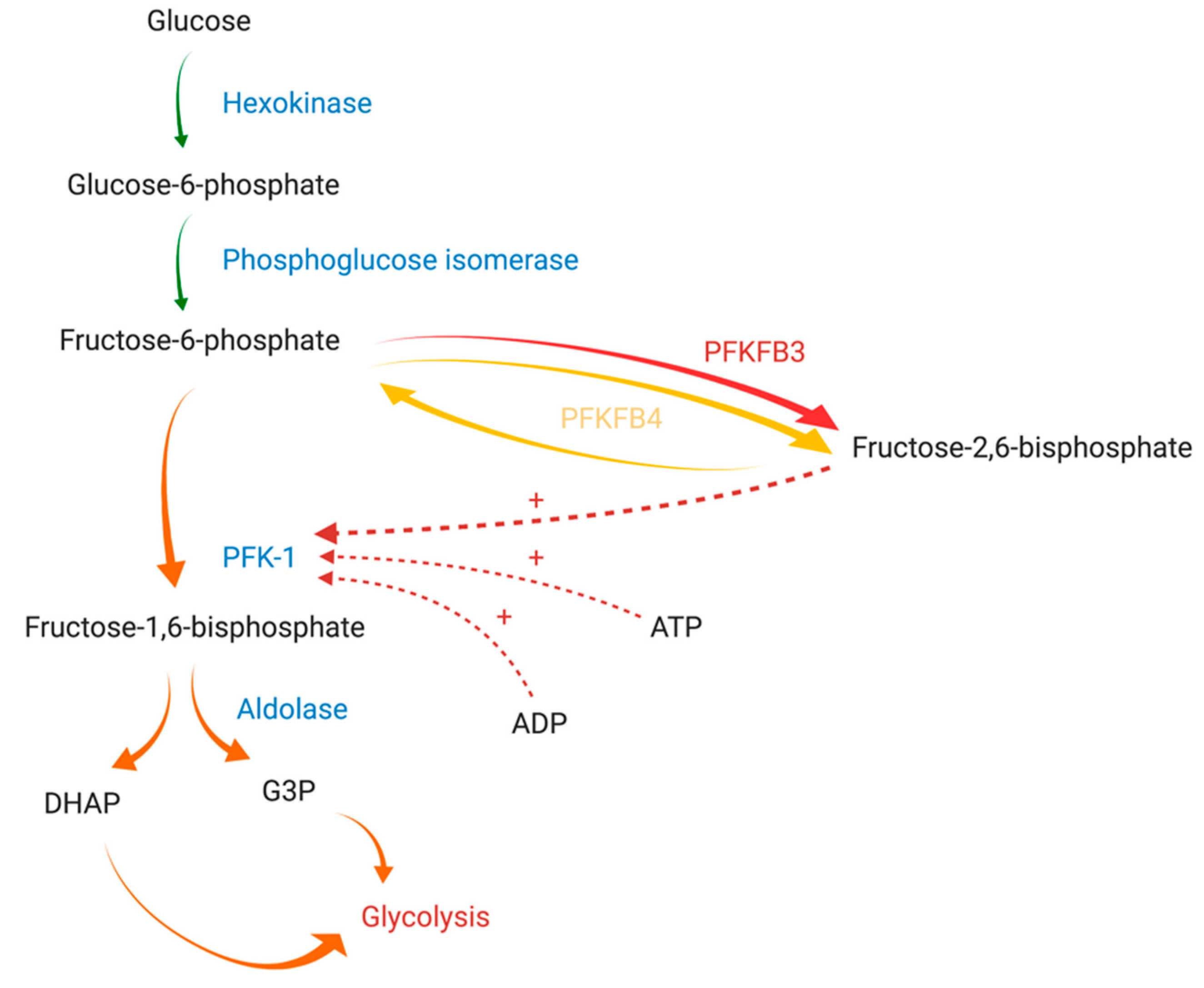
2. PFKFB Genes and Proteins
2.1. PFKFB1
2.2. PFKFB2
2.3. PFKFB3
2.4. PFKFB4
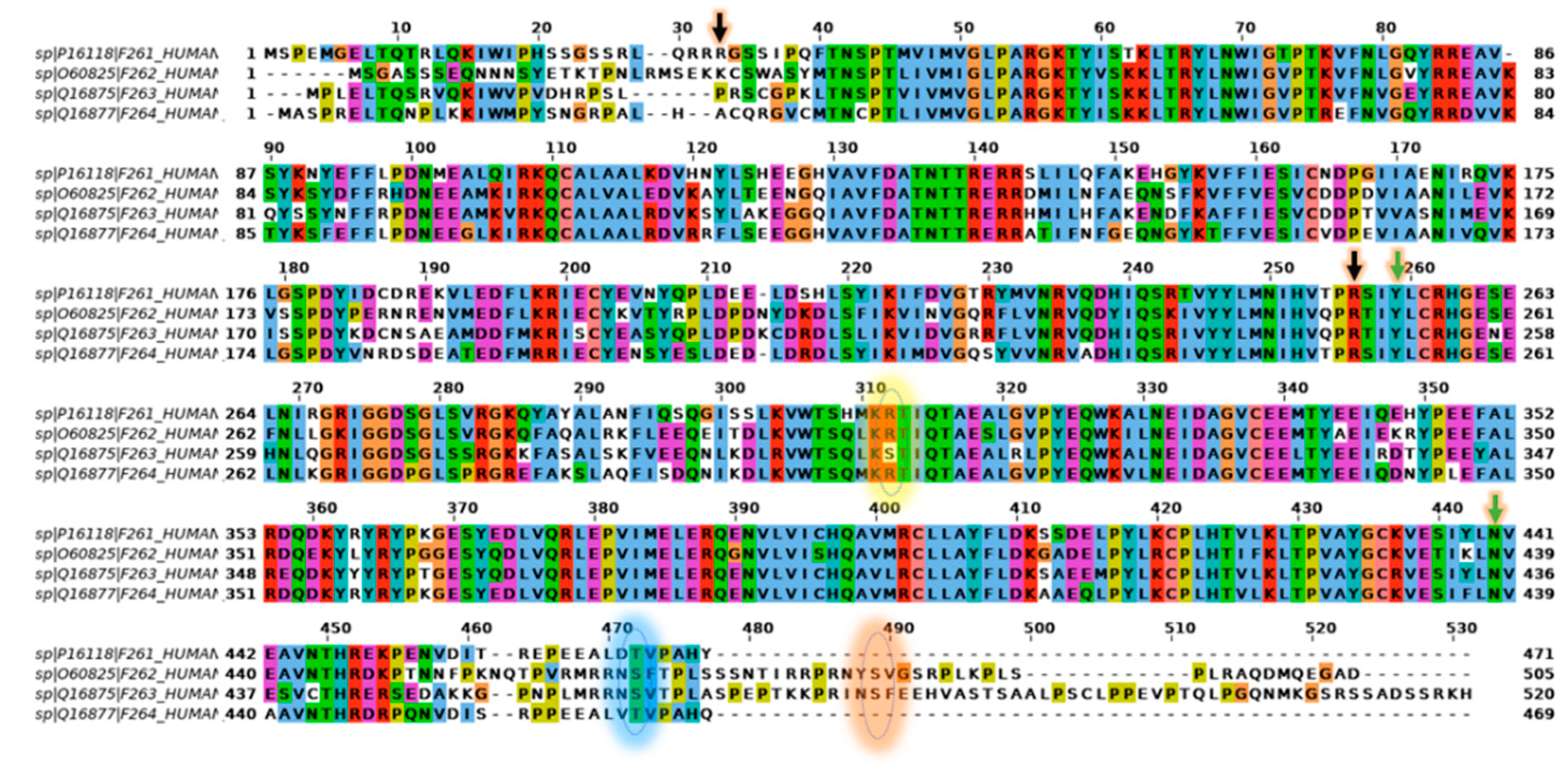
2.5. Comparison of PFKFB1-4 Amino Acid Sequence
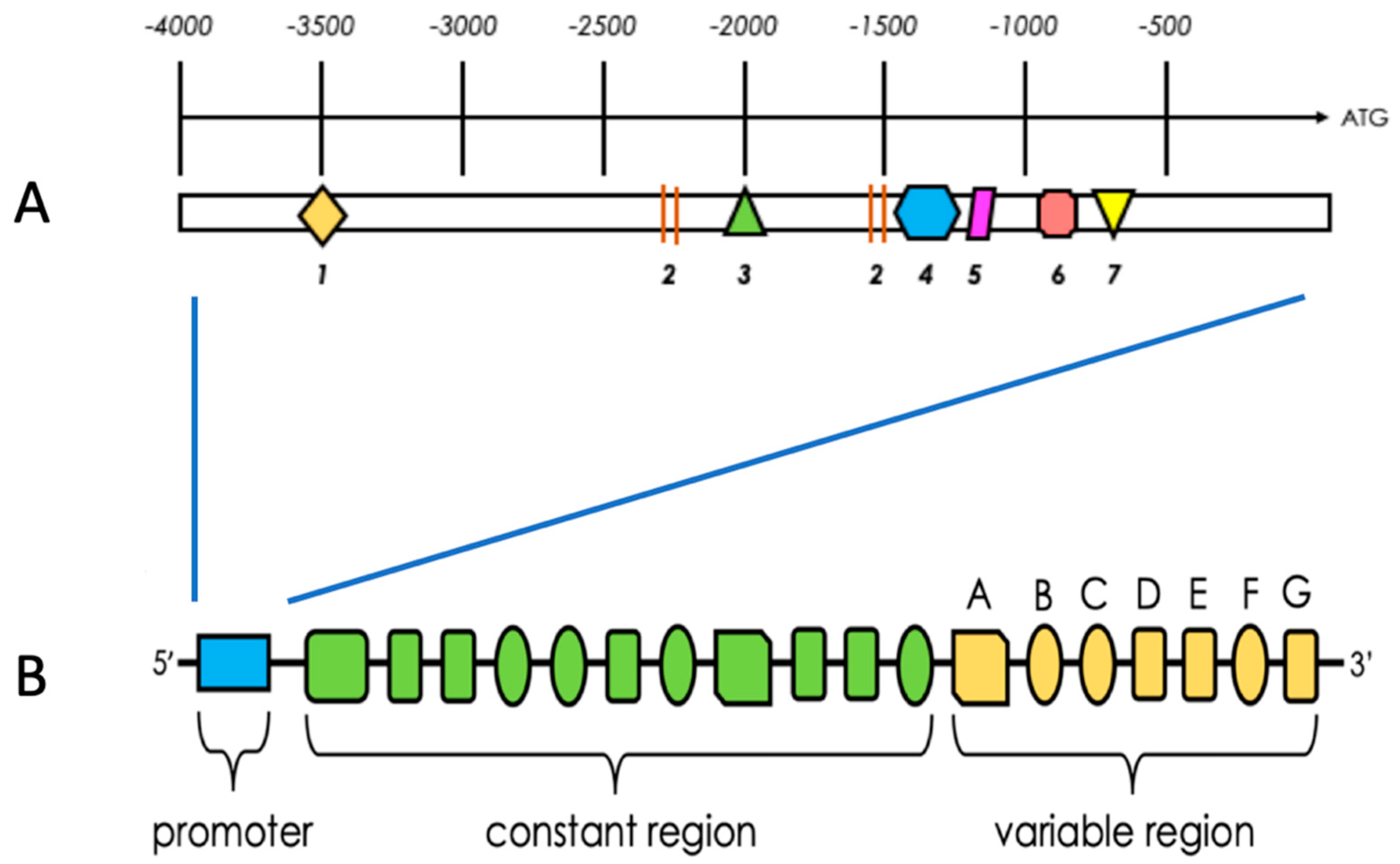
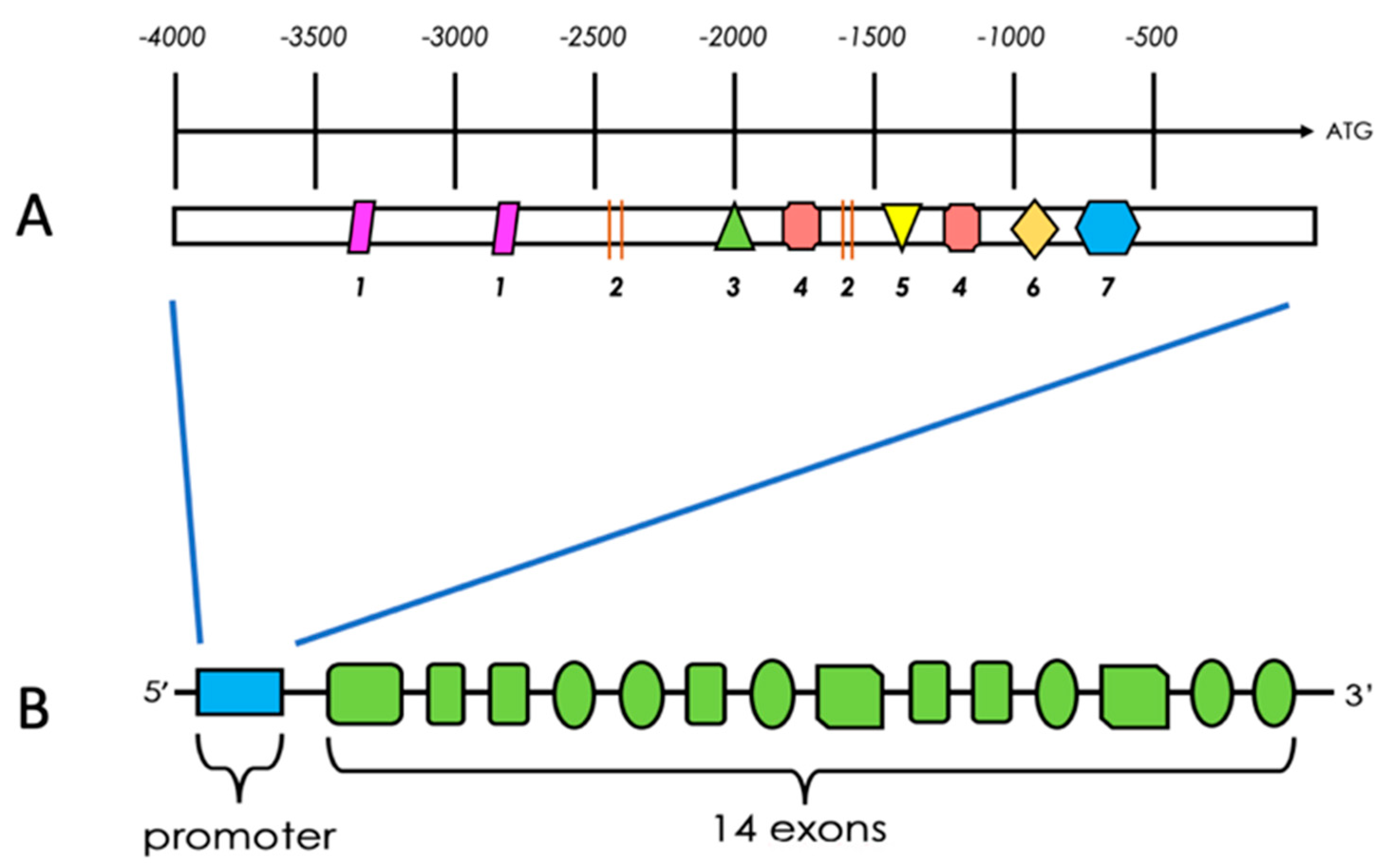
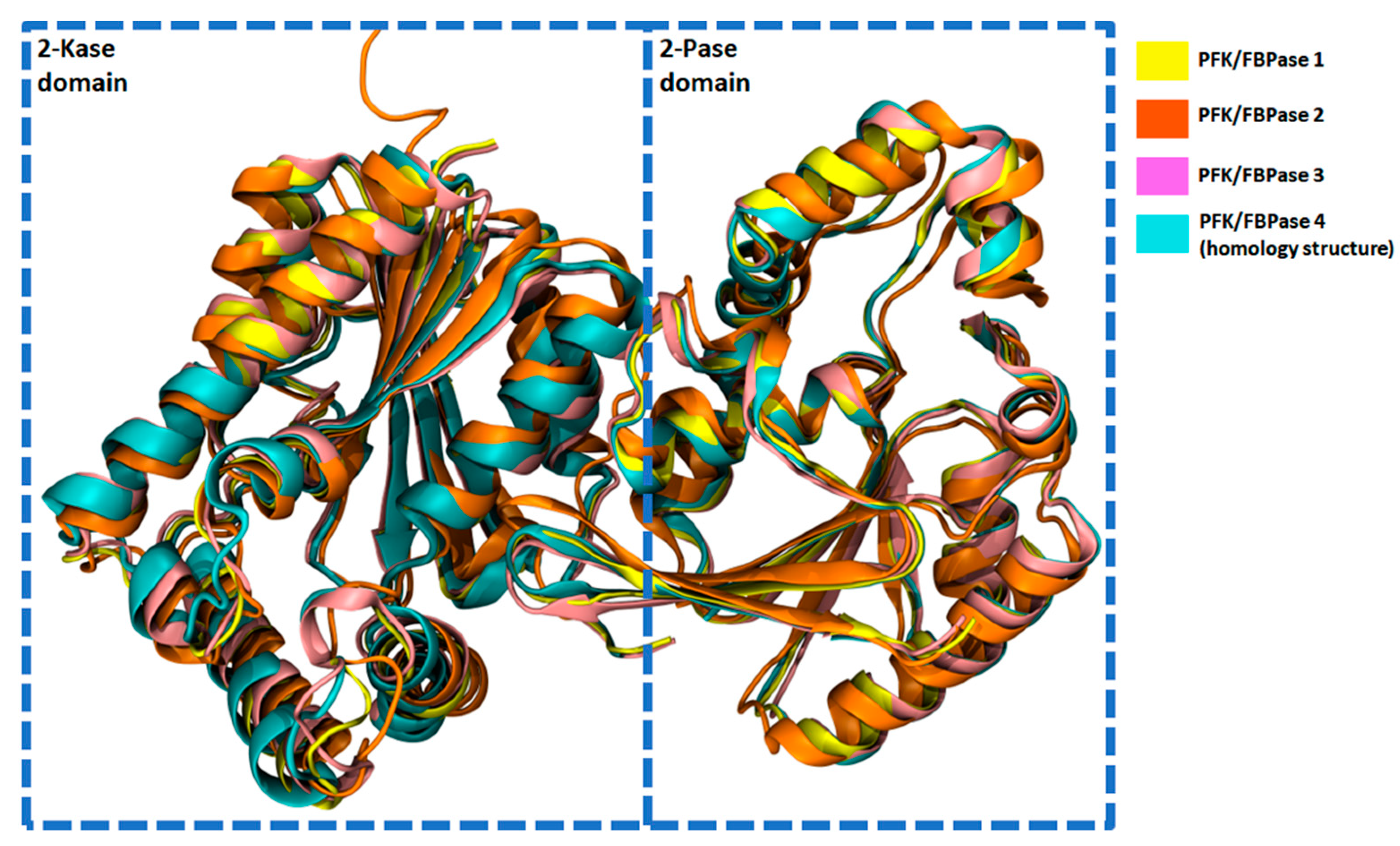
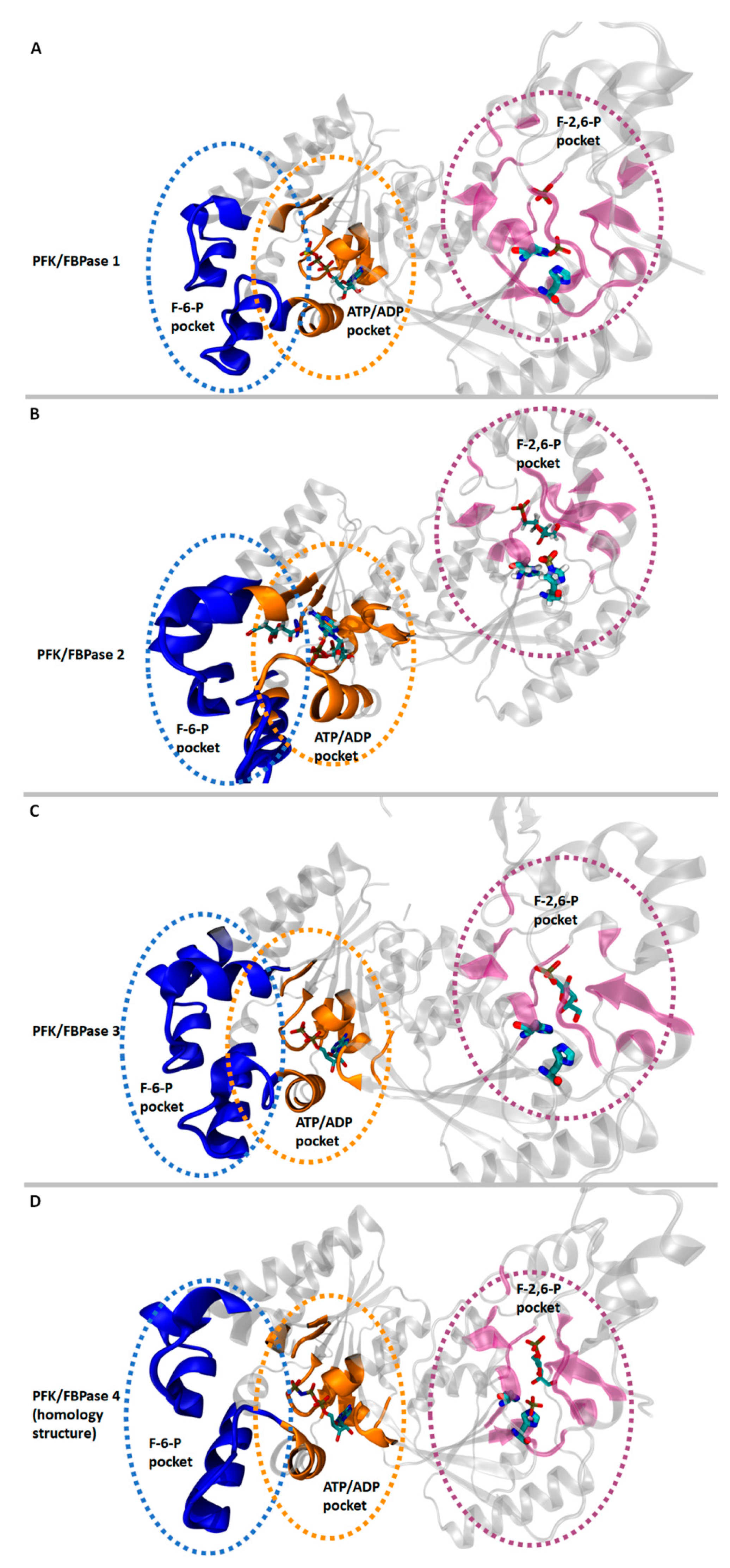
2.6. Structural Characteristics of PFKFB 1-4
2.7. Regulation of PFKFB Expression
2.7.1. Ras-Dependent Regulation of PFKFB Expression
2.7.2. mTOR-Dependent Regulation of PFKFB Expression
2.7.3. Steroid-Dependent Regulation of PFKFB Expression
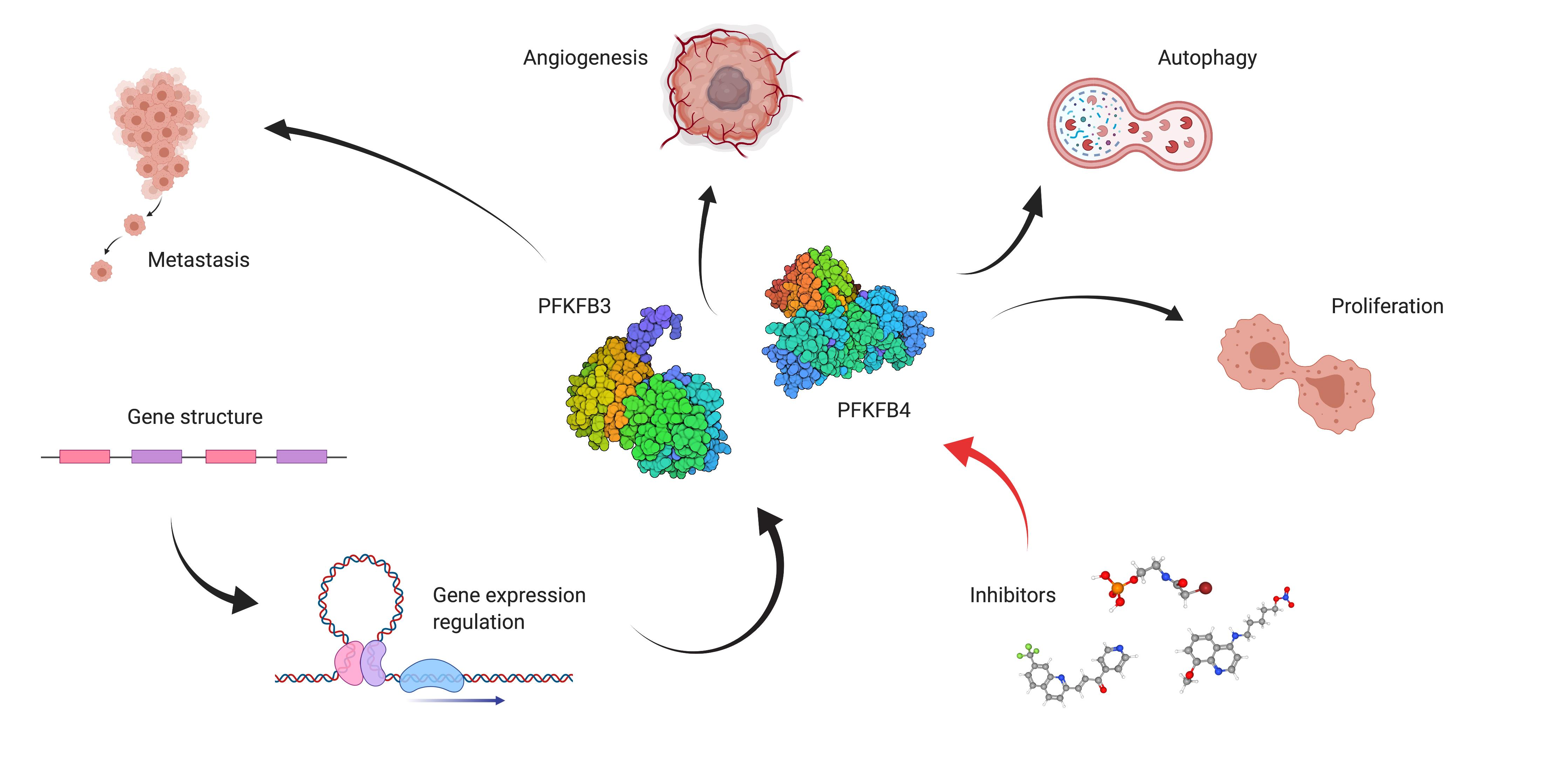
3. PFKFB3 and PFKFB4 in Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isoenzyme | Cancer Type | Research Environment and the Study Material and/or Cell Line | Reference | |
|---|---|---|---|---|
| PFKFB3 | Breast cancer | HMEC, MCF-10A, SKBR3, BT-474 | In vitro | O’Neal et al. [59] |
| HER2+ patient samples | In vitro | Novellasdemunt et al. [72] | ||
| MCF-7, T-47D | In vitro | Imbert-Fernandez et al. [58] | ||
| MCF-7, T-47D, SUM159 | In vitro | Ge et al. [73] | ||
| Breast cancer patient samples, MDA-MB-231, MDA-MB-438, HUVEC | In vitro | Peng et al. [65] | ||
| Melanoma | 451LU, WM983 | In vitro | Warrier et al. [74] | |
| A375 | In vitro/in vivo | Telang et al. [75] | ||
| DB-1, SK-MEL-5 | In vitro | Mendoza et al. [76] | ||
| Gastric cancer | MKN45, AGS, BCG823, GES-1 | In vivo/in vitro | Zhu et al. [77] | |
| MKN45, NUGC3 | In vitro | Bobarykina et al. [28] | ||
| MKN45, NUGC3 | In vitro | Minchenko et al. [67] | ||
| Pancreatic cancer | Panc1 | In vitro | Minchenko et al. [67] | |
| Panc1 | In vitro | Bobarykina et al. [28] | ||
| Panc1 | In vitro | Yalcin et al. [78] | ||
| Colon adenocarcinoma | Colorectal cancer patient samples, SW480, SW1116 | In vivo/in vitro | Han et al. [79] | |
| HCT-116 | In vitro | Klarer et al. [80] | ||
| FFPE tissue samples, SW620 | In vitro | Atsumi et al. [81] | ||
| Ovarian cancer | HeyA8, HeyA8MDR, OVCAR5, OV90 | In vitro | Mondal et al. [82] | |
| Lung cancer | LLC1, H522 | In vitro | Clem et al. [83] | |
| H522, H1437, PC9, HCC827 | In vitro | Lypova et al. [84] | ||
| Bladder cancer | T24, HUVEC | In vitro | Hu et al. [85] | |
| Glioblastoma | U87 | In vitro | Mendoza et al. [76] | |
| Glioblastoma patient samples | In vitro | Kessler et al. [86] | ||
| Glioblastoma patient samples | In vitro | Fleischer et al. [87] | ||
| Glioblastoma patient samples, U87 | In vitro | Zscharnack et al. [88] | ||
| Head and neck carcinoma | Cal27, FaDu, HNSCC patient samples | In vitro | Li et al. [89] | |
| Astrocytoma | Astrocytoma patient samples | In vitro | Kessler et al. [86] | |
| Astrocytoma patient samples | In vitro | Zscharnack et al. [88] | ||
| Neuroblastoma | - | Statistical analysis | Trojan et al. [90] | |
| Cervical cancer | OV2008, C13 | In vitro | Mondal et al. [82] | |
| Renal cancer | ACHN | In vitro | Lu et al. [91] | |
| Thyroid cancer | FFPE tissue samples | In vitro | Atsumi et al. [81] | |
| Osteosarcoma | U20S | In vitro | Du et al. [92] | |
| Osteosarcoma patient samples, Saos-2 | In vitro | Zheng et al. [93] | ||
| Acute myeloid leukemia | THP-1, OCI-AML3 | In vitro | Feng et al. [56] | |
| Esophageal carcinoma | KYSE30, KYSE150 | In vitro/statistical analysis | Liu et al. [94] | |
| PFKFB4 | Breast cancer | MDA-MB-231, T47D, breast cancer patient samples | In vitro | Gao et al. [69] |
| Breast cancer patient samples | In vitro | Yao et al. [95] | ||
| MDA-MB-231, MCF7, SUM159, MDA-MB-468, breast cancer patient samples | In vitro | Gao et al. [96] | ||
| MDA-MB-231, MCF-7, MCF-7-ERE-MAR-Luc, MCF-10A | In vitro/in vivo | Dasgupta et al. [63] | ||
| Ovarian cancer | SKOV3, UPN-251, OC316, OVCAR-3, A2780 | In vitro | Taylor et al. [68] | |
| Gastric cancer | MKN45, NUGC3 | In vitro | Bobarykina et al. [28] | |
| Pancreatic cancer | Panc1 | In vitro | Bobarykina et al. [28] | |
| Neuroblastoma | - | Statistical analysis | Trojan et al. [90] | |
| Prostate cancer | PC-3, LNCaP | In vitro | Li et al. [97] | |
| DU145, PC-3, LNCaP | In vitro | Ros et al. [98] | ||
| Glioblastoma | NCH421k, NCH441, NCH644 | In vitro | Goidts et al. [99] | |
| Bladder cancer | Bladder cancer patient samples | In vitro | Yun et al. [100] | |
| Lung adenocarcinoma | Lung adenocarcinoma patient samples, H460 | In vitro | Chesney et al. [101] | |
Influence of PFKFB3 and PFKFB4 on Carcinogenesis
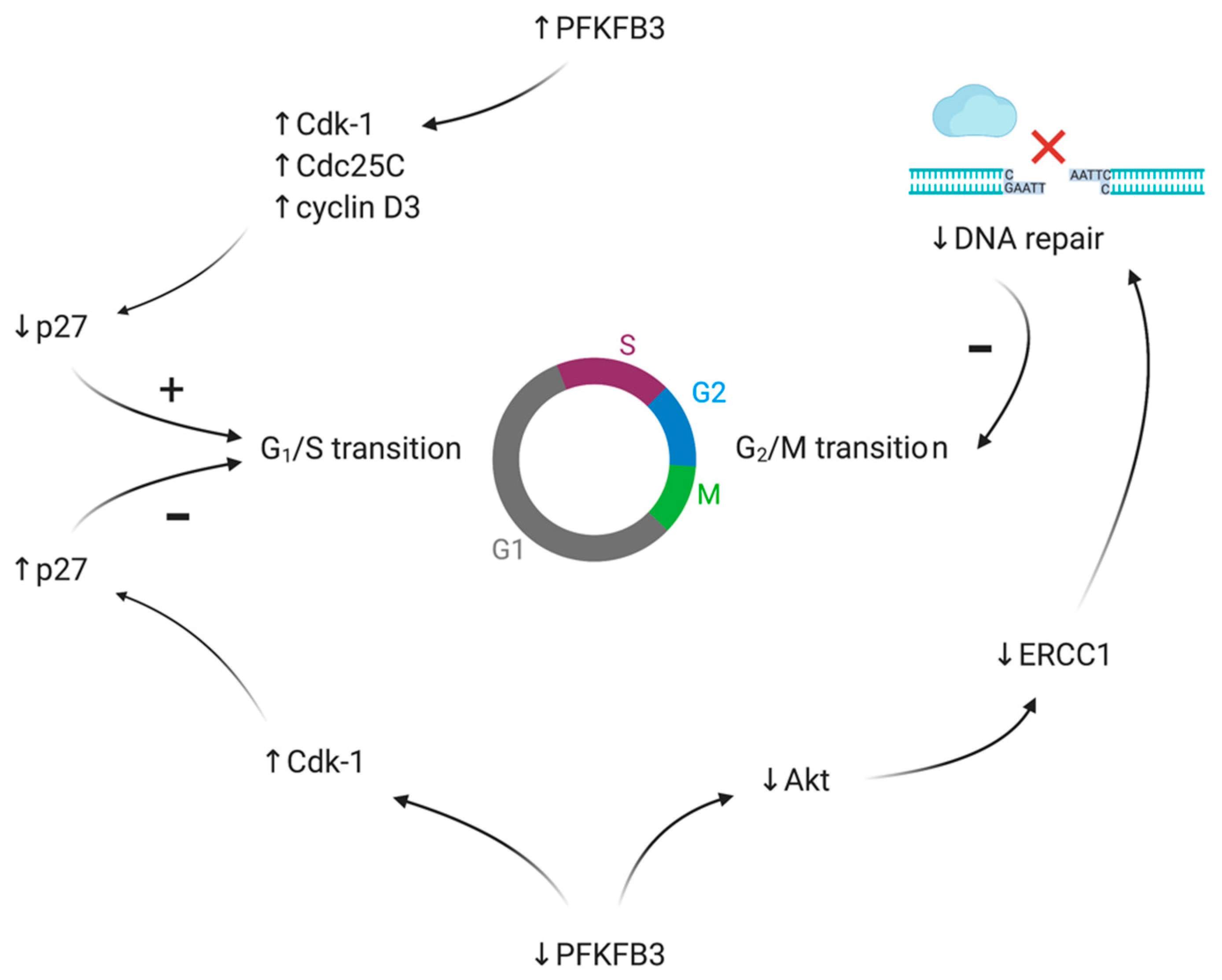
4. Proliferation, Invasiveness and Migration
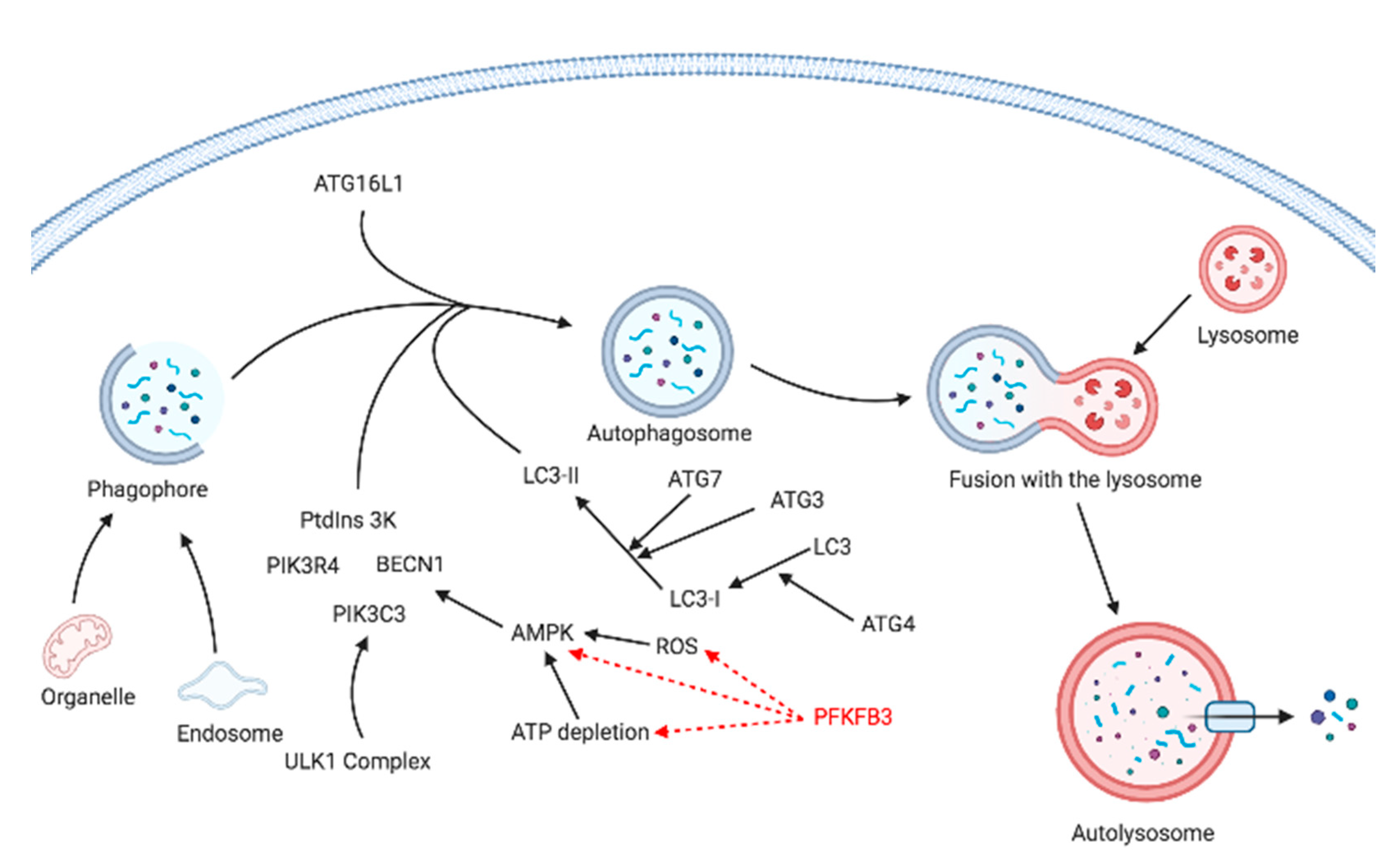
5. Autophagy
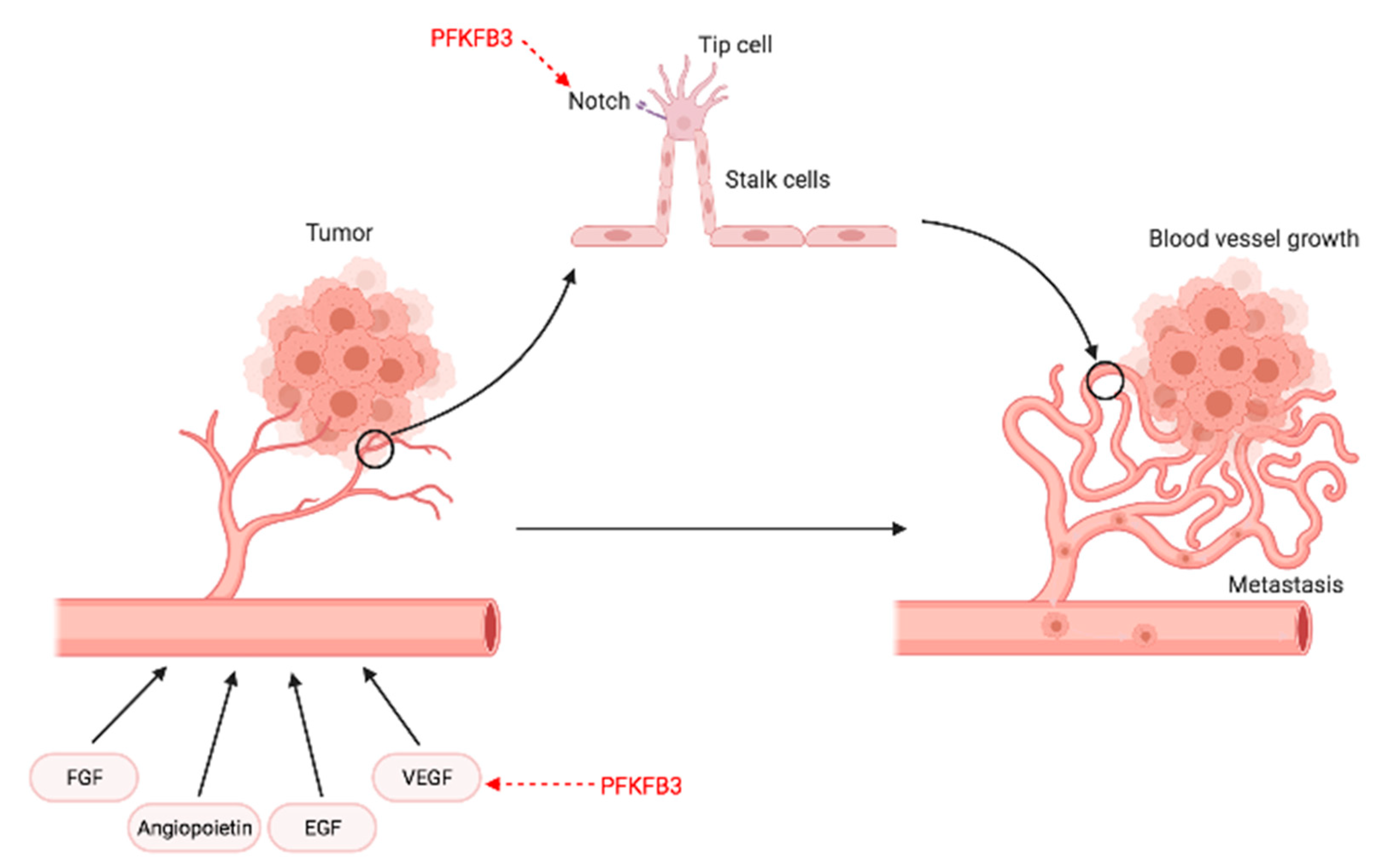
6. Angiogenesis
7. Targeting PFK-2 Isozymes in Malignancies
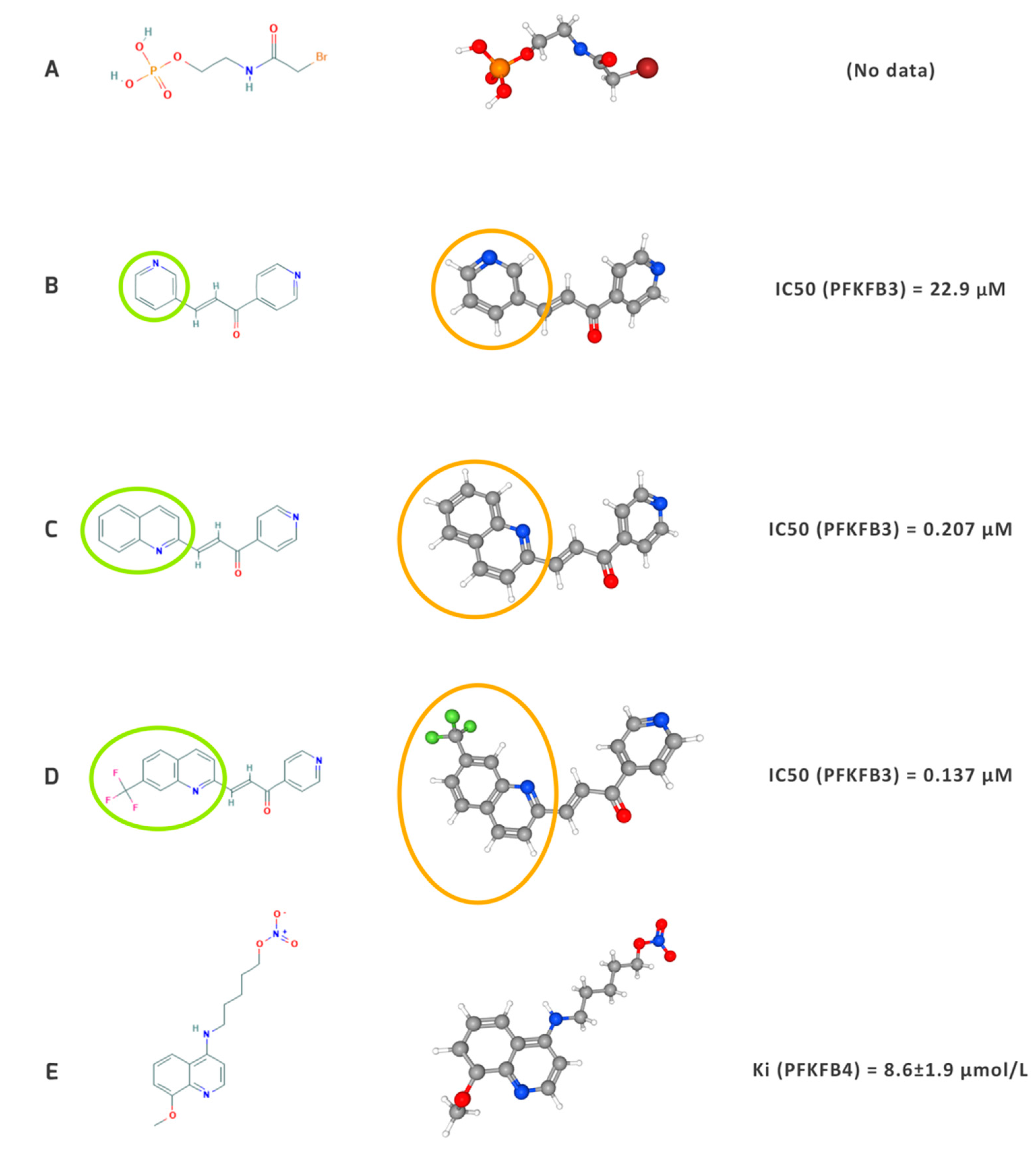
7.1. Outline of the Development of Inhibitors
7.2. Chemosensitivity, Chemoresistance, and Potential Combined Therapies for Malignancies
| Inhibitor | Combined Therapy | Type of Neoplasm | Study |
|---|---|---|---|
| 3PO | imatinib | chronic myeloid leukemia | Zhu Y, et al. [15] |
| PFK15 | imatinib | chronic myeloid leukemia | Zhu Y, et al. [15] |
| PFK15 | rapamycin | acute myeloid leukemia | Feng Y & Wu L [56] |
| PFK158 | vemurafenib | melanoma | Lu L et al. [10] |
| PFK158 | erlotinib | non-small cell lung cancer cell | Lypova N, et al. [84] |
| PFK158 | antiestrogen | breast cancer | Imbert-Fernandez Y, et al. [58] |
| PFK15 | cisplatin | cervical cancer | Li FL, et al. [163] |
| PFK158 | carboplatin | ovarian cancer | Mondal S, et al. [82] |
| 3PO | paclitaxel | breast cancer | Domenech E, et al. [105] |
