
Tumor Suppressor Protein p53 and Inhibitor of Apoptosis Proteins in Colorectal Cancer—A Promising Signaling Network for Therapeutic Interventions

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Biology and Functions of p53, a Brief Introduction
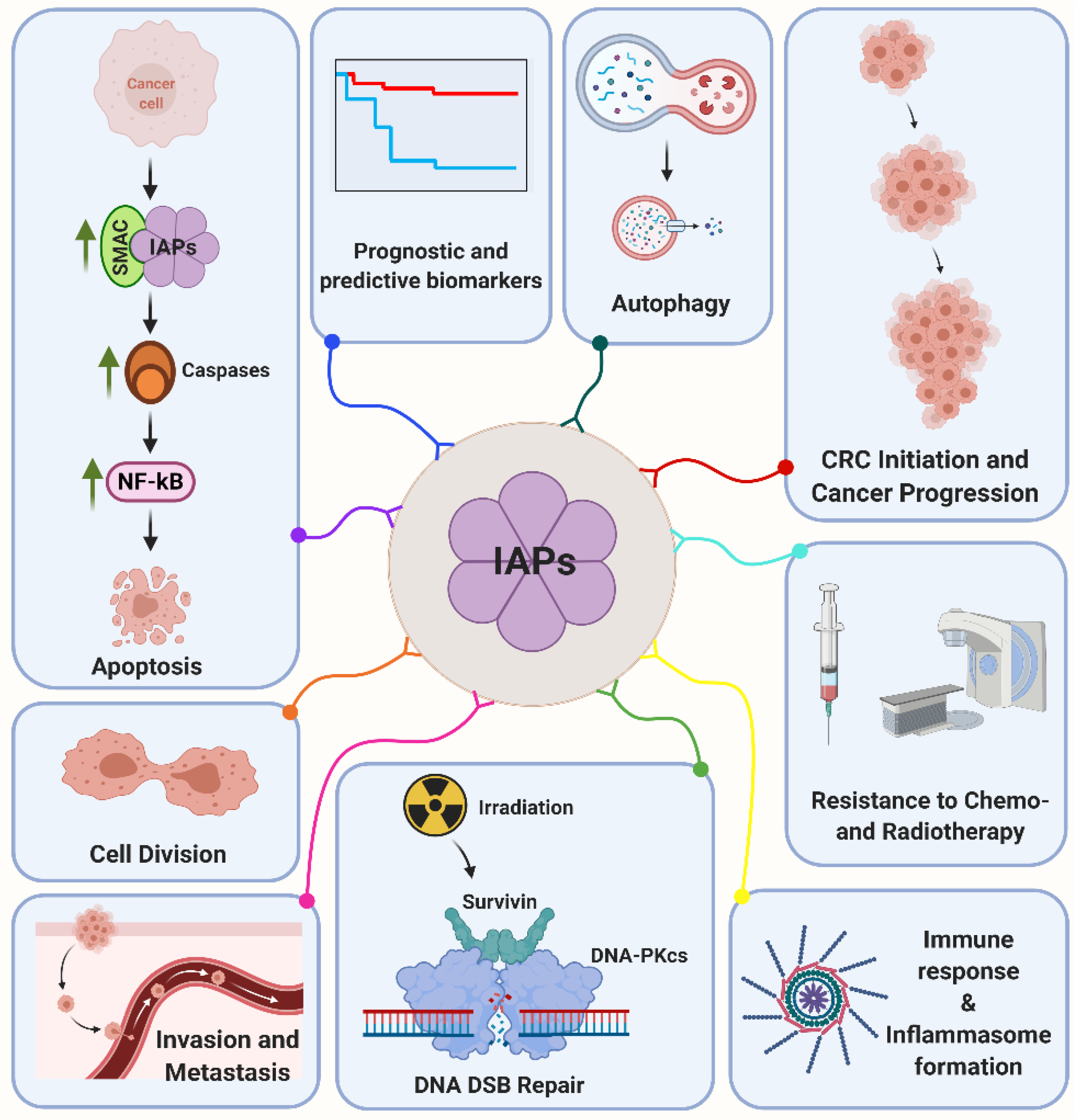
3. Structure and Function of the Inhibitor of Apoptosis Protein Family (IAP)
3.1. cIAP1 and cIAP2
3.2. XIAP
3.3. Survivin
3.4. BRUCE/Apollon
3.5. LIVIN
3.6. NAIP and hILP-2
4. Prognostic Relevance of IAP Expression in CRC
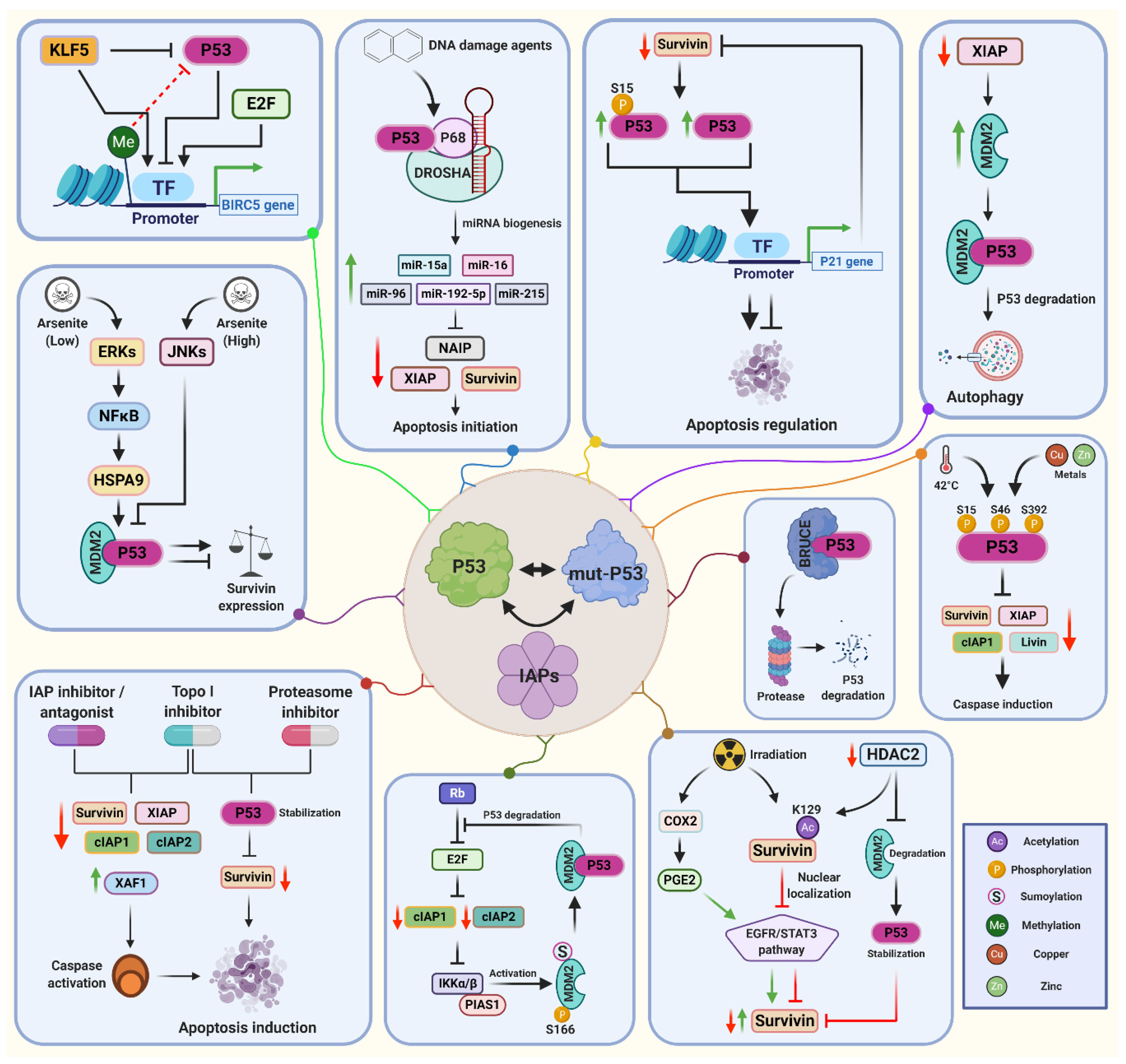
5. Molecular Regulation of IAPs by p53
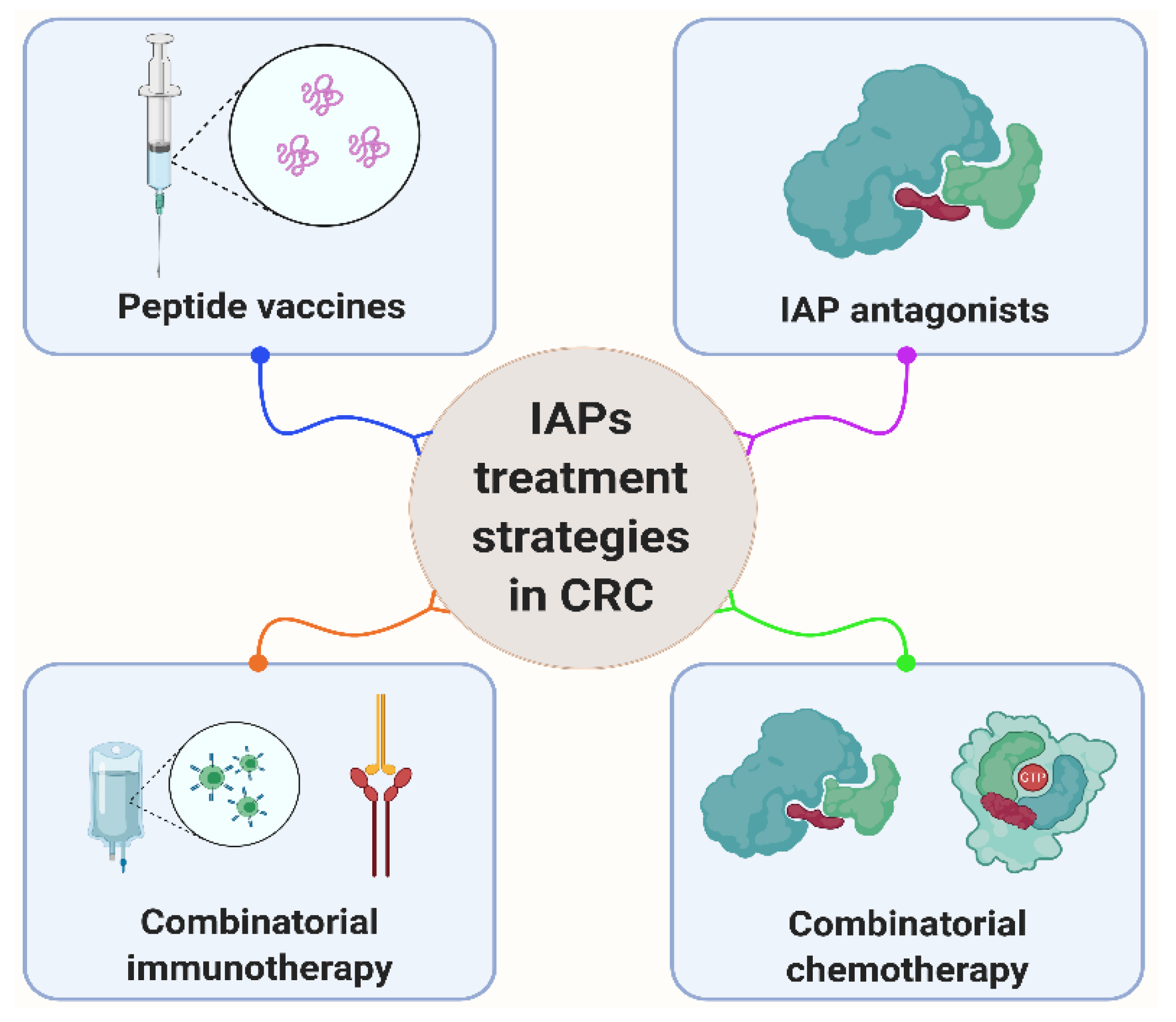
6. Clinical Treatment Potential by IAPs and p53
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, K. Colorectal cancer development and advances in screening. Clin. Interv. Aging 2016, 11, 967–976. [Google Scholar]
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 2019, 11, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Solomon, H.; Dinowitz, N.; Pateras, I.S.; Cooks, T.; Shetzer, Y.; Molchadsky, A.; Charni, M.; Rabani, S.; Koifman, G.; Tarcic, O.; et al. Mutant p53 gain of function underlies high expression levels of colorectal cancer stem cells markers. Oncogene 2018, 37, 1669–1684. [Google Scholar] [CrossRef] [Green Version]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gijn, W.; Marijnen, C.A.; Nagtegaal, I.D.; Kranenbarg, E.M.; Putter, H.; Wiggers, T.; Rutten, H.J.; Pahlman, L.; Glimelius, B.; van de Velde, C.J. Preoperative radiotherapy combined with total mesorectal excision for resectable rectal cancer: 12-year follow-up of the multicentre, randomised controlled TME trial. Lancet Oncol. 2011, 12, 575–582. [Google Scholar] [CrossRef]
- Rodel, C.; Liersch, T.; Becker, H.; Fietkau, R.; Hohenberger, W.; Hothorn, T.; Graeven, U.; Arnold, D.; Lang-Welzenbach, M.; Raab, H.R.; et al. Preoperative chemoradiotherapy and postoperative chemotherapy with fluorouracil and oxaliplatin versus fluorouracil alone in locally advanced rectal cancer: Initial results of the German CAO/ARO/AIO-04 randomised phase 3 trial. Lancet Oncol. 2012, 13, 679–687. [Google Scholar] [CrossRef]
- Fokas, E.; Glynne-Jones, R.; Appelt, A.; Beets-Tan, R.; Beets, G.; Haustermans, K.; Marijnen, C.; Minsky, B.D.; Ludmir, E.; Quirke, P.; et al. Outcome measures in multimodal rectal cancer trials. Lancet Oncol. 2020, 21, e252–e264. [Google Scholar] [CrossRef]
- Fokas, E.; Fietkau, R.; Hartmann, A.; Hohenberger, W.; Grutzmann, R.; Ghadimi, M.; Liersch, T.; Strobel, P.; Grabenbauer, G.G.; Graeven, U.; et al. Neoadjuvant rectal score as individual-level surrogate for disease-free survival in rectal cancer in the CAO/ARO/AIO-04 randomized phase III trial. Ann. Oncol. 2018, 29, 1521–1527. [Google Scholar] [CrossRef]
- Fokas, E.; Strobel, P.; Fietkau, R.; Ghadimi, M.; Liersch, T.; Grabenbauer, G.G.; Hartmann, A.; Kaufmann, M.; Sauer, R.; Graeven, U.; et al. Tumor Regression Grading After Preoperative Chemoradiotherapy as a Prognostic Factor and Individual-Level Surrogate for Disease-Free Survival in Rectal Cancer. J. Natl. Cancer Inst. 2017, 109, djx095. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, M.S.; Bishr, M.K.; Almutairi, F.M.; Ali, A.G. Inhibitors of apoptosis: Clinical implications in cancer. Apoptosis 2017, 22, 1487–1509. [Google Scholar] [CrossRef]
- Alvarez-Gonzalez, R. Genomic maintenance: The p53 poly(ADP-ribosyl)ation connection. Sci. Signal. 2007, 2007, pe68. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, S.; Hartwig, A. PARP1 Is Required for ATM-Mediated p53 Activation and p53-Mediated Gene Expression after Ionizing Radiation. Chem. Res. Toxicol. 2020, 33, 1933–1940. [Google Scholar] [CrossRef]
- Christmann, M.; Kaina, B. Epigenetic regulation of DNA repair genes and implications for tumor therapy. Mutat. Res. 2019, 780, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [PubMed]
- Brazda, V.; Fojta, M. The Rich World of p53 DNA Binding Targets: The Role of DNA Structure. Int. J. Mol. Sci. 2019, 20, 5605. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Fry, E.A.; Frazier, D.P. Transcription factors that interact with p53 and Mdm2. Int. J. Cancer 2016, 138, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Murray, D.; Mirzayans, R. Cellular Responses to Platinum-Based Anticancer Drugs and UVC: Role of p53 and Implications for Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 5766. [Google Scholar] [CrossRef]
- Dobbelstein, M.; Levine, A.J. Mdm2: Open questions. Cancer Sci. 2020, 111, 2203–2211. [Google Scholar] [CrossRef]
- Achanta, G.; Pelicano, H.; Feng, L.; Plunkett, W.; Huang, P. Interaction of p53 and DNA-PK in response to nucleoside analogues: Potential role as a sensor complex for DNA damage. Cancer Res. 2001, 61, 8723–8729. [Google Scholar] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Birnbaum, M.J.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. J. Virol. 1994, 68, 2521–2528. [Google Scholar] [CrossRef] [Green Version]
- Crook, N.E.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J. Virol. 1993, 67, 2168–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, K.; Fujibuchi, W.; Ishida, K.; Naitoh, T.; Ogawa, H.; Ando, T.; Yazaki, N.; Watanabe, K.; Haneda, S.; Shibata, C.; et al. Inhibitor of apoptosis protein family as diagnostic markers and therapeutic targets of colorectal cancer. Surg. Today 2011, 41, 175–182. [Google Scholar] [CrossRef]
- Hrdinka, M.; Yabal, M. Inhibitor of apoptosis proteins in human health and disease. Genes Immun. 2019, 20, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Xavier, R.J. Leucine-rich repeat (LRR) proteins: Integrators of pattern recognition and signaling in immunity. Autophagy 2011, 7, 1082–1084. [Google Scholar] [CrossRef] [Green Version]
- LaCasse, E.C.; Baird, S.; Korneluk, R.G.; MacKenzie, A.E. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene 1998, 17, 3247–3259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothe, M.; Pan, M.G.; Henzel, W.J.; Ayres, T.M.; Goeddel, D.V. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 1995, 83, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Busca, A.; Konarski, Y.; Gajanayaka, N.; O’Hara, S.; Angel, J.; Kozlowski, M.; Kumar, A. cIAP1/2-TRAF2-SHP-1-Src-MyD88 Complex Regulates Lipopolysaccharide-Induced IL-27 Production through NF-kappaB Activation in Human Macrophages. J. Immunol. 2018, 200, 1593–1606. [Google Scholar] [PubMed]
