
Large-Scale Proteomic Analysis of Follicular Lymphoma Reveals Extensive Remodeling of Cell Adhesion Pathway and Identifies Hub Proteins Related to the Lymphomagenesis

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
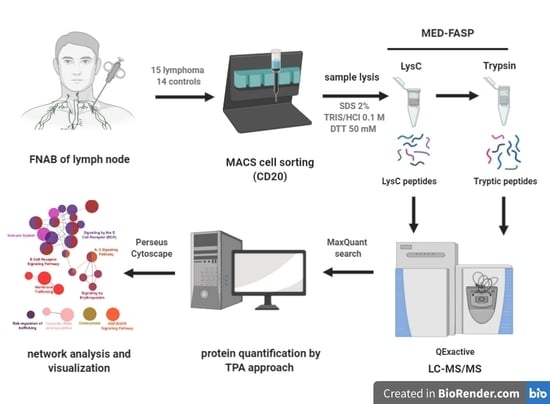
2.1. General Description of Analyzed Material
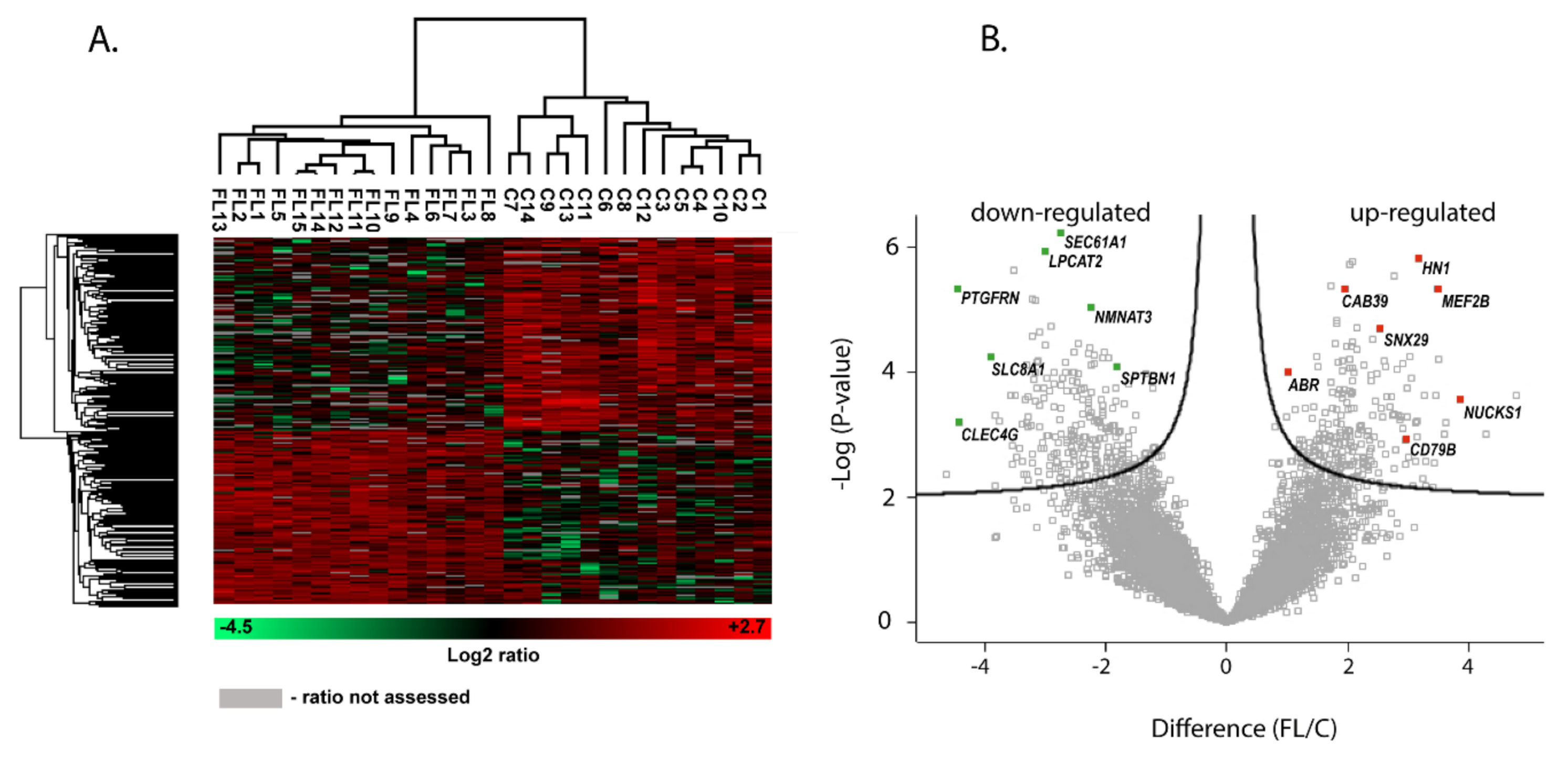
2.2. Identification of DAPs
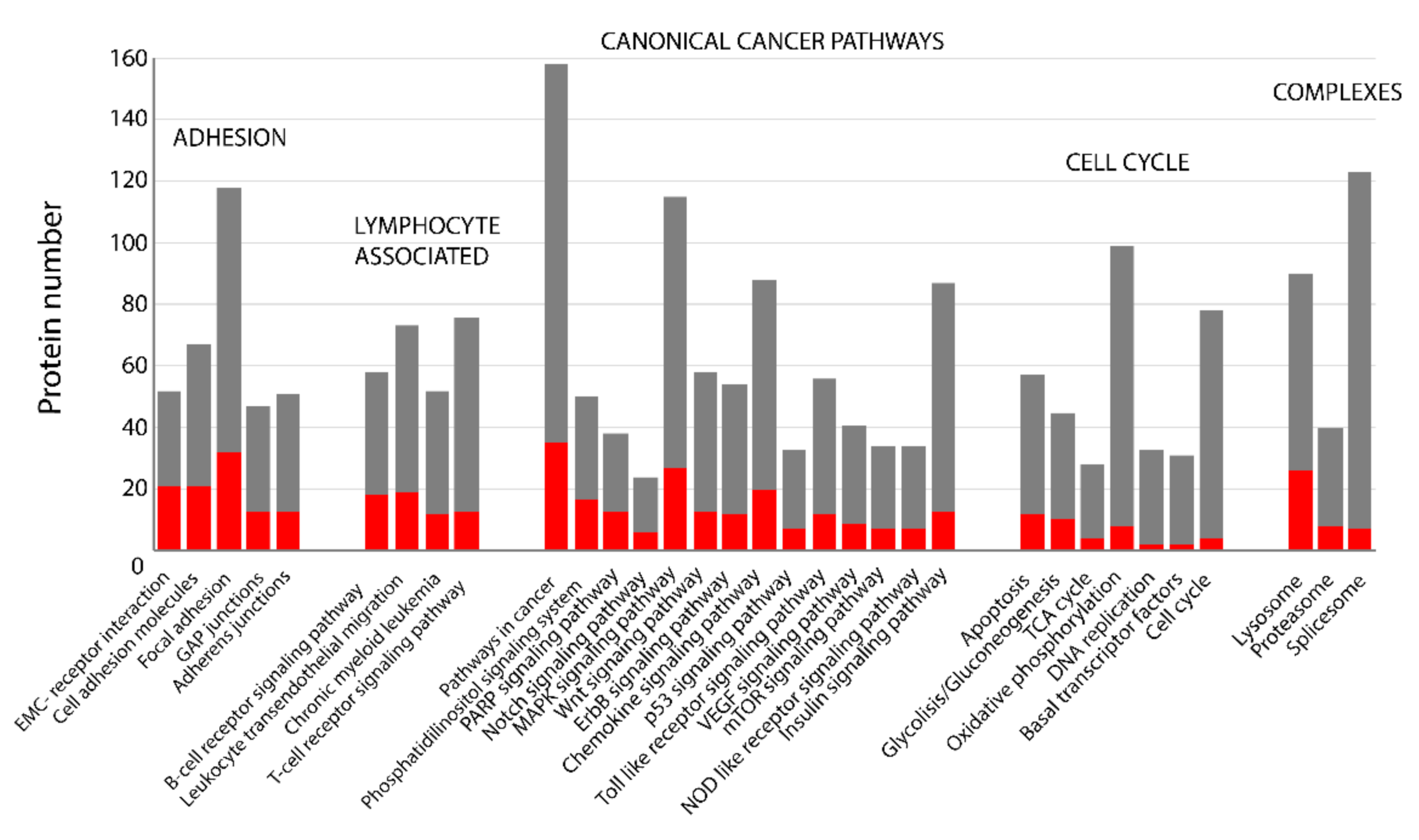
2.3. KEEG Pathway Analysis
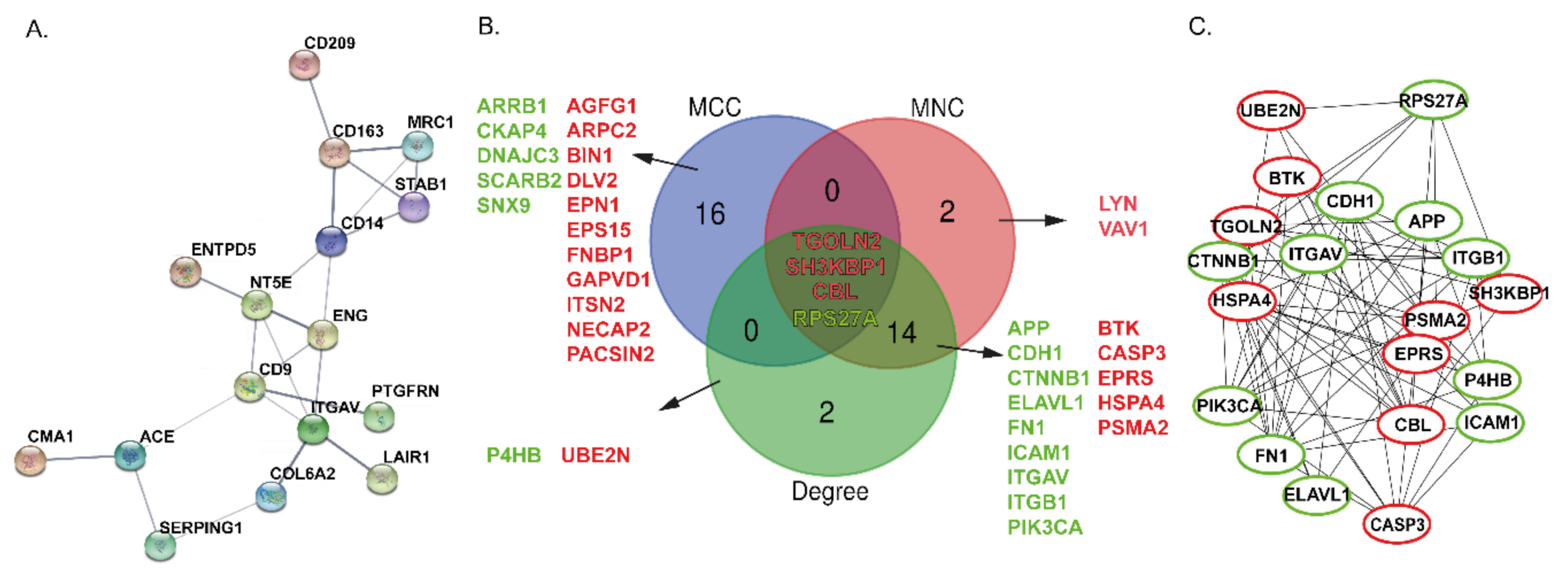
2.4. Protein-Protein Interaction Networks and Hub Proteins in FL
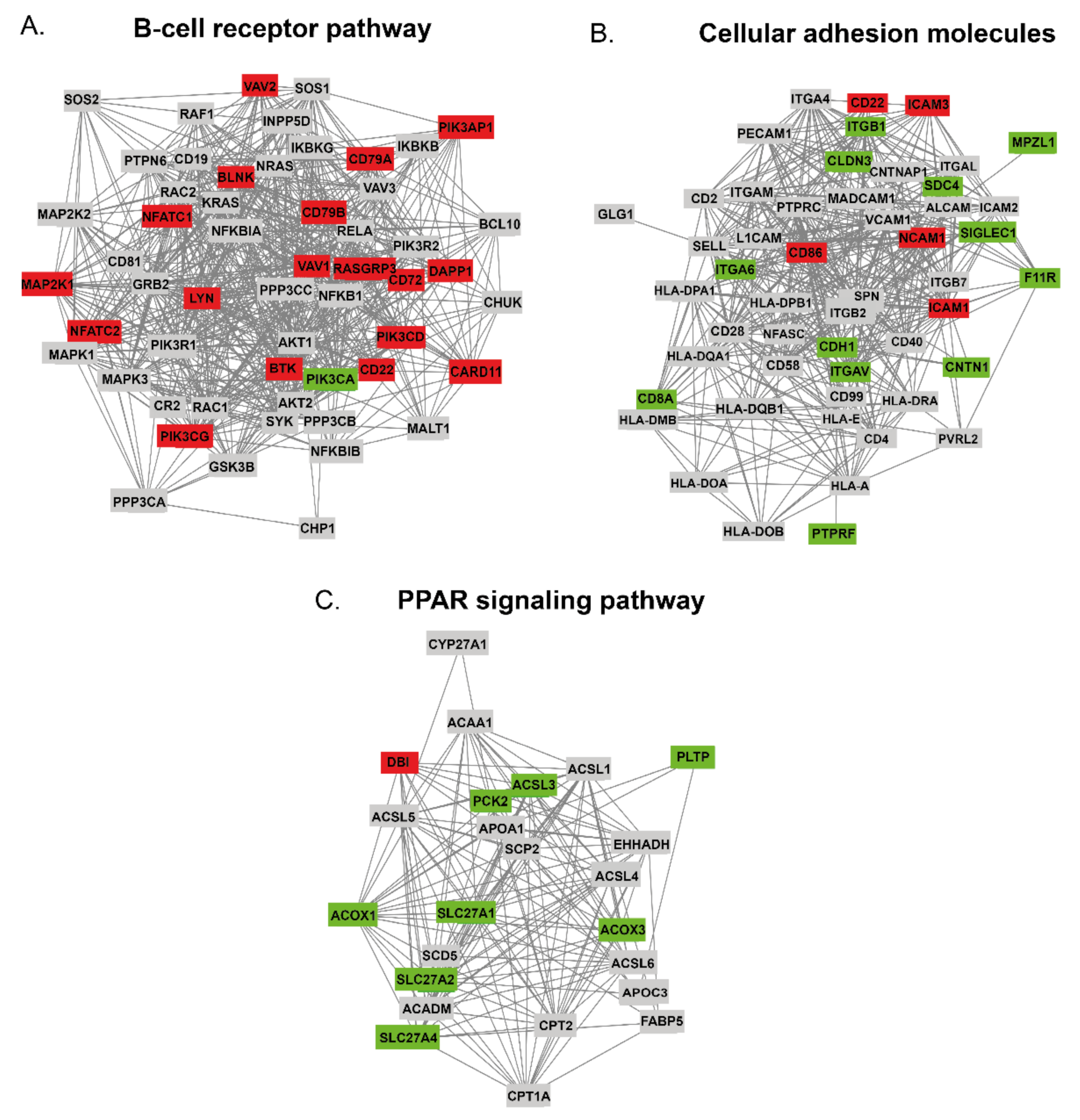
2.4.1. The DAPs in BCR Pathway
2.4.2. The DAPs in CAMs Pathway
2.4.3. The DAPs in PPAR Signaling Pathway
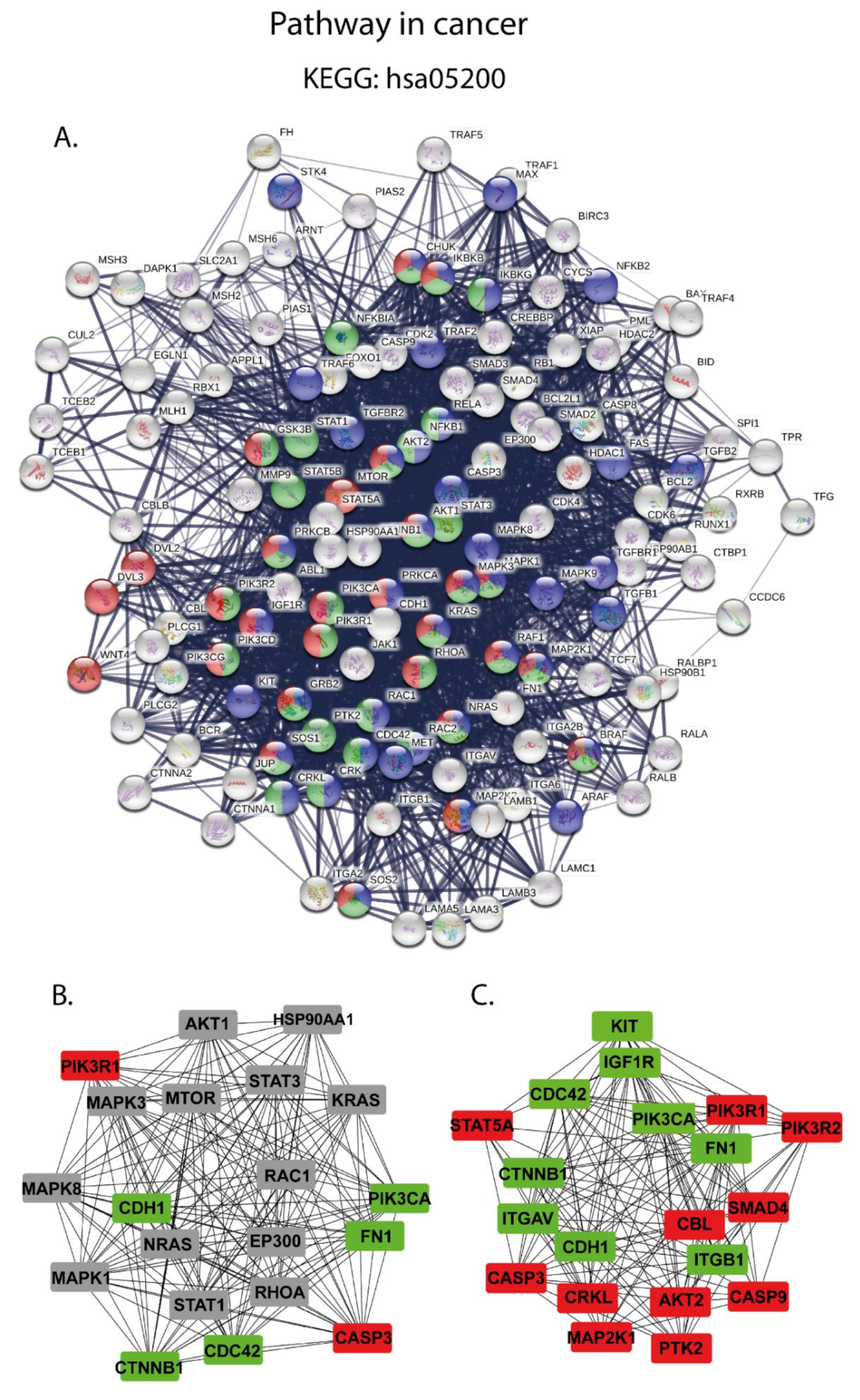
2.4.4. Pathway in Cancer Analysis
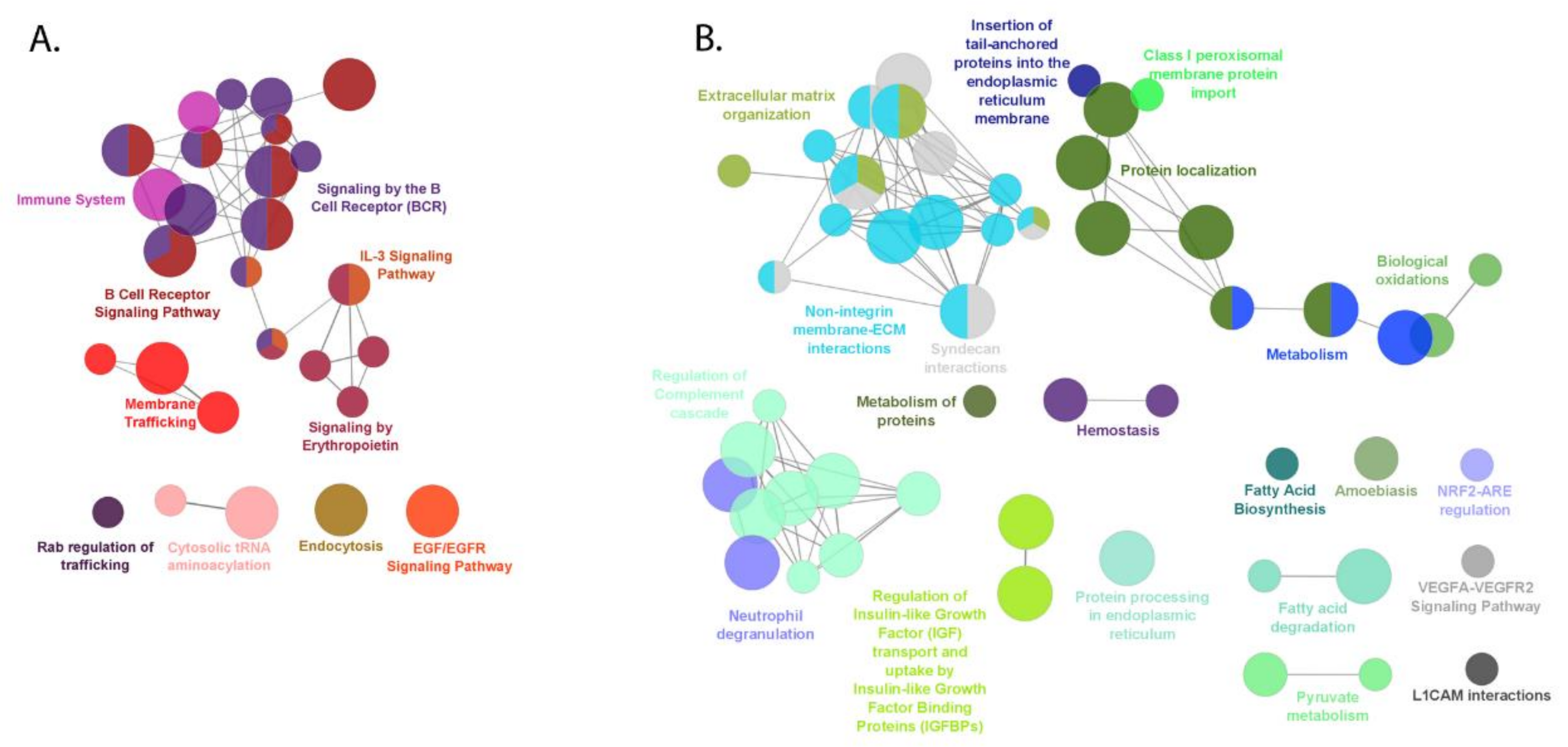
2.5. Pathway Enrichment Analysis by ClueGO
2.5.1. Up-Regulated Proteins in FL
2.5.2. Down-Regulated Proteins in FL
2.6. Flow Cytometric Validation of CD9, ICAM1 and CD79B Expression
3. Discussion
4. Materials and Methods
4.1. Collection of FNAB Samples and Clinical Information
4.2. Magnetic Isolation
4.3. MED FASP
4.4. LC-MS/MS
4.5. MS/MS Data Analysis
4.6. Bioinformatic Analysis
4.7. Flow Cytometry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, J.R.; Armitage, J.O.; Weisenburger, D.D. Epidemiology of the non-Hodgkin’s lymphomas: Distributions of the major subtypes differ by geographic locations. Non-Hodgkin’s Lymphoma Classification Project. Ann. Oncol. 1998, 9, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef]
- Shaffer, A.L., 3rd; Young, R.M.; Staudt, L.M. Pathogenesis of human B cell lymphomas. Annu. Rev. Immunol. 2012, 30, 565–610. [Google Scholar] [CrossRef] [PubMed]
- Cancer Stat Facts: NHL—Follicular Lymphoma. SEER. Available online: https://seer.cancer.gov/statfacts/html/follicular.html (accessed on 12 May 2020).
- Carbone, A.; Roulland, S.; Gloghini, A.; Younes, A.; von Keudell, G.; López-Guillermo, A.; Fitzgibbon, J. Follicular lymphoma. Nat. Rev. Dis. Primers 2019, 5, 83. [Google Scholar] [CrossRef]
- Szumera-Ciećkiewicz, A.; Wojciechowska, U.; Didkowska, J.; Poleszczuk, J.; Rymkiewicz, G.; Paszkiewicz-Kozik, E.; Sokół, K.; Prochorec-Sobieszek, M.; Walewski, J. Population-based epidemiological data of follicular lymphoma in Poland: 15 years of observation. Sci. Rep. 2020, 10, 14610. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Miyata-Takata, T.; Sato, Y.; Yoshino, T. Pathology of follicular lymphoma. J. Clin. Exp. Hematop. 2014, 54, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Huet, S.; Sujobert, P.; Salles, G. From genetics to the clinic: A translational perspective on follicular lymphoma. Nat. Rev. Cancer 2018, 18, 224–239. [Google Scholar] [CrossRef]
- Casulo, C.; Burack, W.R.; Friedberg, J.W. Transformed follicular non-Hodgkin lymphoma. Blood 2015, 125, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Ott, G.; Rosenwald, A. Molecular pathogenesis of follicular lymphoma. Haematologica 2008, 93, 1773–1776. [Google Scholar] [CrossRef] [Green Version]
- Lossos, I.S.; Gascoyne, R.D. Transformation of follicular lymphoma. Best Pract. Res. Clin. Haematol. 2011, 24, 147–163. [Google Scholar] [CrossRef] [Green Version]