| PFK15/siRNA | oxaliplatin | colon cancer | Yan S, et al. [70] |
| 3PO | VEGF inhibitors | endothelial cells | Schoors S, et al. [138] |
7.2.1. Influence on Hematological Malignancies
7.2.2. Gynecological and Breast Cancers
7.2.3. Influence on Lung Cancer (NSCLC and SCLC)
7.2.4. Influence on Other Solid Tumors
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. THE METABOLISM OF TUMORS IN THE BODY. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Wang, Y.; Qu, C.; Liu, T.; Wang, C. PFKFB3 inhibitors as potential anticancer agents: Mechanisms of action, current developments, and structure-activity relationships. Eur. J. Med. Chem. 2020, 203, 112612. [Google Scholar] [CrossRef] [PubMed]
- Kishton, R.J.; Rathmell, J.C. Novel therapeutic targets of tumor metabolism. Cancer J. 2015, 21, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortunato, S.; Bononi, G.; Granchi, C.; Minutolo, F. An Update on Patents Covering Agents That Interfere with the Cancer Glycolytic Cascade. ChemMedChem 2018, 13, 2251–2265. [Google Scholar] [CrossRef] [PubMed]
- Morris, A. Inhibiting glycolysis in tumour cells. Nat. Rev. Endocrinol. 2018, 14, 323. [Google Scholar] [CrossRef]
- Bartrons, R.; Simon-Molas, H.; Rodríguez-García, A.; Castaño, E.; Navarro-Sabaté, À.; Manzano, A.; Martinez-Outschoorn, U.E. Fructose 2,6-bisphosphate in cancer cell metabolism. Front. Oncol. 2018, 8, 331. [Google Scholar] [CrossRef]
- Houddane, A.; Bultot, L.; Novellasdemunt, L.; Johanns, M.; Gueuning, M.A.; Vertommen, D.; Coulie, P.G.; Bartrons, R.; Hue, L.; Rider, M.H. Role of Akt/PKB and PFKFB isoenzymes in the control of glycolysis, cell proliferation and protein synthesis in mitogen-stimulated thymocytes. Cell. Signal. 2017, 34, 23–37. [Google Scholar] [CrossRef]
- Lu, L.; Chen, Y.; Zhu, Y. The molecular basis of targeting PFKFB3 as a therapeutic strategy against cancer. Oncotarget 2017, 8, 62793–62802. [Google Scholar] [CrossRef] [Green Version]
- Yi, M.; Ban, Y.; Tan, Y.; Xiong, W.; Li, G.; Xiang, B. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 and 4: A pair of valves for fine-tuning of glucose metabolism in human cancer. Mol. Metab. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Clem, B.; Telang, S.; Clem, A.; Yalcin, A.; Meier, J.; Simmons, A.; Rasku, M.A.; Arumugam, S.; Dean, W.L.; Eaton, J.; et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 2008, 7, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ros, S.; Schulze, A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013, 1, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Pan, H.; Liu, Z.; Xie, J.; Han, W. Roles of PFKFB3 in cancer. Signal Transduct. Target. Ther. 2017, 2, 17044. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Lu, L.; Qiao, C.; Shan, Y.; Li, H.; Qian, S.; Hong, M.; Zhao, H.; Li, J.; Yang, Z.; et al. Targeting PFKFB3 sensitizes chronic myelogenous leukemia cells to tyrosine kinase inhibitor. Oncogene 2018, 37, 2837–2849. [Google Scholar] [CrossRef]
- Chesney, J. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase and tumor cell glycolysis. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 535–539. [Google Scholar] [CrossRef]
- Gómez, M.; Manzano, A.; Navarro-Sabaté, À.; Duran, J.; Obach, M.; Perales, J.C.; Bartrons, R. Specific expression of pfkfb4 gene in spermatogonia germ cells and analysis of its 5′-flanking region. FEBS Lett. 2005, 579, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Okar, D.A.; Manzano, A.; Navarro-Sabatè, A.; Riera, L.; Bartrons, R.; Lange, A.J. PFK-2/FBPase-2: Maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem. Sci. 2001, 26, 30–35. [Google Scholar] [CrossRef]
- Rider, M.H.; Bertrand, L.; Vertommen, D.; Michels, P.A.; Rousseau, G.G.; Hue, L. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: Head-to-head with a bifunctional enzyme that controls glycolysis. Biochem. J. 2004, 381, 561–579. [Google Scholar] [CrossRef] [Green Version]
- Bartrons, R.; Rider, M.H. Fructose 2,6-bisphosphate: The last milestone of the 20th century in metabolic control? Metabolic reprogramming of tumor cells View project Fructose 2,6-bisphosphate: The last milestone of the 20th century in metabolic control? Artic. Biochem. J. 2010. [Google Scholar] [CrossRef]
- Pilkis, S.J.; Claus, T.H.; Kurland, I.J.; Lange, A.J. 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatase: A Metabolic Signaling Enzyme. Annu. Rev. Biochem. 1995, 64, 799–835. [Google Scholar] [CrossRef]
- Lee, Y.H.; Li, Y.; Uyeda, K.; Hasemann, C.A. Tissue-specific structure/function differentiation of the liver isoform of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. J. Biol. Chem. 2003, 278, 523–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luck, K.; Kim, D.K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef]
- Gene: PFKFB1 (ENSG00000158571)-Summary-Homo_Sapiens-Ensembl Genome Browser 102. Available online: http://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000158571;r=X:54932961-54998534 (accessed on 11 February 2021).
- Minchenko, O.; Opentanova, I.; Caro, J. Hypoxic regulation of the 6-phosphofructo-2-kinase/fructose-2,6- bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS Lett. 2003, 554, 264–270. [Google Scholar] [CrossRef]
- Heine-Suner, D.; Diaz-Guillen, M.A.; Lange, A.J.; Rodriguez de Cordoba, S. Sequence and structure of the human 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase heart isoform gene (PFKFB2). Eur. J. Biochem. 1998, 254, 103–110. [Google Scholar] [CrossRef]