- Mao, A.P.; Li, S.; Zhong, B.; Li, Y.; Yan, J.; Li, Q.; Teng, C.; Shu, H.B. Virus-triggered ubiquitination of TRAF3/6 by cIAP1/2 is essential for induction of interferon-beta (IFN-beta) and cellular antiviral response. J. Biol. Chem. 2010, 285, 9470–9476. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.-G.; Li, J.-P.; Pang, X.-M.; Chen, C.-Y.; Xiang, H.-Y.; Feng, L.-B.; Su, S.-Y.; Li, S.-H.; Zhang, L.; Liu, J.-L. MicroRNA-29c Correlates with Neuroprotection Induced by FNS by Targeting Both Birc2 and Bak1 in Rat Brain after Stroke. CNS Neurosci. Ther. 2015, 21, 496–503. [Google Scholar] [CrossRef]
- Goncharov, T.; Niessen, K.; de Almagro, M.C.; Izrael-Tomasevic, A.; Fedorova, A.V.; Varfolomeev, E.; Arnott, D.; Deshayes, K.; Kirkpatrick, D.S.; Vucic, D. OTUB1 modulates c-IAP1 stability to regulate signalling pathways. EMBO J. 2013, 32, 1103–1114. [Google Scholar] [CrossRef]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a Signaling Platform that Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- Nicholson, J.; Jevons, S.J.; Groselj, B.; Ellermann, S.; Konietzny, R.; Kerr, M.; Kessler, B.M.; Kiltie, A.E. E3 Ligase cIAP2 Mediates Downregulation of MRE11 and Radiosensitization in Response to HDAC Inhibition in Bladder Cancer. Cancer Res. 2017, 77, 3027–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nudel, K.; Massari, P.; Genco, C.A. Neisseria gonorrhoeae Modulates Cell Death in Human Endocervical Epithelial Cells through Export of Exosome-Associated cIAP2. Infect. Immun. 2015, 83, 3410–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, M.M.; Ferguson Bennit, H.R.; Gonda, A.; Diaz Osterman, C.J.; Hibma, A.; Khan, S.; Wall, N.R. Exosomes Secreted from Human Cancer Cell Lines Contain Inhibitors of Apoptosis (IAP). Cancer Microenviron. 2015, 8, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Bennit, H.F.; Wall, N.R. The emerging role of exosomes in survivin secretion. Histol. Histopathol. 2015, 30, 43–50. [Google Scholar] [PubMed]
- Duckett, C.S.; Nava, V.E.; Gedrich, R.W.; Clem, R.J.; Van Dongen, J.L.; Gilfillan, M.C.; Shiels, H.; Hardwick, J.M.; Thompson, C.B. A conserved family of cellular genes related to the baculovirus iap gene and encoding apoptosis inhibitors. EMBO J. 1996, 15, 2685–2694. [Google Scholar] [CrossRef]
- Silke, J.; Vucic, D. IAP family of cell death and signaling regulators. Methods Enzymol. 2014, 545, 35–65. [Google Scholar]
- Xu, J.; Hua, X.; Yang, R.; Jin, H.; Li, J.; Zhu, J.; Tian, Z.; Huang, M.; Jiang, G.; Huang, H.; et al. XIAP Interaction with E2F1 and Sp1 via its BIR2 and BIR3 domains specific activated MMP2 to promote bladder cancer invasion. Oncogenesis 2019, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delbue, D.; Mendonca, B.S.; Robaina, M.C.; Lemos, L.G.T.; Lucena, P.I.; Viola, J.P.B.; Magalhaes, L.M.; Crocamo, S.; Oliveira, C.A.B.; Teixeira, F.R.; et al. Expression of nuclear XIAP associates with cell growth and drug resistance and confers poor prognosis in breast cancer. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118761. [Google Scholar] [CrossRef]
- Wicki, S.; Gurzeler, U.; Wei-Lynn Wong, W.; Jost, P.J.; Bachmann, D.; Kaufmann, T. Loss of XIAP facilitates switch to TNFalpha-induced necroptosis in mouse neutrophils. Cell Death Dis. 2016, 7, e2422. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Zhao, J.; Bulek, K.; Tang, F.; Chen, X.; Cai, G.; Jia, S.; Fox, P.L.; Huang, E.; Pizarro, T.T.; et al. Inflammation mobilizes copper metabolism to promote colon tumorigenesis via an IL-17-STEAP4-XIAP axis. Nat. Commun. 2020, 11, 900. [Google Scholar] [CrossRef] [Green Version]
- Zilu, S.; Qian, H.; Haibin, W.; Chenxu, G.; Deshuai, L.; Qiang, L.; Linfeng, H.; Jun, T.; Minxuan, X. Effects of XIAP on high fat diet-induced hepatic steatosis: A mechanism involving NLRP3 inflammasome and oxidative stress. Aging 2019, 11, 12177–12201. [Google Scholar] [CrossRef]
- Lu, M.; Qin, X.; Yao, J.; Yang, Y.; Zhao, M.; Sun, L. MiR-134-5p targeting XIAP modulates oxidative stress and apoptosis in cardiomyocytes under hypoxia/reperfusion-induced injury. IUBMB Life 2020, 72, 2154–2166. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, R.B.; Nachbur, U.; Yabal, M.; Wong, W.W.; Fiil, B.K.; Kastirr, M.; Rieser, E.; Rickard, J.A.; Bankovacki, A.; Peschel, C.; et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol. Cell 2012, 46, 746–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andree, M.; Seeger, J.M.; Schull, S.; Coutelle, O.; Wagner-Stippich, D.; Wiegmann, K.; Wunderlich, C.M.; Brinkmann, K.; Broxtermann, P.; Witt, A.; et al. BID-dependent release of mitochondrial SMAC dampens XIAP-mediated immunity against Shigella. EMBO J. 2014, 33, 2171–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Wu, Z.; Mei, Y.; Wu, M. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J. 2013, 32, 2204–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.; Ghislat, G.; Luo, S.; Renna, M.; Siddiqi, F.; Rubinsztein, D.C. XIAP and cIAP1 amplifications induce Beclin 1-dependent autophagy through NFkappaB activation. Hum. Mol. Genet. 2015, 24, 2899–2913. [Google Scholar] [CrossRef] [Green Version]
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.S.; Lee, D.H.; Kim, D.H.; Chung, J.H.; Lee, S.J.; Lee, T.H. cIAP1, cIAP2, and XIAP act cooperatively via nonredundant pathways to regulate genotoxic stress-induced nuclear factor-kappaB activation. Cancer Res. 2009, 69, 1782–1791. [Google Scholar] [CrossRef] [Green Version]
- Moussata, D.; Amara, S.; Siddeek, B.; Decaussin, M.; Hehlgans, S.; Paul-Bellon, R.; Mornex, F.; Gerard, J.P.; Romestaing, P.; Rodel, F.; et al. XIAP as a radioresistance factor and prognostic marker for radiotherapy in human rectal adenocarcinoma. Am. J. Pathol. 2012, 181, 1271–1278. [Google Scholar] [CrossRef]
- Hehlgans, S.; Petraki, C.; Reichert, S.; Cordes, N.; Rodel, C.; Rodel, F. Double targeting of Survivin and XIAP radiosensitizes 3D grown human colorectal tumor cells and decreases migration. Radiother. Oncol. 2013, 108, 32–39. [Google Scholar] [CrossRef]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef]
- Altieri, D.C. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer 2003, 3, 46–54. [Google Scholar] [CrossRef]