- Al-Tourah, A.J.; Gill, K.K.; Chhanabhai, M.; Hoskins, P.J.; Klasa, R.J.; Savage, K.J.; Sehn, L.H.; Shenkier, T.N.; Gascoyne, R.D.; Connors, J.M. Population-based analysis of incidence and outcome of transformed non-Hodgkin’s lymphoma. J. Clin. Oncol. 2008, 26, 5165–5169. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. WHO Classification of Tumours, 4th ed.; Revised; World Health Organization: Geneva, Switzerland, 2017; pp. 266–285. [Google Scholar]
- Wagner-Johnston, N.D.; Link, B.K.; Byrtek, M.; Dawson, K.L.; Hainsworth, J.; Flowers, C.R.; Friedberg, J.W.; Bartlett, N.L. Outcomes of transformed follicular lymphoma in the modern era: A report from the National LymphoCare Study (NLCS). Blood 2015, 126, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Rummel, M.J.; Niederle, N.; Maschmeyer, G.; Banat, G.A.; von Grünhagen, U.; Losem, C.; Kofahl-Krause, D.; Heil, G.; Welslau, M.; Balser, C.; et al. Study group indolent Lymphomas (StiL). Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: An open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013, 381, 1203–1210. [Google Scholar] [CrossRef]
- Seymour, J.F.; Marcus, R.; Davies, A.; Gallop-Evans, E.; Grigg, A.; Haynes, A.; Herold, M.; Illmer, T.; Nilsson-Ehle, H.; Sökler, W.; et al. Association of early disease progression and very poor survival in the GALLIUM study in follicular lymphoma: Benefit of obinutuzumab in reducing the rate of early progression. Haematologica 2020, 105, 1465. [Google Scholar] [CrossRef] [PubMed]
- Bachy, E.; Seymour, J.F.; Feugier, P.; Offner, F.; López-Guillermo, A.; Belada, D.; Xerri, L.; Catalano, J.V.; Brice, P.; Lemonnier, F.; et al. Sustained progression-free survival benefit of rituximab maintenance in patients with follicular lymphoma: Long-term results of the PRIMA study. J. Clin. Oncol. 2019, 37, 2815–2824. [Google Scholar] [CrossRef] [PubMed]
- Walewski, J.; Paszkiewicz-Kozik, E.; Michalski, W.; Rymkiewicz, G.; Szpila, T.; Butrym, A.; Giza, A.; Zaucha, J.M.; Kalinka-Warzocha, E.; Wieczorkiewicz, A.; et al. First-line R-CVP versus R-CHOP induction immunochemotherapy for indolent lymphoma with rituximab maintenance. A multicentre, phase III randomized study by the Polish Lymphoma Research Group PLRG4. Br. J. Haematol. 2020, 188, 898–906. [Google Scholar] [CrossRef] [Green Version]
- Deeb, S.J.; D’Souza, R.C.; Cox, J.; Schmidt-Supprian, M.; Mann, M. Super-SILAC allows classification of diffuse large B-cell lymphoma subtypes by their protein expression profiles. Mol. Cell. Proteom. 2012, 11, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Deeb, S.J.; Tyanova, S.; Hummel, M.; Schmidt-Supprian, M.; Cox, J.; Mann, M. Machine Learning-based Classification of Diffuse Large B-cell Lymphoma Patients by Their Protein Expression Profiles. Mol. Cell. Proteom. 2015, 14, 2947–2960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornecker, L.M.; Muller, L.; Bertrand, F.; Paul, N.; Pichot, A.; Herbrecht, R.; Chenard, M.P.; Mauvieux, L.; Vallat, L.; Bahram, S.; et al. Multi-omics dataset to decipher the complexity of drug resistance in diffuse large B-cell lymphoma. Sci. Rep. 2019, 9, 895. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Nijland, M.; Rutgers, B.; Veenstra, R.; Langendonk, M.; van der Meeren, L.E.; Kluin, P.M.; Li, G.; Diepstra, A.; Chiu, J.F.; et al. Proteomics Based Identification of Proteins with Deregulated Expression in B Cell Lymphomas. PLoS ONE 2016, 11, e0146624. [Google Scholar] [CrossRef] [Green Version]