- Bobarykina, A.Y.; Minchenko, D.O.; Opentanova, I.L.; Moenner, M.; Caro, J.; Esumi, H.; Minchenko, O.H. Hypoxic regulation of PFKFB-3 and PFKFB-4 gene expression in gastric and pancreatic cancer cell lines and expression of PFKFB genes in gastric cancers. Acta Biochim. Pol. 2006, 53, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Houles, T.; Gravel, S.P.; Lavoie, G.; Shin, S.; Savall, M.; Meant, A.; Grondin, B.; Gaboury, L.; Yoon, S.O.; St-Pierre, J.; et al. RSK regulates PFK-2 activity to promote metabolic rewiring in melanoma. Cancer Res. 2018, 78, 2191–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsin, A.S.; Bertrand, L.; Rider, M.H.; Deprez, J.; Beauloye, C.; Vincent, M.F.; Van den Berghe, G.; Carling, D.; Hue, L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol. 2000, 10, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Chesney, J.; Mitchell, R.; Benigni, F.; Bacher, M.; Spiegel, L.; Al-Abed, Y.; Han, J.H.; Metz, C.; Bucala, R. An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element: Role in tumor cell glycolysis and the Warburg effect. Proc. Natl. Acad. Sci. USA 1999, 96, 3047–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartrons, R.; Rodríguez-García, A.; Simon-Molas, H.; Castaño, E.; Manzano, A.; Navarro-Sabaté, À. The potential utility of PFKFB3 as a therapeutic target. Expert Opin. Ther. Targets 2018, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, R.; Kato, M.; Okamura, N.; Nakagawa, T.; Komada, Y.; Tominaga, N.; Shimojo, M.; Fukasawa, M. Characterization of a Human Placental Fructose-6-Phosphate, 2-Kinase/Fructose- 2,6-Bisphosphatase. J. Biochem. 1997, 122, 122–128. [Google Scholar] [CrossRef]
- Minchenko, D.O.; Mykhalchenko, V.G.; Tsuchihara, K.; Kanehara, S.; Yavorovsky, O.P.; Zavgorodny, I.V.; Paustovsky, Y.O.; Komisarenko, S.V.; Esumi, H.; Minchenko, O.H. Alternative splice variants of RAT 6-phosphofructo-2-kinase/fructose-2,6- bisphosphatase-4 mRNA. Ukr. Biokhimichnyi Zhurnal 2008, 80, 66–73. [Google Scholar]
- Minchenko, O.H.; Ochiai, A.; Opentanova, I.L.; Ogura, T.; Minchenko, D.O.; Caro, J.; Komisarenko, S.V.; Esumi, H. Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 in the human breast and colon malignant tumors. Biochimie 2005, 87, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Okar, D.A.; Live, D.H.; Devany, M.H.; Lange, A.J. Mechanism of the bisphosphatase reaction of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase probed by 1H-15N NMR spectroscopy. Biochemistry 2000, 39, 9754–9762. [Google Scholar] [CrossRef]
- Crochet, R.B.; Kim, J.-D.; Lee, H.; Yim, Y.-S.; Kim, S.-G.; Neau, D.; Lee, Y.-H. Crystal structure of heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB2) and the inhibitory influence of citrate on substrate binding. Proteins Struct. Funct. Bioinform. 2017, 85, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Cavalier, M.C.; Kim, S.G.; Neau, D.; Lee, Y.H. Molecular basis of the fructose-2,6-bisphosphatase reaction of PFKFB3: Transition state and the C-terminal function. Proteins Struct. Funct. Bioinform. 2012, 80, 1143–1153. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.G.; Manes, N.P.; El-Maghrabi, M.R.; Lee, Y.H. Crystal structure of the hypoxia-inducible form of 6-phosphofructo-2- kinase/fructose-2,6-bisphosphatase (PFKFB3): A possible new target for cancer therapy. J. Biol. Chem. 2006, 281, 2939–2944. [Google Scholar] [CrossRef] [Green Version]
- Yuen, M.H.; Mizuguchi, H.; Lee, Y.-H.; Cook, P.F.; Uyeda, K.; Hasemann, C.A. Crystal Structure of the H256A Mutant of Rat Testis Fructose-6-phosphate,2-kinase/Fructose-2,6-bisphosphatase. J. Biol. Chem. 1999, 274, 2176–2184. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [Green Version]
- Clamp, M.; Cuff, J.; Searle, S.M.; Barton, G.J. The Jalview Java alignment editor. Bioinformatics 2004, 20, 426–427. [Google Scholar] [CrossRef] [Green Version]
- Manes, N.P.; El-Maghrabi, M.R. The kinase activity of human brain 6-phosphofructo-2-kinase/fructose-2,6- bisphosphatase is regulated via inhibition by phosphoenolpyruvate. Arch. Biochem. Biophys. 2005, 438, 125–136. [Google Scholar] [CrossRef]
- Moon, J.S.; Jin, W.J.; Kwak, J.H.; Kim, H.J.; Yun, M.J.; Kim, J.W.; Park, S.W.; Kim, K.S. Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6- bisphosphatase 2 in prostate cancer cells. Biochem. J. 2011, 433, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Delano, W.L. PyMOL: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Telang, S.; Yalcin, A.; Clem, A.L.; Bucala, R.; Lane, A.N.; Eaton, J.W.; Chesney, J. Ras transformation requires metabolic control by 6-phosphofructo-2-kinase. Oncogene 2006, 25, 7225–7234. [Google Scholar] [CrossRef] [Green Version]
- Chesney, J.; Telang, S. Regulation of Glycolytic and Mitochondrial Metabolism by Ras. Curr. Pharm. Biotechnol. 2013, 14, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Marston, H.; Turnbull, A.; Langdon, S.P. A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget 2015, 6, 25677–25695. [Google Scholar] [CrossRef] [Green Version]