- Barrera-Vazquez, O.S.; Cancio-Lonches, C.; Hernandez-Gonzalez, O.; Chavez-Munguia, B.; Villegas-Sepulveda, N.; Gutierrez-Escolano, A.L. The feline calicivirus leader of the capsid protein causes survivin and XIAP downregulation and apoptosis. Virology 2019, 527, 146–158. [Google Scholar] [CrossRef]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep. 2006, 7, 988–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998, 396, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Vader, G.; Kauw, J.J.W.; Medema, R.H.; Lens, S.M.A. Survivin mediates targeting of the chromosomal passenger complex to the centromere and midbody. EMBO Rep. 2006, 7, 85–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Holloway, M.P.; Ma, L.; Cooper, Z.A.; Riolo, M.; Samkari, A.; Elenitoba-Johnson, K.S.; Chin, Y.E.; Altura, R.A. Acetylation directs survivin nuclear localization to repress STAT3 oncogenic activity. J. Biol. Chem. 2010, 285, 36129–36137. [Google Scholar] [CrossRef] [Green Version]
- Engelsma, D.; Rodriguez, J.A.; Fish, A.; Giaccone, G.; Fornerod, M. Homodimerization antagonizes nuclear export of survivin. Traffic 2007, 8, 1495–1502. [Google Scholar] [CrossRef]
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Mu, Y.; Cao, C.; Zeng, F.; Schneider, S.; Tan, J.; Price, J.; Chen, J.; Freeman, M.; Hallahan, D.E. Survivin as a therapeutic target for radiation sensitization in lung cancer. Cancer Res. 2004, 64, 2840–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarti, A.; Zhai, G.G.; Zhang, M.; Malhotra, R.; Latham, D.E.; Delaney, M.A.; Robe, P.; Nestler, U.; Song, Q.; Loeffler, J. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene 2004, 23, 7494–7506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodel, F.; Hoffmann, J.; Distel, L.; Herrmann, M.; Noisternig, T.; Papadopoulos, T.; Sauer, R.; Rodel, C. Survivin as a radioresistance factor, and prognostic and therapeutic target for radiotherapy in rectal cancer. Cancer Res. 2005, 65, 4881–4887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capalbo, G.; Dittmann, K.; Weiss, C.; Reichert, S.; Hausmann, E.; Rodel, C.; Rodel, F. Radiation-induced survivin nuclear accumulation is linked to DNA damage repair. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 226–234. [Google Scholar] [CrossRef]
- Wang, X.; Beitler, J.J.; Huang, W.; Chen, G.; Qian, G.; Magliocca, K.; Patel, M.R.; Chen, A.Y.; Zhang, J.; Nannapaneni, S.; et al. Honokiol Radiosensitizes Squamous Cell Carcinoma of the Head and Neck by Downregulation of Survivin. Clin. Cancer Res. 2018, 24, 858–869. [Google Scholar] [CrossRef] [Green Version]
- Gullulu, O.; Hehlgans, S.; Mayer, B.E.; Gossner, I.; Petraki, C.; Hoffmann, M.; Dombrowsky, M.J.; Kunzmann, P.; Hamacher, K.; Strebhardt, K.; et al. A spatial and functional interaction of a heterotetramer Survivin-DNA-PKcs complex in DNA damage response. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Hauser, H.P.; Bardroff, M.; Pyrowolakis, G.; Jentsch, S. A giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J. Cell Biol. 1998, 141, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Hitz, C.; Vogt-Weisenhorn, D.; Ruiz, P.; Wurst, W.; Floss, T. Progressive loss of the spongiotrophoblast layer of Birc6/Bruce mutants results in embryonic lethality. Genesis 2005, 42, 91–103. [Google Scholar] [CrossRef]
- Hao, Y.; Sekine, K.; Kawabata, A.; Nakamura, H.; Ishioka, T.; Ohata, H.; Katayama, R.; Hashimoto, C.; Zhang, X.; Noda, T.; et al. Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat. Cell Biol. 2004, 6, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Sekine, K.; Hao, Y.; Suzuki, Y.; Takahashi, R.; Tsuruo, T.; Naito, M. HtrA2 cleaves Apollon and induces cell death by IAP-binding motif in Apollon-deficient cells. Biochem. Biophys. Res. Commun. 2005, 330, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, R.; Ohata, H.; Ohoka, N.; Kawabata, A.; Naito, M. APOLLON protein promotes early mitotic CYCLIN A degradation independent of the spindle assembly checkpoint. J. Biol. Chem. 2014, 289, 3457–3467. [Google Scholar] [CrossRef] [Green Version]
- Pohl, C.; Jentsch, S. Final stages of cytokinesis and midbody ring formation are controlled by BRUCE. Cell 2008, 132, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Ge, C.; Che, L.; Ren, J.; Pandita, R.K.; Lu, J.; Li, K.; Pandita, T.K.; Du, C. BRUCE regulates DNA double-strand break response by promoting USP8 deubiquitination of BRIT1. Proc. Natl. Acad. Sci. USA 2015, 112, E1210–E1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, C.; Vilfranc, C.L.; Che, L.; Pandita, R.K.; Hambarde, S.; Andreassen, P.R.; Niu, L.; Olowokure, O.; Shah, S.; Waltz, S.E.; et al. The BRUCE-ATR Signaling Axis Is Required for Accurate DNA Replication and Suppression of Liver Cancer Development. Hepatology 2019, 69, 2608–2622. [Google Scholar] [CrossRef]
- Sun, J.G.; Liao, R.X.; Zhang, S.X.; Duan, Y.Z.; Zhuo, W.L.; Wang, X.X.; Wang, Z.X.; Li, D.Z.; Chen, Z.T. Role of inhibitor of apoptosis protein Livin in radiation resistance in nonsmall cell lung cancer. Cancer Biother. Radiopharm. 2011, 26, 585–592. [Google Scholar] [CrossRef]
- Wu, S.Q.; Xu, Q.B.; Sheng, W.Y.; Su, L.Y.; Zhu, L.W. A novel role for Livin in the response to ultraviolet B radiation and pterygium development. Int. J. Mol. Med. 2020, 45, 1103–1111. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Yang, D.; Wang, S.; Che, X.; Wang, J.; Li, X.; Zhang, Z.; Chen, X.; Song, X. Livin regulates prostate cancer cell invasion by impacting the NF-kappaB signaling pathway and the expression of FN and CXCR4. IUBMB Life 2012, 64, 274–283. [Google Scholar] [CrossRef]
- Li, F.; Yin, X.; Luo, X.; Li, H.Y.; Su, X.; Wang, X.Y.; Chen, L.; Zheng, K.; Ren, G.S. Livin promotes progression of breast cancer through induction of epithelial-mesenchymal transition and activation of AKT signaling. Cell Signal. 2013, 25, 1413–1422. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Lin, Y.J.; Wu, C.P.; Lee, H.T.; Shyu, W.C.; Wang, C.C. Livin contributes to tumor hypoxia-induced resistance to cytotoxic therapies in glioblastoma multiforme. Clin. Cancer Res. 2015, 21, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Kasof, G.M.; Gomes, B.C. Livin, a novel inhibitor of apoptosis protein family member. J. Biol. Chem. 2001, 276, 3238–3246. [Google Scholar] [CrossRef] [Green Version]
- Nachmias, B.; Ashhab, Y.; Bucholtz, V.; Drize, O.; Kadouri, L.; Lotem, M.; Peretz, T.; Mandelboim, O.; Ben-Yehuda, D. Caspase-mediated cleavage converts Livin from an antiapoptotic to a proapoptotic factor: Implications for drug-resistant melanoma. Cancer Res. 2003, 63, 6340–6349. [Google Scholar] [PubMed]