- Pickard, L.; Palladino, G.; Okosun, J. Follicular lymphoma genomics. Leuk. Lymphoma 2020, 61, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Pei, R.; Sha, K.; Chen, L.; Wang, T.; Lu, Y. Comprehensive characterization of driver genes in diffuse large B cell lymphoma. Oncol. Lett. 2020, 20, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Crockett, D.K.; Jenson, S.D.; Lim, M.S.; Elenitoba-Johnson, K.S. Quantitative proteomic and transcriptional analysis of the response to the p38 mitogen-activated protein kinase inhibitor SB203580 in transformed follicular lymphoma cells. Mol. Cell. Proteom. 2004, 3, 820–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinkauf, M.; Christopeit, M.; Hiddemann, W.; Dreyling, M. Proteome- and microarray-based expression analysis of lymphoma cell lines identifies a p53-centered cluster of differentially expressed proteins in mantle cell and follicular lymphoma. Electrophoresis 2007, 28, 4416–4426. [Google Scholar] [CrossRef]
- Jansen, C.; Feuth, T.; Raemaekers, J.M.; Rijntjes, J.; Meijer, J.W.; Westenend, P.J.; van Baarlen, J.; van Krieken, J.H.; Hebeda, K.M.; Groenen, P.J. Protein profiling in pathology: Analysis and evaluation of 239 frozen tissue biopsies for diagnosis of B-cell lymphomas. Proteom. Clin. Appl. 2010, 4, 519–527. [Google Scholar] [CrossRef]
- Rolland, D.; Basrur, V.; Conlon, K.; Wolfe, T.; Fermin, D.; Nesvizhskii, A.I.; Lim, M.S.; Elenitoba-Johnson, K.S. Global phosphoproteomic profiling reveals distinct signatures in B-cell non-Hodgkin lymphomas. Am. J. Pathol. 2014, 184, 1331–1342. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsen, M.; Madsen, C.; Kamper, P.; Hamilton-Dutoit, S.J.; Bendix, K.; d’Amore, F.; Honoré, B. Histologically transformed follicular lymphoma exhibits protein profiles different from both non-transformed follicular and de novo diffuse large B-cell lymphoma. Blood Cancer J. 2015, 5, e293. [Google Scholar] [CrossRef] [Green Version]
- Wiśniewski, J.R.; Ostasiewicz, P.; Duś, K.; Zielińska, D.F.; Gnad, F.; Mann, M. Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma. Mol. Syst. Biol. 2012, 8, 611. [Google Scholar]
- Wiśniewski, J.R.; Hein, M.Y.; Cox, J.; Mann, M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell Proteom. 2014, 13, 3497–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Xu, L.; Li, Z.; Hou, K.Z.; Che, X.F.; Liu, B.F.; Liu, Y.P.; Qu, X.J. Integrated bioinformatics analysis for the identification of potential key genes affecting the pathogenesis of clear cell renal cell carcinoma. Oncol. Lett. 2020, 20, 1573–1584. [Google Scholar] [CrossRef]
- Bende, R.J.; Smit, L.A.; van Noesel, C.J. Molecular pathways in follicular lymphoma. Leukemia 2007, 21, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Young, R.M.; Staudt, L.M. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat. Rev. Drug Discov. 2013, 12, 229–243. [Google Scholar] [CrossRef]
- Niemann, C.U.; Wiestner, A. B-cell receptor signaling as a driver of lymphoma development and evolution. Semin. Cancer Biol. 2013, 23, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Laganà, A.S.; Vitale, S.G.; Nigro, A.; Sofo, V.; Salmeri, F.M.; Rossetti, P.; Rapisarda, A.M.; La Vignera, S.; Condorelli, R.A.; Rizzo, G. Pleiotropic Actions of Peroxisome Proliferator-Activated Receptors (PPARs) in Dysregulated Metabolic Homeostasis, Inflammation and Cancer: Current Evidence and Future Perspectives. Int. J. Mol. Sci. 2016, 17, 999. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Efremov, D.G.; Turkalj, S.; Laurenti, L. Mechanisms of B Cell Receptor Activation and Responses to B Cell Receptor Inhibitors in B Cell Malignancies. Cancers 2020, 12, 1396. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Khiabanian, H.; Fangazio, M.; Vasishtha, M.; Messina, M.; Holmes, A.B.; Ouillette, P.; Trifonov, V.; Rossi, D.; Tabbò, F. Genetics of follicular lymphoma transformation. Cell Rep. 2014, 6, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, K.R.; Steffensen, R.; Haunstrup, T.M.; Bødker, J.S.; Dybkær, K.; Baech, J.; Bøgsted, M.; Johnsen, H.E. Inherited variation in immune response genes in follicular lymphoma and diffuse large B-cell lymphoma. Leuk. Lymphoma 2015, 56, 3257–3266. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef] [PubMed]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2017, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Paul, J.; Soujon, M.; Wengner, A.; Zitzmann-Kolbe, S.; Sturz, A.; Haike, K.; Keng Magdalene, K.H.; Tan, S.H.; Lange, M.; Tan, S.Y. Simultaneous Inhibition of PI3Kδ and PI3Kα Induces ABC-DLBCL Regression by Blocking BCR-Dependent and -Independent Activation of NF-κB and AKT. Cancer Cell 2017, 31, 64–78. [Google Scholar]
- Rossi, D.; Ciardullo, C.; Gaidano, G. Genetic aberrations of signaling pathways in lymphomagenesis: Revelations from next generation sequencing studies. Semin. Cancer Biol. 2013, 23, 422–430. [Google Scholar] [CrossRef]
- Bedsaul, J.R.; Carter, N.M.; Deibel, K.E.; Hutcherson, S.M.; Jones, T.A.; Wang, Z.; Yang, C.; Yang, Y.K.; Pomerantz, J.L. Mechanisms of Regulated and Dysregulated CARD11 Signaling in Adaptive Immunity and Disease. Front. Immunol. 2018, 9, 2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bognar, M.K.; Vincendeau, M.; Erdmann, T.; Seeholzer, T.; Grau, M.; Linnemann, J.R.; Ruland, J.; Scheel, C.H.; Lenz, P.; Ott, G.; et al. Oncogenic CARMA1 couples NF-κB and β-catenin signaling in diffuse large B-cell lymphomas. Oncogene 2016, 35, 4269–4281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelan, J.D.; Young, R.M.; Webster, D.E.; Roulland, S.; Wright, G.W.; Kasbekar, M.; Shaffer, A.L., 3rd; Ceribelli, M.; Wang, J.Q.; Schmitz, R.; et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 2018, 560, 387–391. [Google Scholar] [CrossRef]
- Myklebust, J.H.; Brody, J.; Kohrt, H.E.; Kolstad, A.; Czerwinski, D.K.; Wälchli, S.; Green, M.R.; Trøen, G.; Liestøl, K.; Beiske, K.; et al. Distinct patterns of B-cell receptor signaling in non-Hodgkin lymphomas identified by single-cell profiling. Blood 2017, 129, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R. Vav family exchange factors: An integrated regulatory and functional view. Small GTPases 2014, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Oberley, M.J.; Wang, D.S.; Yang, D.T. Vav1 in hematologic neoplasms, a mini review. Am. J. Blood Res. 2012, 2, 1–8. [Google Scholar]