- Blum, R.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Kloog, Y. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1α, causing glycolysis shutdown and cell death. Cancer Res. 2005, 65, 999–1006. [Google Scholar]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Li, X.; Zhang, J. mTOR signaling in cancer and mtor inhibitors in solid tumor targeting therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wu, L. mTOR up-regulation of PFKFB3 is essential for acute myeloid leukemia cell survival. Biochem. Biophys. Res. Commun. 2017, 483, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Ferri, N.; Cignarella, A.; Trevisi, L.; Bolego, C. The Glycolytic Enzyme PFKFB3 Is Involved in Estrogen-Mediated Angiogenesis via GPER1. J. Pharmacol. Exp. Ther. 2017, 361, 361–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbert-Fernandez, Y.; Clem, B.F.; O’Neal, J.; Kerr, D.A.; Spaulding, R.; Lanceta, L.; Clem, A.L.; Telang, S.; Chesney, J. Estradiol stimulates glucose metabolism via 6-phosphofructo-2-kinase (PFKFB3). J. Biol. Chem. 2014, 289, 9440–9448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neal, J.; Clem, A.; Reynolds, L.; Dougherty, S.; Imbert-Fernandez, Y.; Telang, S.; Chesney, J.; Clem, B.F. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res. Treat. 2016, 160, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Zawacka-Pankau, J.; Grinkevich, V.V.; Hünten, S.; Nikulenkov, F.; Gluch, A.; Li, H.; Enge, M.; Kel, A.; Selivanova, G. Inhibition of glycolytic enzymes mediated by pharmacologically activated p53: Targeting Warburg effect to fight cancer. J. Biol. Chem. 2011, 286, 41600–41615. [Google Scholar] [CrossRef] [Green Version]
- Cordero-Espinoza, L.; Hagen, T. Increased concentrations of fructose 2,6-bisphosphate contribute to the Warburg effect in phosphatase and tensin homolog (PTEN)-deficient cells. J. Biol. Chem. 2013, 288, 36020–36028. [Google Scholar] [CrossRef] [Green Version]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, S.; Rajapakshe, K.; Zhu, B.; Nikolai, B.C.; Yi, P.; Putluri, N.; Choi, J.M.; Jung, S.Y.; Coarfa, C.; Westbrook, T.F.; et al. Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nature 2018, 556, 249–254. [Google Scholar] [CrossRef]
- Cieslar-Pobuda, A.; Jain, M.V.; Kratz, G.; Rzeszowska-Wolny, J.; Ghavami, S.; Wiechec, E. The expression pattern of PFKFB3 enzyme distinguishes between induced-pluripotent stem cells and cancer stem cells. Oncotarget 2015, 6, 29753–29770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, F.; Li, Q.; Sun, J.Y.; Luo, Y.; Chen, M.; Bao, Y. PFKFB3 is involved in breast cancer proliferation, migration, invasion and angiogenesis. Int. J. Oncol. 2018, 52, 945–954. [Google Scholar] [CrossRef] [Green Version]
- Gu, M.; Li, L.; Zhang, Z.; Chen, J.; Zhang, W.; Zhang, J.; Han, L.; Tang, M.; You, B.; Zhang, Q.; et al. PFKFB3 promotes proliferation, migration and angiogenesis in nasopharyngeal carcinoma. J. Cancer 2017, 8, 3887–3896. [Google Scholar] [CrossRef] [Green Version]
- Minchenko, O.H.; Tsuchihara, K.; Minchenko, D.O.; Bikfalvi, A.; Esumi, H. Mechanisms of regulation of PFKFB expression in pancreatic and gastric cancer cells. World J. Gastroenterol. 2014, 20, 13705–13717. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.; Mannion, D.; Miranda, F.; Karaminejadranjbar, M.; Herrero-Gonzalez, S.; Hellner, K.; Zheng, Y.; Bartholomeusz, G.; Bast, R.C.; Ahmed, A.A. Loss of PFKFB4 induces cell death in mitotically arrested ovarian cancer cells. Oncotarget 2017, 8, 17960–17980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, R.; Liu, Y.; Li, D.; Xun, J.; Zhou, W.; Wang, P.; Liu, C.; Li, X.; Shen, W.; Su, W.; et al. PFKFB4 Promotes Breast Cancer Metastasis via Induction of Hyaluronan Production in a p38-Dependent Manner. Cell. Physiol. Biochem. 2018, 50, 2108–2123. [Google Scholar] [CrossRef]
- Yan, S.; Zhou, N.; Zhang, D.; Zhang, K.; Zheng, W.; Bao, Y.; Yang, W. PFKFB3 inhibition attenuates oxaliplatin-induced autophagy and enhances its cytotoxicity in colon cancer cells. Int. J. Mol. Sci. 2019, 20, 5415. [Google Scholar] [CrossRef] [Green Version]
- Libby, C.J.; Tran, A.N.; Scott, S.E.; Griguer, C.; Hjelmeland, A.B. The pro-tumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 175–188. [Google Scholar] [CrossRef]
- Novellasdemunt, L.; Obach, M.; Millán-ariño, L.; Manzano, A.; Ventura, F.; Rosa, J.L.; Jordan, A.; Navarro-Sabate, À.; Bartrons, R. Progestins activate 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) in breast cancer cells. Biochem. J. 2012, 442, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Lyu, P.; Cao, Z.; Li, J.; Guo, G.; Xia, W.; Gu, Y. Overexpression of miR-206 suppresses glycolysis, proliferation and migration in breast cancer cells via PFKFB3 targeting. Biochem. Biophys. Res. Commun. 2015, 463, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Warrier, G.; Lanceta, L.; Imbert-Fernandez, Y.; Chesney, J.A. Inhibition of glucose metabolism through treatment of BRAF mutated metastatic melanoma with vemurafenib. J. Clin. Oncol. 2019, 37, e21005. [Google Scholar] [CrossRef]
- Telang, S.; O’Neal, J.; Tapolsky, G.; Clem, B.; Kerr, A.; Imbert-Ferndandez, Y.; Chesney, J. Discovery of a PFKFB3 inhibitor for phase I trial testing that synergizes with the B-Raf inhibitor vemurafenib. Cancer Metab. 2014, 2, P14. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, E.E.; Pocceschi, M.G.; Kong, X.; Leeper, D.B.; Caro, J.; Limesand, K.H.; Burd, R. Control of glycolytic flux by AMP-activated protein kinase in tumor cells adapted to low ph1. Transl. Oncol. 2012, 5, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Ye, L.; Zhang, J.; Yu, P.; Wang, H.; Ye, Z.; Tian, J. PFK15, a Small Molecule Inhibitor of PFKFB3, Induces Cell Cycle Arrest, Apoptosis and Inhibits Invasion in Gastric Cancer. PLoS ONE 2016, 11, e0163768. [Google Scholar] [CrossRef]
- Yalcin, A.; Solakoglu, T.H.; Ozcan, S.C.; Guzel, S.; Peker, S.; Celikler, S.; Balaban, B.D.; Sevinc, E.; Gurpinar, Y.; Chesney, J.A. 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase-3 is required for transforming growth factor β1-enhanced invasion of Panc1 cells in vitro. Biochem. Biophys. Res. Commun. 2017, 484, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Meng, Q.; Xi, Q.; Zhang, Y.; Zhuang, Q.; Han, Y.; Jiang, Y.; Ding, Q.; Wu, G. Interleukin-6 stimulates aerobic glycolysis by regulating PFKFB3 at early stage of colorectal cancer. Int. J. Oncol. 2016, 48, 215–224. [Google Scholar] [CrossRef]