- Nachmias, B.; Lazar, I.; Elmalech, M.; Abed-El-Rahaman, I.; Asshab, Y.; Mandelboim, O.; Perlman, R.; Ben-Yehuda, D. Subcellular localization determines the delicate balance between the anti- and pro-apoptotic activity of Livin. Apoptosis 2007, 12, 1129–1142. [Google Scholar] [CrossRef]
- Wang, X.; Xu, J.; Ju, S.; Ni, H.; Zhu, J.; Wang, H. Livin gene plays a role in drug resistance of colon cancer cells. Clin. Biochem. 2010, 43, 655–660. [Google Scholar] [CrossRef]
- Oh, B.Y.; Lee, R.A.; Kim, K.H. siRNA targeting Livin decreases tumor in a xenograft model for colon cancer. World J. Gastroenterol. 2011, 17, 2563–2571. [Google Scholar] [PubMed]
- Roy, N.; Mahadevan, M.S.; McLean, M.; Shutler, G.; Yaraghi, Z.; Farahani, R.; Baird, S.; Besner-Johnston, A.; Lefebvre, C.; Kang, X.; et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 1995, 80, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Abadia-Molina, F.; Moron-Calvente, V.; Baird, S.D.; Shamim, F.; Martin, F.; MacKenzie, A. Neuronal apoptosis inhibitory protein (NAIP) localizes to the cytokinetic machinery during cell division. Sci. Rep. 2017, 7, 39981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karki, R.; Lee, E.; Place, D.; Samir, P.; Mavuluri, J.; Sharma, B.R.; Balakrishnan, A.; Malireddi, R.K.S.; Geiger, R.; Zhu, Q.; et al. IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 2018, 173, 920–933. [Google Scholar] [CrossRef] [PubMed]
- Lesne, S.; Gabriel, C.; Nelson, D.A.; White, E.; Mackenzie, E.T.; Vivien, D.; Buisson, A. Akt-dependent expression of NAIP-1 protects neurons against amyloid-{beta} toxicity. J. Biol. Chem. 2005, 280, 24941–24947. [Google Scholar] [CrossRef] [Green Version]
- Kano, O.; Tanaka, K.; Kanno, T.; Iwasaki, Y.; Ikeda, J.E. Neuronal apoptosis inhibitory protein is implicated in amyotrophic lateral sclerosis symptoms. Sci. Rep. 2018, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Crocker, S.J.; Wigle, N.; Liston, P.; Thompson, C.S.; Lee, C.J.; Xu, D.; Roy, S.; Nicholson, D.W.; Park, D.S.; MacKenzie, A.; et al. NAIP protects the nigrostriatal dopamine pathway in an intrastriatal 6-OHDA rat model of Parkinson’s disease. Eur. J. Neurosci. 2001, 14, 391–400. [Google Scholar] [CrossRef]
- Richter, B.W.; Mir, S.S.; Eiben, L.J.; Lewis, J.; Reffey, S.B.; Frattini, A.; Tian, L.; Frank, S.; Youle, R.J.; Nelson, D.L.; et al. Molecular cloning of ILP-2, a novel member of the inhibitor of apoptosis protein family. Mol. Cell Biol. 2001, 21, 4292–4301. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Zhou, W.; Zhu, X.; Xiang, S.; Wang, S.; Peng, Y.; Lu, B.; Tang, P.; Chen, Q.; Wu, M.; et al. Inhibitor of apoptosis proteinlike protein2: A novel growth accelerator for breast cancer cells. Oncol. Rep. 2018, 40, 2047–2055. [Google Scholar] [PubMed]
- De Oliveira Lima, F.; De Oliveira Costa, H.; Barrezueta, L.F.; Fujiyama Oshima, C.T.; Silva, J.A., Jr.; Gomes, T.S.; Pinheiro, N., Jr.; Neto, R.A.; Franco, M. Immunoexpression of inhibitors of apoptosis proteins and their antagonist SMAC/DIABLO in colorectal carcinoma: Correlation with apoptotic index, cellular proliferation and prognosis. Oncol. Rep. 2009, 22, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krajewska, M.; Kim, H.; Kim, C.; Kang, H.; Welsh, K.; Matsuzawa, S.; Tsukamoto, M.; Thomas, R.G.; Assa-Munt, N.; Piao, Z.; et al. Analysis of apoptosis protein expression in early-stage colorectal cancer suggests opportunities for new prognostic biomarkers. Clin. Cancer Res. 2005, 11, 5451–5461. [Google Scholar] [CrossRef] [Green Version]
- Karasawa, H.; Miura, K.; Fujibuchi, W.; Ishida, K.; Kaneko, N.; Kinouchi, M.; Okabe, M.; Ando, T.; Murata, Y.; Sasaki, H.; et al. Down-regulation of cIAP2 enhances 5-FU sensitivity through the apoptotic pathway in human colon cancer cells. Cancer Sci. 2009, 100, 903–913. [Google Scholar] [CrossRef]
- Xiang, G.; Wen, X.; Wang, H.; Chen, K.; Liu, H. Expression of X-linked inhibitor of apoptosis protein in human colorectal cancer and its correlation with prognosis. J. Surg. Oncol. 2009, 100, 708–712. [Google Scholar]
- Kanwar, J.R.; Kamalapuram, S.K.; Kanwar, R.K. Survivin signaling in clinical oncology: A multifaceted dragon. Med. Res. Rev. 2013, 33, 765–789. [Google Scholar] [CrossRef] [PubMed]
- Rodel, F.; Sprenger, T.; Kaina, B.; Liersch, T.; Rodel, C.; Fulda, S.; Hehlgans, S. Survivin as a prognostic/predictive marker and molecular target in cancer therapy. Curr. Med. Chem. 2012, 19, 3679–3688. [Google Scholar] [CrossRef]
- Kim, P.J.; Plescia, J.; Clevers, H.; Fearon, E.R.; Altieri, D.C. Survivin and molecular pathogenesis of colorectal cancer. Lancet 2003, 362, 205–209. [Google Scholar] [CrossRef]
- Li, W.; Lee, M.R.; Kim, T.; Kim, Y.W.; Cho, M.Y. Activated STAT3 may participate in tumor progression through increasing CD133/survivin expression in early stage of colon cancer. Biochem. Biophys. Res. Commun. 2018, 497, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.M.; Farma, J.M.; Coppola, D.; Hakam, A.; Fulp, W.J.; Chen, D.T.; Siegel, E.M.; Yeatman, T.J.; Shibata, D. Expression of the antiapoptotic protein survivin in colon cancer. Clin. Colorectal Cancer 2011, 10, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Sohn, I.; Do, I.G.; Jang, J.; Kim, S.H.; Jung, I.H.; Park, J.O.; Park, Y.S.; Talasaz, A.; Lee, J.; et al. Transcriptome analysis of CD133-positive stem cells and prognostic value of survivin in colorectal cancer. Cancer Genom. Proteom. 2014, 11, 259–266. [Google Scholar] [CrossRef]
- Huang, Y.J.; Qi, W.X.; He, A.N.; Sun, Y.J.; Shen, Z.; Yao, Y. The prognostic value of survivin expression in patients with colorectal carcinoma: A meta-analysis. Jpn. J. Clin. Oncol. 2013, 43, 988–995. [Google Scholar] [CrossRef] [Green Version]
- Sprenger, T.; Rodel, F.; Beissbarth, T.; Conradi, L.C.; Rothe, H.; Homayounfar, K.; Wolff, H.A.; Ghadimi, B.M.; Yildirim, M.; Becker, H.; et al. Failure of downregulation of survivin following neoadjuvant radiochemotherapy in rectal cancer is associated with distant metastases and shortened survival. Clin. Cancer Res. 2011, 17, 1623–1631. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Ahn, S.; Kim, K.; Cho, M.S.; Kim, K.H.; Lee, R.A.; Nam, E.M. Prognostic Significance of Survivin Expression and Combined Analysis with Cancer Stem Cell and Epithelial-Mesenchymal Transition-related Markers in Patients with Rectal Cancer Undergoing Preoperative Chemoradiotherapy. Anticancer Res. 2018, 38, 6881–6889. [Google Scholar] [CrossRef]