- Prieto-Sánchez, R.M.; Hernández, J.A.; García, J.L.; Gutiérrez, N.C.; San Miguel, J.; Bustelo, X.R.; Hernández, J.M. Overexpression of the VAV proto-oncogene product is associated with B-cell chronic lymphocytic leukaemia displaying loss on 13q. Br. J. Haematol. 2006, 133, 642–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollmann, A.; Aloyz, R.; Baker, K.; Dirnhofer, S.; Owens, T.; Sladek, R.; Tzankov, A. Vav-1 expression correlates with NFκB activation and CD40-mediated cell death in diffuse large B-cell lymphoma cell lines. Hematol. Oncol. 2010, 28, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Ligresti, G.; Militello, L.; Steelman, L.S.; Cavallaro, A.; Basile, F.; Nicoletti, F.; Stivala, F.; McCubrey, J.A.; Libra, M. PIK3CA mutations in human solid tumors: Role in sensitivity to various therapeutic approaches. Cell Cycle 2009, 8, 1352–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abubaker, J.; Bavi, P.P.; Al-Harbi, S.; Siraj, A.K.; Al-Dayel, F.; Uddin, S.; Al-Kuraya, K. PIK3CA mutations are mutually exclusive with PTEN loss in diffuse large B-cell lymphoma. Leukemia 2007, 21, 2368–2370. [Google Scholar] [CrossRef] [Green Version]
- Jacob, M.C.; Agrawal, S.; Chaperot, L.; Giroux, C.; Gressin, R.; Le Marc’Hadour, F.; Favre, M.; Sotto, J.J.; Bensa, J.C.; Plumas, J. Quantification of cellular adhesion molecules on malignant B cells from non-Hodgkin’s lymphoma. Leukemia 1999, 13, 1428–1433. [Google Scholar] [CrossRef] [Green Version]
- Figenschau, S.L.; Knutsen, E.; Urbarova, I.; Fenton, C.; Elston, B.; Perander, M.; Mortensen, E.S.; Fenton, K.A. ICAM1 expression is induced by proinflammatory cytokines and associated with TLS formation in aggressive breast cancer subtypes. Sci. Rep. 2018, 8, 11720. [Google Scholar] [CrossRef] [Green Version]
- Di, D.; Chen, L.; Wang, L.; Sun, P.; Liu, Y.; Xu, Z.; Ju, J. Downregulation of human intercellular adhesion molecule-1 attenuates the metastatic ability in human breast cancer cell lines. Oncol. Rep. 2016, 35, 1541–1548. [Google Scholar] [CrossRef]
- Strell, C.; Entschladen, F. Extravasation of leukocytes in comparison to tumor cells. Cell Commun. Signal. 2008, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terol, M.J.; López-Guillermo, A.; Bosch, F.; Villamor, N.; Cid, M.C.; Rozman, C.; Campo, E.; Montserrat, E. Expression of the adhesion molecule ICAM-1 in non-Hodgkin’s lymphoma: Relationship with tumor dissemination and prognostic importance. J. Clin. Oncol. 1998, 16, 35–40. [Google Scholar] [CrossRef]
- Schniederjan, S.D.; Li, S.; Saxe, D.F.; Lechowicz, M.J.; Lee, K.L.; Terry, P.D.; Mann, K.P. A novel flow cytometric antibody panel for distinguishing Burkitt lymphoma from CD10+ diffuse large B-cell lymphoma. Am. J. Clin. Pathol. 2010, 133, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Wu, D.; Li, F.; Zhang, P.; Feng, Y.; He, A. Identification of key biomarkers associated with cell adhesion in multiple myeloma by integrated bioinformatics analysis. Cancer Cell Int. 2020, 20, 262. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Lei, S.; Zhang, Q.; Ma, L.; Li, N.; Wang, J. Deregulation of cell adhesion molecules is associated with progression and poor outcomes in endometrial cancer: Analysis of The Cancer Genome Atlas data. Oncol. Lett. 2020, 19, 1906–1914. [Google Scholar] [CrossRef] [Green Version]
- Eieland, A.K.; Normann, K.R.; Sundaram, A.Y.M.; Nyman, T.A.; Øystese, K.A.B.; Lekva, T.; Berg, J.P.; Bollerslev, J.; Olarescu, N.C. Distinct Pattern of Endoplasmic Reticulum Protein Processing and Extracellular Matrix Proteins in Functioning and Silent Corticotroph Pituitary Adenomas. Cancers 2020, 12, 2980. [Google Scholar] [CrossRef]
- Collins, M.; Ling, V.; Carreno, B.M. The B7 family of immune-regulatory ligands. Genome Biol. 2005, 6, 223. [Google Scholar] [CrossRef] [Green Version]