- Klarer, A.C.; O’Neal, J.; Imbert-Fernandez, Y.; Clem, A.; Ellis, S.R.; Clark, J.; Clem, B.; Chesney, J.; Telang, S. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab. 2014, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsumi, T.; Chesney, J.; Metz, C.; Leng, L.; Donnelly, S.; Makita, Z.; Mitchell, R.; Bucala, R. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002, 62, 5881–5887. [Google Scholar]
- Mondal, S.; Roy, D.; Sarkar Bhattacharya, S.; Jin, L.; Jung, D.; Zhang, S.; Kalogera, E.; Staub, J.; Wang, Y.; Xuyang, W.; et al. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int. J. Cancer 2019, 144, 178–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clem, B.F.; O’Neal, J.; Tapolsky, G.; Clem, A.L.; Imbert-Fernandez, Y.; Kerr, D.A.; Klarer, A.C.; Redman, R.; Miller, D.M.; Trent, J.O.; et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol. Cancer Ther. 2013, 12, 1461–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lypova, N.; Telang, S.; Chesney, J.; Imbert-Fernandez, Y. Increased 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 activity in response to EGFR signaling contributes to non-small cell lung cancer cell survival. J. Biol. Chem. 2019, 294, 10530–10543. [Google Scholar] [CrossRef]
- Hu, K.Y.; Wang, D.G.; Liu, P.F.; Cao, Y.W.; Wang, Y.H.; Yang, X.C.; Hu, C.X.; Sun, L.J.; Niu, H.T. Targeting of MCT1 and PFKFB3 influences cell proliferation and apoptosis in bladder cancer by altering the tumor microenvironment. Oncol. Rep. 2016, 36, 945–951. [Google Scholar] [CrossRef]
- Kessler, R.; Bleichert, F.; Warnke, J.P.; Eschrich, K. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3) is up-regulated in high-grade astrocytomas. J. Neurooncol. 2008, 86, 257–264. [Google Scholar] [CrossRef]
- Fleischer, M.; Kessler, R.; Klammer, A.; Warnke, J.P.; Eschrich, K. LOH on 10p14-p15 targets the PFKFB3 gene locus in human glioblastomas. Genes Chromosom. Cancer 2011, 50, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Zscharnack, K.; Kessler, R.; Bleichert, F.; Warnke, J.P.; Eschrich, K. The PFKFB3 splice variant UBI2K4 is downregulated in high-grade astrocytomas and impedes the growth of U87 glioblastoma cells. Neuropathol. Appl. Neurobiol. 2009, 35, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Yang, J.G.; Liu, Z.J.; Wang, W.M.; Yu, Z.L.; Ren, J.G.; Chen, G.; Zhang, W.; Jia, J. Blockage of glycolysis by targeting PFKFB3 suppresses tumor growth and metastasis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 7. [Google Scholar] [CrossRef] [Green Version]
- Trojan, S.E.; Markiewicz, M.J.; Leśkiewicz, K.; Kocemba-Pilarczyk, K.A. The influence of PFK-II overexpression on neuroblastoma patients’ survival may be dependent on the particular isoenzyme expressed, PFKFB3 or PFKFB4. Cancer Cell Int. 2019, 19, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Yan, S.; Sun, H.; Wang, W.; Li, Y.; Yang, X.; Jiang, X.; Che, Y.; Xi, Z. Akt inhibition attenuates rasfonin-induced autophagy and apoptosis through the glycolytic pathway in renal cancer cells. Cell Death Dis. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Du, J.Y.; Wang, L.F.; Wang, Q.; Yu, L.D. MiR-26b inhibits proliferation, migration, invasion and apoptosis induction via the downregulation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 driven glycolysis in osteosarcoma cells. Oncol. Rep. 2015, 33, 1890–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.D.; Zhou, F.L.; Lin, N. MicroRNA-26b inhibits osteosarcoma cell migration and invasion by down-regulating PFKFB3 expression. Genet. Mol. Res. 2015, 14, 16872–16879. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Z.X.; Wu, Q.N.; Lu, Y.X.; Wong, C.W.; Miao, L.; Wang, Y.; Wang, Z.; Jin, Y.; He, M.M.; et al. Long noncoding RNA AGPG regulates PFKFB3-mediated tumor glycolytic reprogramming. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Yao, L.; Wang, L.; Cao, Z.G.; Hu, X.; Shao, Z.M. High expression of metabolic enzyme PFKFB4 is associated with poor prognosis of operable breast cancer. Cancer Cell Int. 2019, 19, 165. [Google Scholar] [CrossRef]
- Gao, R.; Li, D.; Xun, J.; Zhou, W.; Li, J.; Wang, J.; Liu, C.; Li, X.; Shen, W.; Qiao, H.; et al. CD44ICD promotes breast cancer stemness via PFKFB4-mediated glucose metabolism. Theranostics 2018, 8, 6248–6262. [Google Scholar] [CrossRef]
- Li, W.; Qian, L.; Lin, J.; Huang, G.; Hao, N.; Wei, X.; Wang, W.; Liang, J. CD44 regulates prostate cancer proliferation, invasion and migration via PDK1 and PFKFB4. Oncotarget 2017, 8, 65143–65151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ros, S.; Santos, C.R.; Moco, S.; Baenke, F.; Kelly, G.; Howell, M.; Zamboni, N.; Schulze, A. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2012, 2, 328–343. [Google Scholar] [CrossRef] [Green Version]
- Goidts, V.; Bageritz, J.; Puccio, L.; Nakata, S.; Zapatka, M.; Barbus, S.; Toedt, G.; Campos, B.; Korshunov, A.; Momma, S.; et al. RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene 2012, 31, 3235–3243. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.J.; Jo, S.W.; Ha, Y.S.; Lee, O.J.; Kim, W.T.; Kim, Y.J.; Lee, S.C.; Kim, W.J. PFKFB4 as a prognostic marker in non-muscle-invasive bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2012, 30, 893–899. [Google Scholar] [CrossRef]