- Mehrotra, S.; Languino, L.R.; Raskett, C.M.; Mercurio, A.M.; Dohi, T.; Altieri, D.C. IAP regulation of metastasis. Cancer Cell 2010, 17, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Ning, Y.; Hanna, D.L.; Zhang, W.; Mendez, A.; Yang, D.; El-Khoueiry, R.; Matsusaka, S.; Sunakawa, Y.; Stremitzer, S.; Parekh, A.; et al. Cytokeratin-20 and Survivin-Expressing Circulating Tumor Cells Predict Survival in Metastatic Colorectal Cancer Patients by a Combined Immunomagnetic qRT-PCR Approach. Mol. Cancer Ther. 2015, 14, 2401–2408. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.Y.; Zhang, H.; Adell, G.; Olsson, B.; Sun, X.F. Livin expression is an independent factor in rectal cancer patients with or without preoperative radiotherapy. Radiat. Oncol. 2013, 8, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myung, D.S.; Park, Y.L.; Chung, C.Y.; Park, H.C.; Kim, J.S.; Cho, S.B.; Lee, W.S.; Lee, K.H.; Lee, J.H.; Joo, Y.E. Expression of Livin in colorectal cancer and its relationship to tumor cell behavior and prognosis. PLoS ONE 2013, 8, e73262. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Weng, S.; Tang, W.; Xue, R.; Chen, S.; Cai, G.; Cai, Y.; Shen, X.; Zhang, S.; Dong, L. Overexpression of BIRC6 Is a Predictor of Prognosis for Colorectal Cancer. PLoS ONE 2015, 10, e0125281. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Xue, R.; Weng, S.; Wu, J.; Fang, Y.; Wang, Y.; Ji, L.; Hu, T.; Liu, T.; Huang, X.; et al. BIRC6 promotes hepatocellular carcinogenesis: Interaction of BIRC6 with p53 facilitating p53 degradation. Int. J. Cancer 2015, 136, E475–E487. [Google Scholar] [CrossRef] [Green Version]
- Van Houdt, W.J.; Emmink, B.L.; Pham, T.V.; Piersma, S.R.; Verheem, A.; Vries, R.G.; Fratantoni, S.A.; Pronk, A.; Clevers, H.; Borel Rinkes, I.H.; et al. Comparative proteomics of colon cancer stem cells and differentiated tumor cells identifies BIRC6 as a potential therapeutic target. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef]
- Lu, C.D.; Altieri, D.C.; Tanigawa, N. Expression of a novel antiapoptosis gene, survivin, correlated with tumor cell apoptosis and p53 accumulation in gastric carcinomas. Cancer Res. 1998, 58, 1808–1812. [Google Scholar] [PubMed]
- Formby, B.; Wiley, T.S. Bcl-2, survivin and variant CD44 v7-v10 are downregulated and p53 is upregulated in breast cancer cells by progesterone: Inhibition of cell growth and induction of apoptosis. Mol. Cell. Biochem. 1999, 202, 53–61. [Google Scholar] [CrossRef]
- Mirza, A.; McGuirk, M.; Hockenberry, T.N.; Wu, Q.; Ashar, H.; Black, S.; Wen, S.F.; Wang, L.; Kirschmeier, P.; Bishop, W.R.; et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 2002, 21, 2613–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, W.H.; Biade, S.; Zilfou, J.T.; Chen, J.; Murphy, M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem. 2002, 277, 3247–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badr, E.A.; Assar, M.F.; Eltorgoman, A.M.A.; Labeeb, A.Z.; Breaka, G.A.; Elkhouly, E.A. A correlation between BCL-2 modifying factor, p53 and livin gene expressions in cancer colon patients. Biochem. Biophys. Rep. 2020, 22, 100747. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.; Liu, T.; Samadashwily, G.; Li, F.; Grossman, D. Survivin repression by p53, Rb and E2F2 in normal human melanocytes. Carcinogenesis 2008, 29, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.K.; Hwang, C.S.; Choe, T.B.; Hong, S.I.; Yi, J.Y.; Hwang, S.G.; Lee, H.G.; Oh, S.T.; Lee, Y.H.; Park, I.C. Selective inhibition of histone deacetylase 2 induces p53-dependent survivin downregulation through MDM2 proteasomal degradation. Oncotarget 2015, 6, 26528–26540. [Google Scholar] [CrossRef] [PubMed]
- Nabilsi, N.H.; Broaddus, R.R.; Loose, D.S. DNA methylation inhibits p53-mediated survivin repression. Oncogene 2009, 28, 2046–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Gu, L.; Findley, H.W.; Chen, C.; Dong, J.T.; Yang, L.; Zhou, M. KLF5 Interacts with p53 in regulating survivin expression in acute lymphoblastic leukemia. J. Biol. Chem. 2006, 281, 14711–14718. [Google Scholar] [CrossRef] [Green Version]
- Punga, T.; Akusjarvi, G. Adenovirus 2 E1B-55K protein relieves p53-mediated transcriptional repression of the survivin and MAP4 promoters. FEBS Lett. 2003, 552, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Wang, X.; Li, Z.; Li, B.; Ma, F.; Peng, L.; Zhang, Y.; Xu, A.; Jiang, B. microRNA-16 represses colorectal cancer cell growth in vitro by regulating the p53/survivin signaling pathway. Oncol. Rep. 2013, 29, 1652–1658. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.; Tang, H.; Wei, W.; Li, J.; Ji, L.; Ge, J. Aberrant Expression of MicroRNA-15a and MicroRNA-16 Synergistically Associates with Tumor Progression and Prognosis in Patients with Colorectal Cancer. Gastroenterol. Res. Pract. 2014, 2014, 364549. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Jackstadt, R.; Siemens, H.; Li, H.; Kirchner, T.; Hermeking, H. p53-induced miR-15a/16-1 and AP4 form a double-negative feedback loop to regulate epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer Res. 2014, 74, 532–542. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhao, W.; Wei, P.; Zuo, W.; Zhu, S. Tumor suppressor p53 induces miR-15a processing to inhibit neuronal apoptosis inhibitory protein (NAIP) in the apoptotic response DNA damage in breast cancer cell. Am. J. Transl. Res. 2017, 9, 683–691. [Google Scholar] [PubMed]
- Kim, S.A.; Kim, I.; Yoon, S.K.; Lee, E.K.; Kuh, H.J. Indirect modulation of sensitivity to 5-fluorouracil by microRNA-96 in human colorectal cancer cells. Arch. Pharm. Res. 2015, 38, 239–248. [Google Scholar] [CrossRef]
- Ye, M.; Zhang, J.; Miao, Q.; Yao, L. Curcumin promotes apoptosis by activating the p53-miR-192-5p/215-XIAP pathway in non-small cell lung cancer. Cancer Lett. 2015, 357, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Ling, X.; Liu, W.; Das, G.M.; Li, F. Transcriptional inhibition of p21WAF1/CIP1 gene (CDKN1) expression by survivin is at least partially p53-dependent: Evidence for survivin acting as a transcription factor or co-factor. Biochem. Biophys. Res. Commun. 2012, 421, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Beltrami, E.; Plescia, J.; Wilkinson, J.C.; Duckett, C.S.; Altieri, D.C. Acute ablation of survivin uncovers p53-dependent mitotic checkpoint functions and control of mitochondrial apoptosis. J. Biol. Chem. 2004, 279, 2077–2084. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xing, M.; Zhu, M.; Wang, X.; Wang, M.; Zhou, S.; Li, N.; Wu, R.; Zhou, M. Glycogen synthase kinase 3beta induces apoptosis in cancer cells through increase of survivin nuclear localization. Cancer Lett. 2008, 272, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, J.C.; Altieri, D.C. Activation of p53-dependent apoptosis by acute ablation of glycogen synthase kinase-3beta in colorectal cancer cells. Clin. Cancer Res. 2005, 11, 4580–4588. [Google Scholar] [CrossRef] [Green Version]