- De Charette, M.; Houot, R. Hide or defend, the two strategies of lymphoma immune evasion: Potential implications for immunotherapy. Haematologica 2018, 103, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ma, Q.; Chen, X.; Guo, K.; Li, J.; Zhang, M. Clinical significance of B7-H1 and B7-1 expressions in pancreatic carcinoma. World J. Surg. 2010, 34, 1059–1065. [Google Scholar] [CrossRef]
- Schultze, J.L.; Cardoso, A.A.; Freeman, G.J.; Seamon, M.J.; Daley, J.; Pinkus, G.S.; Gribben, J.G.; Nadler, L.M. Follicular lymphomas can be induced to present alloantigen efficiently: A conceptual model to improve their tumor immunogenicity. Proc. Natl. Acad. Sci. USA 1995, 92, 8200–8204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Fu, X.; Chang, Y.; Li, X.; Wu, X.; Li, L.; Zhang, L.; Sun, Z.; Zhang, X.; Zhang, M. B7-CD28 gene family expression is associated with prognostic and immunological characteristics of diffuse large B-cell lymphoma. Aging 2019, 11, 3939–3957. [Google Scholar] [CrossRef]
- Sims, M.A.; Field, S.D.; Barnes, M.R.; Shaikh, N.; Ellington, K.; Murphy, K.E.; Spurr, N.; Campbell, D.A. Cloning and characterisation of ITGAV, the genomic sequence for human cell adhesion protein (vitronectin) receptor alpha subunit, CD51. Cytogenet. Cell Genet. 2000, 89, 268–271. [Google Scholar] [CrossRef]
- Wang, H.; Chen, H.; Jiang, Z.; Lin, Y.; Wang, X.; Xiang, J.; Peng, J. Integrin subunit alpha V promotes growth, migration, and invasion of gastric cancer cells. Pathol. Res. Pract. 2019, 215, 152531. [Google Scholar] [CrossRef] [PubMed]
- Waisberg, J.; De Souza Viana, L.; Affonso Junior, R.J.; Silva, S.R.; Denadai, M.V.; Margeotto, F.B.; De Souza, C.S.; Matos, D. Overexpression of the ITGAV gene is associated with progression and spread of colorectal cancer. Anticancer Res. 2014, 34, 5599–5607. [Google Scholar]
- Van der Horst, G.; van den Hoogen, C.; Buijs, J.T.; Cheung, H.; Bloys, H.; Pelger, R.C.; Lorenzon, G.; Heckmann, B.; Feyen, J.; Pujuguet, P. Targeting of alpha(v)-integrins in stem/progenitor cells and supportive microenvironment impairs bone metastasis in human prostate cancer. Neoplasia 2011, 13, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheuk, I.W.; Siu, M.T.; Ho, J.C.; Chen, J.; Shin, V.Y.; Kwong, A. ITGAV targeting as a therapeutic approach for treatment of metastatic breast cancer. Am. J. Cancer Res. 2020, 10, 211–223. [Google Scholar] [PubMed]
- Blonska, M.; Zhu, Y.; Chuang, H.H.; You, M.J.; Kunkalla, K.; Vega, F.; Lin, X. Jun-regulated genes promote interaction of diffuse large B-cell lymphoma with the microenvironment. Blood 2015, 125, 981–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 30 October 2020).
- Yoon, S.O.; Zhang, X.; Freedman, A.S.; Zahrieh, D.; Lossos, I.S.; Li, L.; Choi, Y.S. Down-regulation of CD9 expression and its correlation to tumor progression in B lymphomas. Am. J. Pathol. 2010, 177, 377–386. [Google Scholar] [CrossRef]
- Dong, T.; Liu, Z.; Zhao, S.; Hu, C.; Liu, Y.; Ma, W.; Zhang, Q. The Expression of CD9 and PIK3CD is Associated with Prognosis of Follicular Lymphoma. J. Cancer 2015, 6, 1222–1229. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.Z.; Ansell, S.M. The tumor microenvironment in follicular lymphoma. Clin. Adv. Hematol. Oncol. 2012, 10, 810–818. [Google Scholar] [PubMed]