- Chesney, J.; Clark, J.; Klarer, A.C.; Imbert-Fernandez, Y.; Lane, A.N.; Telang, S. Fructose-2,6-bisphosphate synthesis by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 (PFKFB4) is required for the glycolytic response to hypoxia and tumor growth. Oncotarget 2014, 5, 6670–6686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalcin, A.; Telang, S.; Clem, B.; Chesney, J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp. Mol. Pathol. 2009, 86, 174–179. [Google Scholar] [CrossRef]
- Calvo, M.N.; Bartrons, R.; Castaño, E.; Perales, J.C.; Navarro-Sabaté, A.; Manzano, A. PFKFB3 gene silencing decreases glycolysis, induces cell-cycle delay and inhibits anchorage-independent growth in HeLa cells. FEBS Lett. 2006, 580, 3308–3314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalcin, A.; Clem, B.F.; Simmons, A.; Lane, A.; Nelson, K.; Clem, A.L.; Brock, E.; Siow, D.; Wattenberg, B.; Telang, S.; et al. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J. Biol. Chem. 2009, 284, 24223–24232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doménech, E.; Maestre, C.; Esteban-Martínez, L.; Partida, D.; Pascual, R.; Fernández-Miranda, G.; Seco, E.; Campos-Olivas, R.; Pérez, M.; Megias, D.; et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat. Cell Biol. 2015, 17, 1304–1316. [Google Scholar] [CrossRef]
- Shi, W.K.; Zhu, X.D.; Wang, C.H.; Zhang, Y.Y.; Cai, H.; Li, X.L.; Cao, M.Q.; Zhang, S.Z.; Li, K.S.; Sun, H.C. PFKFB3 blockade inhibits hepatocellular carcinoma growth by impairing DNA repair through AKT article. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotowski, K.; Supplitt, S.; Wiczew, D.; Bartosik, W.; Saczko, J.; Rossowska, J.; Drąg-Zalesińska, M.; Michel, O.; Kulbacka, J. 3PO as a Selective Inhibitor of 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3 in A375 Human Melanoma Cells. Anticancer Res. 2020, 40, 2613–2625. [Google Scholar] [CrossRef]
- Song, X.; Zhang, C.; Zhao, M.; Chen, H.; Liu, X.; Chen, J.; Lonard, D.M.; Qin, L.; Xu, J.; Wang, X.; et al. Steroid Receptor Coactivator-3 (SRC-3/AIB1) as a Novel Therapeutic Target in Triple Negative Breast Cancer and Its Inhibition with a Phospho-Bufalin Prodrug. PLoS ONE 2015, 10, e0140011. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Chen, S.; You, Z.; Xie, C.; Huang, S.; Hu, X. PFKFB4 negatively regulated the expression of histone acetyltransferase GCN5 to mediate the tumorigenesis of thyroid cancer. Dev. Growth Differ. 2020, 62, 129–138. [Google Scholar] [CrossRef]
- Lue, H.W.; Podolak, J.; Kolahi, K.; Cheng, L.; Rao, S.; Garg, D.; Xue, C.H.; Rantala, J.K.; Tyner, J.W.; Thornburg, K.L.; et al. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev. 2017, 31, 2067–2084. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxidants Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R.; et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy 2017, 13, 781–819. [Google Scholar] [CrossRef]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D.; et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, M.A.; Sharifi, M.; Ghafary, S.M.; Mohammadalipour, Z.; Khataee, A.; Rahmati, M.; Hajjaran, S.; Łos, M.J.; Klonisch, T.; Ghavami, S. Photodynamic N-TiO2 nanoparticle treatment induces controlled ROS-mediated autophagy and terminal differentiation of leukemia cells. Sci. Rep. 2016, 6, 1–16. [Google Scholar] [CrossRef]
- Nikseresht, M.; Shahverdi, M.; Dehghani, M.; Abidi, H.; Mahmoudi, R.; Ghalamfarsa, G.; Manzouri, L.; Ghavami, S. Association of single nucleotide autophagy-related protein 5 gene polymorphism rs2245214 with susceptibility to non–small cell lung cancer. J. Cell. Biochem. 2019, 120, 1924–1931. [Google Scholar] [CrossRef]
- Ghavami, S.; Eshragi, M.; Ande, S.R.; Chazin, W.J.; Klonisch, T.; Halayko, A.J.; McNeill, K.D.; Hashemi, M.; Kerkhoff, C.; Los, M. S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res. 2010, 20, 314–331. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Goronzy, J.J.; Weyand, C.M. The glycolytic enzyme PFKFB3/phosphofructokinase regulates autophagy. Autophagy 2014, 10, 382–383. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Fujii, H.; Mohan, S.V.; Goronzy, J.J.; Weyand, C.M. Phosphofructokinase deficiency impairs atp generation, autophagy, and redox balance in rheumatoid arthritis T cells. J. Exp. Med. 2013, 210, 2119–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desideri, E.; Vegliante, R.; Cardaci, S.; Nepravishta, R.; Paci, M.; Ciriolo, M.R. MAPK14/p38α-dependent modulation of glucose metabolism affects ROS levels and autophagy during starvation. Autophagy 2014, 10, 1652–1665. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Qu, J.; Yan, S.; Gao, Q.; Hao, S.; Zhou, D. PFK15, a PFKFB3 antagonist, inhibits autophagy and proliferation in rhabdomyosarcoma cells. Int. J. Mol. Med. 2018, 42, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Wei, X.; Xu, S.; Sun, H.; Wang, W.; Liu, L.; Jiang, X.; Zhang, Y.; Che, Y. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase isoform 3 spatially mediates autophagy through the AMPK signaling pathway. Oncotarget 2017, 8, 80909–80922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Takano, N.; Ishiwata, K.; Ohmura, M.; Nagahata, Y.; Matsuura, T.; Kamata, A.; Sakamoto, K.; Nakanishi, T.; Kubo, A.; et al. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- La Belle Flynn, A.; Calhoun, B.C.; Sharma, A.; Chang, J.C.; Almasan, A.; Schiemann, W.P. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohecker, A.M.; Joshi, S.; Possemato, R.; Abraham, R.T.; Sabatini, D.M.; White, E. Identification of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase as a novel autophagy regulator by high content shRNA screening. Oncogene 2015, 34, 5662–5676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zeng, F.; Sun, Y.; Qiu, Q.; Zhang, J.; Huang, W.; Huang, J.; Huang, X.; Guo, L. Etk interaction with PFKFB4 modulates chemoresistance of small-cell lung cancer by regulating autophagy. Clin. Cancer Res. 2018, 24, 950–962. [Google Scholar] [CrossRef] [Green Version]