- Benson, E.K.; Mungamuri, S.K.; Attie, O.; Kracikova, M.; Sachidanandam, R.; Manfredi, J.J.; Aaronson, S.A. p53-dependent gene repression through p21 is mediated by recruitment of E2F4 repression complexes. Oncogene 2014, 33, 3959–3969. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Quaas, M.; Nickel, A.; Engeland, K. Indirect p53-dependent transcriptional repression of Survivin, CDC25C, and PLK1 genes requires the cyclin-dependent kinase inhibitor p21/CDKN1A and CDE/CHR promoter sites binding the DREAM complex. Oncotarget 2015, 6, 41402–41417. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Kalkum, M.; Overholtzer, M.; Stoffel, A.; Chait, B.T.; Levine, A.J. CIAP1 and the serine protease HTRA2 are involved in a novel p53-dependent apoptosis pathway in mammals. Genes Dev. 2003, 17, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Gu, L.; Li, F.; Zhu, Y.; Woods, W.G.; Findley, H.W. DNA damage induces a novel p53-survivin signaling pathway regulating cell cycle and apoptosis in acute lymphoblastic leukemia cells. J. Pharmacol. Exp. Ther. 2002, 303, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Bassi, L.; Carloni, M.; Meschini, R.; Fonti, E.; Palitti, F. X-irradiated human lymphocytes with unstable aberrations and their preferential elimination by p53/survivin-dependent apoptosis. Int. J. Radiat. Biol. 2003, 79, 943–954. [Google Scholar] [CrossRef]
- Grossman, D.; Kim, P.J.; Blanc-Brude, O.P.; Brash, D.E.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Transgenic expression of survivin in keratinocytes counteracts UVB-induced apoptosis and cooperates with loss of p53. J. Clin. Investig. 2001, 108, 991–999. [Google Scholar] [CrossRef]
- Zhu, N.; Gu, L.; Findley, H.W.; Li, F.; Zhou, M. An alternatively spliced survivin variant is positively regulated by p53 and sensitizes leukemia cells to chemotherapy. Oncogene 2004, 23, 7545–7551. [Google Scholar] [CrossRef] [Green Version]
- Ahamed, M.; Akhtar, M.J.; Raja, M.; Ahmad, I.; Siddiqui, M.K.; AlSalhi, M.S.; Alrokayan, S.A. ZnO nanorod-induced apoptosis in human alveolar adenocarcinoma cells via p53, survivin and bax/bcl-2 pathways: Role of oxidative stress. Nanomedicine 2011, 7, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.F.; He, B.; Li, N.P.; Ma, J.; Gong, G.Z.; Zhang, M. The oncogenic role of NS5A of hepatitis C virus is mediated by up-regulation of survivin gene expression in the hepatocellular cell through p53 and NF-kappaB pathways. Cell Biol. Int. 2011, 35, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, R.; Zhao, Y.; Xu, Y.; Ling, M.; Pang, Y.; Shen, L.; Zhou, Y.; Zhang, J.; Zhou, J.; et al. Opposed arsenite-mediated regulation of p53-survivin is involved in neoplastic transformation, DNA damage, or apoptosis in human keratinocytes. Toxicology 2012, 300, 121–131. [Google Scholar] [CrossRef]
- Burada, F.; Nicoli, E.R.; Ciurea, M.E.; Uscatu, D.C.; Ioana, M.; Gheonea, D.I. Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 2015, 7, 271–284. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Shi, M.; Liu, R.; Yang, Q.H.; Johnson, T.; Skarnes, W.C.; Du, C. The Birc6 (Bruce) gene regulates p53 and the mitochondrial pathway of apoptosis and is essential for mouse embryonic development. Proc. Natl. Acad. Sci. USA 2005, 102, 565–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopergolo, A.; Pennati, M.; Gandellini, P.; Orlotti, N.I.; Poma, P.; Daidone, M.G.; Folini, M.; Zaffaroni, N. Apollon gene silencing induces apoptosis in breast cancer cells through p53 stabilisation and caspase-3 activation. Br. J. Cancer 2009, 100, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Harmse, L.; Gangat, N.; Martins-Furness, C.; Dam, J.; de Koning, C.B. Copper-imidazo[1,2-a]pyridines induce intrinsic apoptosis and modulate the expression of mutated p53, haem-oxygenase-1 and apoptotic inhibitory proteins in HT-29 colorectal cancer cells. Apoptosis 2019, 24, 623–643. [Google Scholar] [CrossRef]
- Vancsik, T.; Forika, G.; Balogh, A.; Kiss, E.; Krenacs, T. Modulated electro-hyperthermia induced p53 driven apoptosis and cell cycle arrest additively support doxorubicin chemotherapy of colorectal cancer in vitro. Cancer Med. 2019, 8, 4292–4303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Barcelo, J.M.; Rao, V.A.; Sordet, O.; Jobson, A.G.; Thibaut, L.; Miao, Z.H.; Seiler, J.A.; Zhang, H.; Marchand, C.; et al. Repair of topoisomerase I-mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 179–229. [Google Scholar]
- Vaziri, S.A.; Hill, J.; Chikamori, K.; Grabowski, D.R.; Takigawa, N.; Chawla-Sarkar, M.; Rybicki, L.R.; Gudkov, A.V.; Mekhail, T.; Bukowski, R.M.; et al. Sensitization of DNA damage-induced apoptosis by the proteasome inhibitor PS-341 is p53 dependent and involves target proteins 14-3-3sigma and survivin. Mol. Cancer Ther. 2005, 4, 1880–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steigerwald, C.; Rasenberger, B.; Christmann, M.; Tomicic, M.T. Sensitization of colorectal cancer cells to irinotecan by the Survivin inhibitor LLP3 depends on XAF1 proficiency in the context of mutated p53. Arch. Toxicol. 2018, 92, 2645–2648. [Google Scholar] [CrossRef] [PubMed]
- Tomicic, M.T.; Steigerwald, C.; Rasenberger, B.; Brozovic, A.; Christmann, M. Functional mismatch repair and inactive p53 drive sensitization of colorectal cancer cells to irinotecan via the IAP antagonist BV6. Arch. Toxicol. 2019, 93, 2265–2277. [Google Scholar] [CrossRef]
- Cheng, L.; Zhou, Z.; Flesken-Nikitin, A.; Toshkov, I.A.; Wang, W.; Camps, J.; Ried, T.; Nikitin, A.Y. Rb inactivation accelerates neoplastic growth and substitutes for recurrent amplification of cIAP1, cIAP2 and Yap1 in sporadic mammary carcinoma associated with p53 deficiency. Oncogene 2010, 29, 5700–5711. [Google Scholar] [CrossRef] [Green Version]
- Lau, R.; Niu, M.Y.; Pratt, M.A. cIAP2 represses IKKalpha/beta-mediated activation of MDM2 to prevent p53 degradation. Cell Cycle 2012, 11, 4009–4019. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, Y.Y.; Yang, X.F.; Shen, D.F.; Sun, H.Z.; Huang, K.Q.; Zheng, H.C. Effects and mechanism of STAT3 silencing on the growth and apoptosis of colorectal cancer cells. Oncol. Lett. 2018, 16, 5575–5582. [Google Scholar] [CrossRef]