- Duś-Szachniewicz, K.; Drobczyński, S.; Ziółkowski, P.; Kołodziej, P.; Walaszek, K.M.; Korzeniewska, A.K.; Agrawal, A.; Kupczyk, P.; Woźniak, M. Physiological Hypoxia (Physioxia) Impairs the Early Adhesion of Single Lymphoma Cell to Marrow Stromal Cell and Extracellular Matrix. Optical Tweezers Study. Int. J. Mol. Sci. 2018, 19, 1880. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.X. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Antonosante, A.; d’Angelo, M.; Castelli, V.; Catanesi, M.; Iannotta, D.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. The Involvement of PPARs in the Peculiar Energetic Metabolism of Tumor Cells. Int. J. Mol. Sci. 2018, 19, 1907. [Google Scholar] [CrossRef] [Green Version]
- Lv, S.; Wang, W.; Wang, H.; Zhu, Y.; Lei, C. PPARγ activation serves as therapeutic strategy against bladder cancer via inhibiting PI3K-Akt signaling pathway. BMC Cancer 2019, 19, 204. [Google Scholar] [CrossRef]
- Chang, W.H.; Lai, A.G. The pan-cancer mutational landscape of the PPAR pathway reveals universal patterns of dysregulated metabolism and interactions with tumor immunity and hypoxia. Ann. N. Y. Acad. Sci. 2019, 1448, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.N. Hypothesis of Opposite Interplay Between the Canonical WNT/beta-catenin Pathway and PPAR Gamma in Primary Central Nervous System Lymphomas. Curr. Issues Mol. Biol. 2019, 31, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szumera-Ciećkiewicz, A.; Rymkiewicz, G.; Sokół, K.; Paszkiewicz-Kozik, E.; Borysiuk, A.; Poleszczuk, J.; Bachnio, K.; Bystydzienski, Z.; Woroniecka, R.; Grygalewicz, B.; et al. Significance of CD10 protein expression in the diagnostics of follicular lymphoma: A comparison of conventional immunohistochemistry with flow cytometry supported by the establishment of BCL2 and BCL6 rearrangements. Int. J. Lab. Hematol. 2020, 42, 453–463. [Google Scholar] [CrossRef]
- Wogsland, C.E.; Greenplate, A.R.; Kolstad, A.; Myklebust, J.H.; Irish, J.M.; Huse, K. Mass Cytometry of Follicular Lymphoma Tumors Reveals Intrinsic Heterogeneity in Proteins Including HLA-DR and a Deficit in Nonmalignant Plasmablast and Germinal Center B-Cell Populations. Cytometry B Clin. Cytom. 2017, 92, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.K.; Motaung, B.; du Plessis, N.; Shabangu, A.N.; Loxton, A.G. SU-IRG Consortium. Isolation of B-cells using Miltenyi MACS bead isolation kits. PLoS ONE 2019, 14, e0213832. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Gaugaz, F.Z. Fast and sensitive total protein and Peptide assays for proteomic analysis. Anal. Chem. 2015, 87, 4110–4116. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Mann, M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 2012, 84, 2631–2637. [Google Scholar]
- Wiśniewski, J.R. Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Anal. Chem. 2016, 88, 5438–5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihama, Y.; Rappsilber, J.; Andersen, J.S.; Mann, M. Microcolumns with self-assembled particle frits for proteomics. J. Chromatogr. A 2002, 979, 233–239. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Rakus, D. Multi-enzyme digestion FASP and the ‘Total Protein Approach’-based absolute quantification of the Escherichia coli proteome. J. Proteom. 2014, 109, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar]
- Cytoscape. Available online: https://cytoscape.org, (accessed on 7 May 2020).