- Bhat, T.A.; Singh, R.P. Tumor angiogenesis—A potential target in cancer chemoprevention. Food Chem. Toxicol. 2008, 46, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Ruoslahti, E. Specialization of tumour vasculature. Nat. Rev. Cancer 2002, 2, 83–90. [Google Scholar] [CrossRef]
- Ebos, J.M.L.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated Metastasis after Short-Term Treatment with a Potent Inhibitor of Tumor Angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Loges, S.; Mazzone, M.; Hohensinner, P.; Carmeliet, P. Silencing or Fueling Metastasis with VEGF Inhibitors: Antiangiogenesis Revisited. Cancer Cell 2009, 15, 167–170. [Google Scholar] [CrossRef] [Green Version]
- Yi, M.; Wang, W.; Chen, S.; Peng, Y.; Li, J.; Cai, J.; Zhou, Y.; Peng, Q.; Ban, Y.; Zeng, Z.; et al. Dual-functionality of RASSF1A overexpression in A375 cells is mediated by activation of IL-6/STAT3 regulatory loop. Mol. Biol. Rep. 2018, 45, 1277–1287. [Google Scholar] [CrossRef]
- Li, J.; Wang, W.; Chen, S.; Cai, J.; Ban, Y.; Peng, Q.; Zhou, Y.; Zeng, Z.; Li, X.; Xiong, W.; et al. FOXA1 reprograms the TGF-β-stimulated transcriptional program from a metastasis promoter to a tumor suppressor in nasopharyngeal carcinoma. Cancer Lett. 2019, 442, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yetkin-Arik, B.; Vogels, I.M.C.; Nowak-Sliwinska, P.; Weiss, A.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; An, X.; Guo, X.; Habtetsion, T.G.; Wang, Y.; Xu, X.; Kandala, S.; Li, Q.; Li, H.; Zhang, C.; et al. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1231–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquière, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conradi, L.-C.; Brajic, A.; Cantelmo, A.R.; Bouché, A.; Kalucka, J.; Pircher, A.; Brüning, U.; Teuwen, L.-A.; Vinckier, S.; Ghesquière, B.; et al. Tumor vessel disintegration by maximum tolerable PFKFB3 blockade. Angiogenesis 2017, 20, 599–613. [Google Scholar] [CrossRef]
- Li, J.; Mao, X.-H.; Tian, T.; Wang, W.-M.; Su, T.; Jiang, C.-H.; Hu, C.-Y. Role of PFKFB3 and CD163 in Oral Squamous Cell Carcinoma Angiogenesis. Curr. Med. Sci. 2019, 39, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, R.; Kitajima, S.; Hartman, F.C.; Uyedag, K. Hexose Phosphate Binding Sites of Fructose-6-phosphate,2-kinase:Fructose-2,6-bisphosphatase 3-bromo-1,4-dihydroxy-2-butanone 1,4-bisphosphate. J. Biol. Chem. 1984, 259, 14023–14028. [Google Scholar] [CrossRef]
- Harada, Y.; Tominaga, N.; Watanabe, M.; Shimokawa, R.; Ishiguro, M.; Sakakibara, R. Inhibition of fructose-6-phosphate,2-kinase by N-bromoacetylethanolamine phosphate in vitro and in vivo. J. Biochem. 1997, 121, 724–730. [Google Scholar] [CrossRef]
- Yuen, M.H.; Wang, X.L.; Mizuguchi, H.; Uyeda, K.; Hasemann, C.A. A Switch in the Kinase Domain of Rat Testis 6-Phosphofructo-2-kinase/Fructose-2,6-bisphosphatase. Biochemistry 1999, 38, 12333–12342. [Google Scholar] [CrossRef] [PubMed]
- Pisarsky, L.; Bill, R.; Fagiani, E.; Dimeloe, S.; Goosen, R.W.; Hagmann, J.; Hess, C.; Christofori, G. Targeting Metabolic Symbiosis to Overcome Resistance to Anti-angiogenic Therapy. Cell Rep. 2016, 15, 1161–1174. [Google Scholar] [CrossRef] [Green Version]
- Lea, M.A.; Altayyar, M.; desBordes, C. Inhibition of Growth of Bladder Cancer Cells by 3-(3-Pyridinyl)-1-(4-pyridinyl)-2-propen-1-one in Combination with Other Compounds Affecting Glucose Metabolism. Anticancer Res. 2015, 35, 5889–5899. [Google Scholar]
- Akter, S.; Clem, B.F.; Lee, H.J.; Chesney, J.; Bae, Y. Block copolymer micelles for controlled delivery of glycolytic enzyme inhibitors. Pharm. Res. 2012, 29, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.; Kim, J.D.; Neau, D.; Sehgal, I.; Lee, Y.H. Structure-based development of small molecule PFKFB3 inhibitors: A framework for potential cancer therapeutic agents targeting the Warburg effect. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Sarkar Bhattacharya, S.; Thirusangu, P.; Jin, L.; Roy, D.; Jung, D.; Xiao, Y.; Staub, J.; Roy, B.; Molina, J.R.; Shridhar, V. PFKFB3 inhibition reprograms malignant pleural mesothelioma to nutrient stress-induced macropinocytosis and ER stress as independent binary adaptive responses. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Phase 1 Safety Study of ACT-PFK-158, 2HCl in Patients with Advanced Solid Malignancies—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT02044861 (accessed on 12 April 2020).
- Boyd, S.; Brookfield, J.L.; Critchlow, S.E.; Cumming, I.A.; Curtis, N.J.; Debreczeni, J.; Degorce, S.L.; Donald, C.; Evans, N.J.; Groombridge, S.; et al. Structure-Based Design of Potent and Selective Inhibitors of the Metabolic Kinase PFKFB3. J. Med. Chem. 2015, 58, 3611–3625. [Google Scholar] [CrossRef]
- Lea, M.A.; Guzman, Y.; Desbordes, C. Inhibition of growth by combined treatment with inhibitors of lactate dehydrogenase and either phenformin or inhibitors of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3. Anticancer Res. 2016, 36, 1479–1488. [Google Scholar] [PubMed]