- Steinauer, K.K.; Gibbs, I.; Ning, S.; French, J.N.; Armstrong, J.; Knox, S.J. Radiation induces upregulation of cyclooxygenase-2 (COX-2) protein in PC-3 cells. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 325–328. [Google Scholar] [CrossRef]
- Chun, K.S.; Langenbach, R. The prostaglandin E2 receptor, EP2, regulates survivin expression via an EGFR/STAT3 pathway in UVB-exposed mouse skin. Mol. Carcinog 2011, 50, 439–448. [Google Scholar] [CrossRef]
- Barjaktarovic, Z.; Kempf, S.J.; Sriharshan, A.; Merl-Pham, J.; Atkinson, M.J.; Tapio, S. Ionizing radiation induces immediate protein acetylation changes in human cardiac microvascular endothelial cells. J. Radiat. Res. 2015, 56, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Hehlgans, S.; Oppermann, J.; Reichert, S.; Fulda, S.; Rodel, C.; Rodel, F. The SMAC mimetic BV6 sensitizes colorectal cancer cells to ionizing radiation by interfering with DNA repair processes and enhancing apoptosis. Radiat. Oncol. 2015, 10, 198. [Google Scholar] [CrossRef] [Green Version]
- Fichtner, M.; Bozkurt, E.; Salvucci, M.; McCann, C.; McAllister, K.A.; Halang, L.; Dussmann, H.; Kinsella, S.; Crawford, N.; Sessler, T.; et al. Molecular subtype-specific responses of colon cancer cells to the SMAC mimetic Birinapant. Cell Death Dis. 2020, 11, 1020. [Google Scholar] [CrossRef]
- Knoll, G.; Ehrenschwender, M. The non-peptidomimetic IAP antagonist ASTX660 sensitizes colorectal cancer cells for extrinsic apoptosis. FEBS Open Bio. 2021. [Google Scholar] [CrossRef] [PubMed]
- Perimenis, P.; Galaris, A.; Voulgari, A.; Prassa, M.; Pintzas, A. IAP antagonists Birinapant and AT-406 efficiently synergise with either TRAIL, BRAF, or BCL-2 inhibitors to sensitise BRAFV600E colorectal tumour cells to apoptosis. BMC Cancer 2016, 16, 624. [Google Scholar] [CrossRef] [Green Version]
- Ehrenschwender, M.; Bittner, S.; Seibold, K.; Wajant, H. XIAP-targeting drugs re-sensitize PIK3CA-mutated colorectal cancer cells for death receptor-induced apoptosis. Cell Death Dis. 2014, 5, e1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Shi, K.J.; An, J.J.; Ci, Y.L.; Li, F.; Hui, K.Y.; Yang, Y.; Xu, C.M. The LEF1/CYLD axis and cIAPs regulate RIP1 deubiquitination and trigger apoptosis in selenite-treated colorectal cancer cells. Cell Death Dis. 2014, 5, e1085. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Li, X.; Li, Q.; Liu, H.; Shi, Y.; Zhuo, H.; Li, C.; Zhu, H. Silencing Livin improved the sensitivity of colon cancer cells to 5-fluorouracil by regulating crosstalk between apoptosis and autophagy. Oncol. Lett. 2018, 15, 7707–7715. [Google Scholar] [CrossRef] [PubMed]
- AlShamaileh, H.; Wang, T.; Xiang, D.; Yin, W.; Tran, P.H.; Barrero, R.A.; Zhang, P.Z.; Li, Y.; Kong, L.; Liu, K.; et al. Aptamer-mediated survivin RNAi enables 5-fluorouracil to eliminate colorectal cancer stem cells. Sci. Rep. 2017, 7, 5898. [Google Scholar] [CrossRef] [PubMed]
- Gillissen, B.; Richter, A.; Overkamp, T.; Essmann, F.; Hemmati, P.G.; Preissner, R.; Belka, C.; Daniel, P.T. Targeted therapy of the XIAP/proteasome pathway overcomes TRAIL-resistance in carcinoma by switching apoptosis signaling to a Bax/Bak-independent ‘type I’ mode. Cell Death Dis. 2013, 4, e643. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Faehling, V.; Rauscher, B.; Betge, J.; Ebert, M.P.; Boutros, M. Multi-omics integration identifies a selective vulnerability of colorectal cancer subtypes to YM155. Int. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Men, K.; Zhang, Y.; Zhang, R.; Yang, L.; Duan, X. Local and systemic delivery of mRNA encoding survivin-T34A by lipoplex for efficient colon cancer gene therapy. Int. J. Nanomed. 2019, 14, 2733–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, W.; Che, X.; Li, G.; Wang, A.; Wang, Y.; Shi, X.; Hou, K.; Zhang, X.; Qu, X.; Liu, Y. Sur-X, a novel peptide, kills colorectal cancer cells by targeting survivin-XIAP complex. J. Exp. Clin. Cancer Res. 2020, 39, 82. [Google Scholar] [CrossRef]
- Gu, L.; Zhang, H.; Liu, T.; Zhou, S.; Du, Y.; Xiong, J.; Yi, S.; Qu, C.K.; Fu, H.; Zhou, M. Discovery of Dual Inhibitors of MDM2 and XIAP for Cancer Treatment. Cancer Cell 2016, 30, 623–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, B.Z.; Mak, D.H.; Schober, W.D.; Koller, E.; Pinilla, C.; Vassilev, L.T.; Reed, J.C.; Andreeff, M. Simultaneous activation of p53 and inhibition of XIAP enhance the activation of apoptosis signaling pathways in AML. Blood 2010, 115, 306–314. [Google Scholar] [CrossRef]
- Huang, S.W.; Chyuan, I.T.; Shiue, C.; Yu, M.C.; Hsu, Y.F.; Hsu, M.J. Lovastatin-mediated MCF-7 cancer cell death involves LKB1-AMPK-p38MAPK-p53-survivin signalling cascade. J. Cell Mol. Med. 2020, 24, 1822–1836. [Google Scholar] [CrossRef]
- Phiboonchaiyanan, P.P.; Petpiroon, N.; Sritularak, B.; Chanvorachote, P. Phoyunnanin E Induces Apoptosis of Non-small Cell Lung Cancer Cells via p53 Activation and Down-regulation of Survivin. Anticancer Res. 2018, 38, 6281–6290. [Google Scholar] [CrossRef]
- Yang, J.; Li, L.; Xu, C.; Yang, D.; Wang, S.; Yuan, S. CT1042, a novel anticancer agent, exhibits effects by activating p53 and inhibiting survivin. Oncol. Rep. 2018, 39, 2759–2768. [Google Scholar]
- Ellebaek, E.; Engell-Noerregaard, L.; Iversen, T.Z.; Froesig, T.M.; Munir, S.; Hadrup, S.R.; Andersen, M.H.; Svane, I.M. Metastatic melanoma patients treated with dendritic cell vaccination, Interleukin-2 and metronomic cyclophosphamide: Results from a phase II trial. Cancer Immunol. Immunother. 2012, 61, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Kameshima, H.; Tsuruma, T.; Torigoe, T.; Takahashi, A.; Hirohashi, Y.; Tamura, Y.; Tsukahara, T.; Ichimiya, S.; Kanaseki, T.; Iwayama, Y.; et al. Immunogenic enhancement and clinical effect by type-I interferon of anti-apoptotic protein, survivin-derived peptide vaccine, in advanced colorectal cancer patients. Cancer Sci. 2011, 102, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Promises and Challenges of Smac Mimetics as Cancer Therapeutics. Clin. Cancer Res. 2015, 21, 5030–5036. [Google Scholar] [CrossRef] [Green Version]