- Mlecnik, B.; Galon, J.; Bindea, G. Comprehensive functional analysis of large lists of genes and proteins. J. Proteom. 2018, 171, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bioinformatics and Evolutionary Genomics Group. Available online: http://bioinformatics.psb.ugent.be/webtools/Venn/ (accessed on 4 November 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | ||||
| Gene Name | Protein Name | Accession | Fold Change | −Log p-Value |
| NUCKS1 | Nuclear ubiquitous casein and cyclin-dependent kinase substrate 1 | Q9H1E3 | 36.535 | 4.47 |
| SLC14A1 | Urea transporter 1 | Q13336 | 34.949 | 3.37 |
| GPALPP1 | GPALPP motifs-containing protein 1 | Q8IXQ4 | 24.970 | 1.69 |
| ADPRH | [Protein ADP-ribosylarginine] hydrolase | P54922 | 21.681 | 3.80 |
| CD72 | B-cell differentiation antigen CD72 | P21854 | 21.000 | 2.29 |
| PRDM15 | PR domain zinc finger protein 15 | P57071 | 20.148 | 1.71 |
| HMGN1 | Non-histone chromosomal protein HMG-14 | P05114 | 19.527 | 3.59 |
| MEF2B | Myocyte-specific enhancer factor 2B | Q02080-2 | 19.051 | 5.72 |
| PLEKHG2 | Pleckstrin homology domain-containing family G member 2 | Q9H7P9 | 16.283 | 2.01 |
| DBNL | Drebrin-like protein | Q9UJU6-2 | 14.342 | 1.61 |
| NAGPA | N-acetylglucosamine-1-phosphodiester alpha-N-acetylglucosaminidase | Q9UK23 | 14.081 | 1.84 |
| SPTY2D1 | Protein SPT2 homolog | Q68D10 | 13.958 | 1.81 |
| SEMA4A | Semaphorin-4A | Q9H3S1 | 13.311 | 3.33 |
| CCDC43 | Coiled-coil domain-containing protein 43 | Q96MW1 | 12.680 | 3.75 |
| ARPP19 | cAMP-regulated phosphoprotein 19 | P56211 | 12.429 | 1.83 |
| RASGRP3 | Ras guanyl-releasing protein 3 | Q8IV61 | 11.771 | 3.17 |
| DDT | D-dopachrome decarboxylase;D-dopachrome decarboxylase-like protein | P30046 | 11.570 | 2.49 |
| SP100 | Nuclear autoantigen Sp-100 | P23497 | 11.558 | 3.38 |
| GGACT | Gamma-glutamylaminecyclotransferase | Q9BVM4 | 11.222 | 3.47 |
| RRM2B | Ribonucleoside-diphosphate reductase subunit M2 B | Q7LG56 | 11.201 | 3.67 |
| (B) | ||||
| Gene Name | Protein Name | Accession | Fold Change | −Log p-Value |
| ITGAV | Integrin alpha-V | P06756 | 0.045 | 3.46 |
| NT5E | 5-nucleotidase | P21589 | 0.047 | 2.93 |
| PLTP | Phospholipid transfer protein | P55058 | 0.048 | 1.81 |
| CNTN1 | Contactin-1 | Q12860 | 0.053 | 2.14 |
| CD163 | Soluble CD163 | Q86VB7 | 0.056 | 6.11 |
| CLDN3 | Claudin-3 | O15551 | 0.059 | 4.36 |
| CD14 | Monocyte differentiation antigen CD14 | P08571 | 0.063 | 4.02 |
| CD9 | CD9 antigen | P21926 | 0.066 | 3.96 |
| CRAT | Carnitine O-acetyltransferase | P43155 | 0.066 | 1.77 |
| STAB1 | Stabilin-1 | Q9NY15 | 0.067 | 4.27 |
| MYO1B | Unconventional myosin-Ib | O437 | 0.068 | 4.28 |
| TMEM119 | Transmembrane protein 119 | Q4V9L6 | 0.070 | 1.84 |
| PTGFRN | Prostaglandin F2 receptor negative regulator | Q9P2B2 | 0.070 | 5.26 |
| SERPING1 | Plasma protease C1 inhibitor | P05155 | 0.071 | 4.23 |
| MACROD1 | O-acetyl-ADP-ribose deacetylase MACROD1 | Q9BQ69 | 0.075 | 5.54 |
| RAB34 | Ras-related protein Rab-34 | Q9BZG1 | 0.077 | 2.58 |
| ACE | Angiotensin-converting enzyme | P12821 | 0.078 | 2.72 |
| ENTPD5 | Ectonucleoside triphosphate diphosphohydrolase 5 | O75356 | 0.080 | 2.77 |
| ENG | Endoglin | P17813 | 0.081 | 3.03 |
| IGHG3 | Ig gamma-3 chain C region | A0A087WXL8 | 0.081 | 3.76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duś-Szachniewicz, K.; Rymkiewicz, G.; Agrawal, A.K.; Kołodziej, P.; Wiśniewski, J.R. Large-Scale Proteomic Analysis of Follicular Lymphoma Reveals Extensive Remodeling of Cell Adhesion Pathway and Identifies Hub Proteins Related to the Lymphomagenesis. Cancers 2021, 13, 630. https://doi.org/10.3390/cancers13040630
Duś-Szachniewicz K, Rymkiewicz G, Agrawal AK, Kołodziej P, Wiśniewski JR. Large-Scale Proteomic Analysis of Follicular Lymphoma Reveals Extensive Remodeling of Cell Adhesion Pathway and Identifies Hub Proteins Related to the Lymphomagenesis. Cancers. 2021; 13(4):630. https://doi.org/10.3390/cancers13040630
Chicago/Turabian StyleDuś-Szachniewicz, Kamila, Grzegorz Rymkiewicz, Anil Kumar Agrawal, Paweł Kołodziej, and Jacek R. Wiśniewski. 2021. "Large-Scale Proteomic Analysis of Follicular Lymphoma Reveals Extensive Remodeling of Cell Adhesion Pathway and Identifies Hub Proteins Related to the Lymphomagenesis" Cancers 13, no. 4: 630. https://doi.org/10.3390/cancers13040630