- Gustafsson, N.M.S.; Färnegårdh, K.; Bonagas, N.; Ninou, A.H.; Groth, P.; Wiita, E.; Jönsson, M.; Hallberg, K.; Lehto, J.; Pennisi, R.; et al. Targeting PFKFB3 radiosensitizes cancer cells and suppresses homologous recombination. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- N-Bromoacetylethanolamine phosphate |C4H9BrNO5P—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/188343 (accessed on 10 May 2020).
- 3-(3-Pyridinyl)-1-(4-pyridinyl)-2-propen-1-one |C13H10N2O—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5720233 (accessed on 11 May 2020).
- 1-(Pyridin-4-yl)-3-(quinolin-2-yl)prop-2-en-1-one |C17H12N2O—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/25142799 (accessed on 11 May 2020).
- CID 71730058 | C18H11F3N2O-PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/71730058 (accessed on 11 May 2020).
- 5-[(8-Methoxyquinolin-4-yl)amino]pentyl nitrate | C15H19N3O4—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/4060327#section=3D-Conformer (accessed on 11 May 2020).
- Chesney, J.; Clark, J.; Lanceta, L.; Trent, J.O.; Telang, S. Targeting the sugar metabolism of tumors with a first-in-class 6-phosphofructo-2-kinase (PFKFB4) inhibitor. Oncotarget 2015, 6, 18001–18011. [Google Scholar] [CrossRef]
- Kotowski, K.; Supplitt, S.; Saczko, J.; Kulbacka, J. Molecular determinants of resistance to treatment with BRAF inhibitors and innovative possibilities of its overcoming. Postępy Biol. Komórki 2019, 46, 147–158. [Google Scholar]
- Xu, R.H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar] [PubMed]
- Liu, H.; Hu, Y.P.; Savaraj, N.; Priebe, W.; Lampidis, T.J. Hypersensitization of tumor cells to glycolytic inhibitors. Biochemistry 2001, 40, 5542–5547. [Google Scholar] [CrossRef]
- Li, F.L.; Liu, J.P.; Bao, R.X.; Yan, G.; Feng, X.; Xu, Y.P.; Sun, Y.P.; Yan, W.; Ling, Z.Q.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Gilliland, D.G. The JAK2V617F tyrosine kinase mutation in myeloproliferative disorders: Status report and immediate implications for disease classification and diagnosis. Mayo Clin. Proc. 2005, 80, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, M.M.; Fernandes, M.S.; Deshpande, A.; Weisberg, E.; Inguilizian, H.V.; Abdel-Wahab, O.; Kung, A.L.; Levine, R.L.; Griffin, J.D.; Sattler, M. The JAK2V617F oncogene requires expression of inducible phosphofructokinase/fructose-bisphosphatase 3 for cell growth and increased metabolic activity. Leukemia 2012, 26, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Kominsky, D.J.; Klawitter, J.; Brown, J.L.; Boros, L.G.; Melo, J.V.; Eckhardt, S.G.; Serkova, N.J. Abnormalities in glucose uptake and metabolism in imatinib-resistant human BCR-ABL-positive cells. Clin. Cancer Res. 2009, 15, 3442–3450. [Google Scholar] [CrossRef] [Green Version]
- Barta, J.A.; Powell, C.A.; Wisnivesky, J.P. Global Epidemiology of Lung Cancer. Ann. Glob. Health 2019, 85. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 749–768. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Qian, L.; Ke, H.; Yao, C.; Tian, W.; Liu, Y.; Zhang, J. Expression of PFKFB3 and Ki67 in lung adenocarcinomas and targeting PFKFB3 as a therapeutic strategy. Mol. Cell. Biochem. 2018, 445, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, N.; Vhora, I.; Patel, K.; Doddapaneni, R.; Mondal, A.; Singh, M. Liposomes co-Loaded with 6-Phosphofructo-2-Kinase/Fructose-2, 6-Biphosphatase 3 (PFKFB3) shRNA Plasmid and Docetaxel for the Treatment of non-small Cell Lung Cancer. Pharm. Res. 2017, 34, 2371–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Dai, W.; Mo, W.; Li, J.; Feng, J.; Wu, L.; Liu, T.; Yu, Q.; Xu, S.; Wang, W.; et al. By inhibiting PFKFB3, aspirin overcomes sorafenib resistance in hepatocellular carcinoma. Int. J. Cancer 2017, 141, 2571–2584. [Google Scholar] [CrossRef] [PubMed]
- Telang, S.; O’Neal, J.; Imbert-Fernandez, Y.; Clem, B.; Lypova, N.; Tapolsky, G.H.; Trent, J.; Chesney, J. Abstract 4478: 6-Phosphofructo-2-Kinase (PFKFB3): At the Crossroads of Resistance to Targeted Cancer Therapies; AACR: Philadelphia, PA, USA, 2015; Volume 75, p. 4478. [Google Scholar]
- Szostak, B.; Machaj, F.; Rosik, J.; Pawlik, A. CTLA4 antagonists in phase I and phase II clinical trials, current status and future perspectives for cancer therapy. Expert Opin. Investig. Drugs 2019, 28, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Song, W.P.; Zheng, S.; Yao, H.J.; Zhou, X.F.; Li, R.; Zhang, C.Y.; Zhao, J.Y.; Wang, L.W.; Shao, R.G.; Li, L. Different transcriptome profiles between human retinoblastoma Y79 cells and an etoposide-resistant subline reveal a chemoresistance mechanism. BMC Ophthalmol. 2020, 20. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, N. Protecting the genome through metabolic enzymes—DNA Repair Conference. In Proceedings of the 6th EU-US Conference on Repair of Endogenous DNA Damage, Udine, Italy, 24–28 September 2017. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotowski, K.; Rosik, J.; Machaj, F.; Supplitt, S.; Wiczew, D.; Jabłońska, K.; Wiechec, E.; Ghavami, S.; Dzięgiel, P. Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers 2021, 13, 909. https://doi.org/10.3390/cancers13040909
Kotowski K, Rosik J, Machaj F, Supplitt S, Wiczew D, Jabłońska K, Wiechec E, Ghavami S, Dzięgiel P. Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers. 2021; 13(4):909. https://doi.org/10.3390/cancers13040909
Chicago/Turabian StyleKotowski, Krzysztof, Jakub Rosik, Filip Machaj, Stanisław Supplitt, Daniel Wiczew, Karolina Jabłońska, Emilia Wiechec, Saeid Ghavami, and Piotr Dzięgiel. 2021. "Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets" Cancers 13, no. 4: 909. https://doi.org/10.3390/cancers13040909