- Cong, H.; Xu, L.; Wu, Y.; Qu, Z.; Bian, T.; Zhang, W.; Xing, C.; Zhuang, C. Inhibitor of Apoptosis Protein (IAP) Antagonists in Anticancer Agent Discovery: Current Status and Perspectives. J. Med. Chem. 2019, 62, 5750–5772. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Dees, E.C.; Olszanski, A.J.; Dhuria, S.V.; Sen, S.; Cameron, S.; Cohen, R.B. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 3103–3110. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target | Inhibitor | Mode of Action | Reference |
|---|---|---|---|
| Targeting IAPs | |||
| cIAP1/2 XIAP | SMAC mimetics: BV6, Birinapant AT-406, Tolinapant | BV6 enhances the cellular radiosensitivity and the number of radiation-induced DNA damage foci in CRC cells. Birinapant alone or in combination with AT-406 increases apoptosis upon oxaliplatin/5-FU treatment. Tolinapant facilitates the activation of extrinsic apoptosis pathway by stimulating TNF-α and TRAIL and caspase activation | [164,165,166,167] |
| cIAP1/2 | siRNA | cIAP1/2 silencing chemosensitizes colorectal cancer cells and triggers caspase-8 activation and apoptosis | [169] |
| XIAP | Sur-X peptide inhibitor, siRNA, BV6, Mithramycin-A | Sur-X peptide inhibitor prevents the Survivin-XIAP interaction and inhibits their antiapoptotic and prometastatic activities; attenuation of XIAP by siRNA, Mithramycin-A or BV6 overcomes TRAIL-dependent apoptosis resistance in CRC cells | [168,172,175] |
| Survivin | YM155, LLP3 protein inhibitor, Sur-X peptide inhibitor, Survivin-T34A gene therapy, EpCAM-aptamer-guided survivin RNAi | YM155 treatment induces apoptosis by modulating ER-stress mediated apoptosis signaling. LLP3 drives the proteolytic degradation of Survivin and sensitizes CRC cells when combined with irinotecan (Topoisomerase inhibitor). Survivin-T34A mRNA encapsulated with a liposome-protamine lipoplex exhibits superior antitumor effect in a CRC mouse model. The aptamer-guided survivin RNAi enhances chemosensitivity, increases apoptosis, inhibits tumor growth in CRC stem cells and improves survival in xenograft mice | [156,171,173,174,175] |
| Livin | siRNA | Silencing of Livin enhances chemosensitivity to 5-FU in CRC cells, regulates crosstalk between apoptosis and autophagy and supresses tumor cell migration and invasion | [112,170] |
| Targeting IAPs and p53 | |||
| MDM2 XIAP | MX69 (dual MDM2 protein and XIAP RNA inhibitor) | MX69 triggers the downregulation of XIAP and degradation of MDM2 enabling activation of p53, induction of apoptosis and inhibition of cell proliferation in vivo | [176] |
| MDM2 XIAP | Nutlin-3 and XIAP antisense oligo- nucleotides | Nutlin-3 induces MDM2 degradation and activation of p53 that results in apoptosis induction in synergy with XIAP attenuation by antisense oligonucleotides | [177] |
| Survivin-p53 axis | Lovastatin (statin) CT-1042 (small molecule), Phoyunnanin-E | Lovastatin, CT-1042 and Phoyunnanin-E are involved in suppression of Survivin, activation of p53 and sensitization of cancer cells to apoptosis | [178,179,180] |
| Identifier | Disease | Treatment | Purpose | Outcome Measures |
|---|---|---|---|---|
| NCT00108875 | melanoma, pancreatic and cervical cancer, CRC | Survivin epitope peptide vaccine | to evaluate the safety, immunological response and clinical outcome | PFS, OS, best response, immunological response |
| NCT02587962 | Phase 1: solid tumors Phase 2: ovarian and cervical cancer, CRC | Birinapant (SMAC-mimetic), Pembrolizumab (PD-1 inhibitor) | to evaluate the safety, tolerability, pharmaco-dynamics and efficacy of combined modality treatment | Blood pressure, electro- cardiogram, enzymes, hemoglobin, physical exam, overall response |
| NCT03871959 | non-MSI-high advanced or metastatic pancreatic, colon and rectum cancer | DEBIO1143 (SMAC-mimetic) Pembrolizumab | to determine the MTD, the recomm. dose for a phase 2 trial and to evaluate efficacy | MTD and the dose for phase 2 of Debio1143 when combined with a fixed dose of Pembrolizumab, duration of response, clinical benefit, tumor response efficacy, PFS |
| NCT01188499 | advanced or metastatic tumors including CRC | Birinapant TL32711 (SMAC-mimetic) combined with standard regimes of chemotherapy | to evaluate dose escalation safety | Number of adverse events as a measure of safety and tolerability, anti tumor effect according to RECIST criteria |
| [182] | advanced or recurrent CRC | HLA-A24-restricted antigenic peptide, Survivin-2B80-88 vaccine, IFA and type 1 interferon (IFNalpha) | to assess whether immunogenicity of the Survivin-peptide could be enhanced with other vaccination protocols | Survivin-2B80-88 plus IFA and IFNalpha resulted in clinical improvement and enhanced levels of peptide-specific cytotoxic T lymphocytes |
| NCT02890069 | CRC, non-small cell lung, renal cancer, triple negative breast cancer | PDR001 (anti-PD1 antibody), LCL161 (SMAC-mimetic), everolimus (mTOR inhib.), Panobinostat (histone deacetylase inhib.), QBM076 (CXCR2 antagonist), HDM201 (p53-MDM2 interaction inhib.) | to identify the doses and schedule for combination therapy and to assess the safety, tolerability, pharmacological and clinical activity of combinations | Dose limiting toxicities, frequency of dose interruptions and reductions, adverse and serious adverse events, changes in laboratory parameters, PFS |
| NCT00197912 NCT00978913 | HLA-A2 positive, advanced melanoma and breast cancer | p53, Survivin and human telomerase peptide-pulsed dendritic cells | to show if autologous dendritic cells pulsed with peptides or tumor lysates can induce an immune response and clinical effects | Tolerability and safety, evaluation of treatment induced immune response and clinical tumor response/duration |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Güllülü, Ö.; Hehlgans, S.; Rödel, C.; Fokas, E.; Rödel, F. Tumor Suppressor Protein p53 and Inhibitor of Apoptosis Proteins in Colorectal Cancer—A Promising Signaling Network for Therapeutic Interventions. Cancers 2021, 13, 624. https://doi.org/10.3390/cancers13040624
Güllülü Ö, Hehlgans S, Rödel C, Fokas E, Rödel F. Tumor Suppressor Protein p53 and Inhibitor of Apoptosis Proteins in Colorectal Cancer—A Promising Signaling Network for Therapeutic Interventions. Cancers. 2021; 13(4):624. https://doi.org/10.3390/cancers13040624
Chicago/Turabian StyleGüllülü, Ömer, Stephanie Hehlgans, Claus Rödel, Emmanouil Fokas, and Franz Rödel. 2021. "Tumor Suppressor Protein p53 and Inhibitor of Apoptosis Proteins in Colorectal Cancer—A Promising Signaling Network for Therapeutic Interventions" Cancers 13, no. 4: 624. https://doi.org/10.3390/cancers13040624