Origin of Subsequent Malignant Neoplasms in Patients with History of Testicular Germ Cell Tumor


 ,
, 

Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
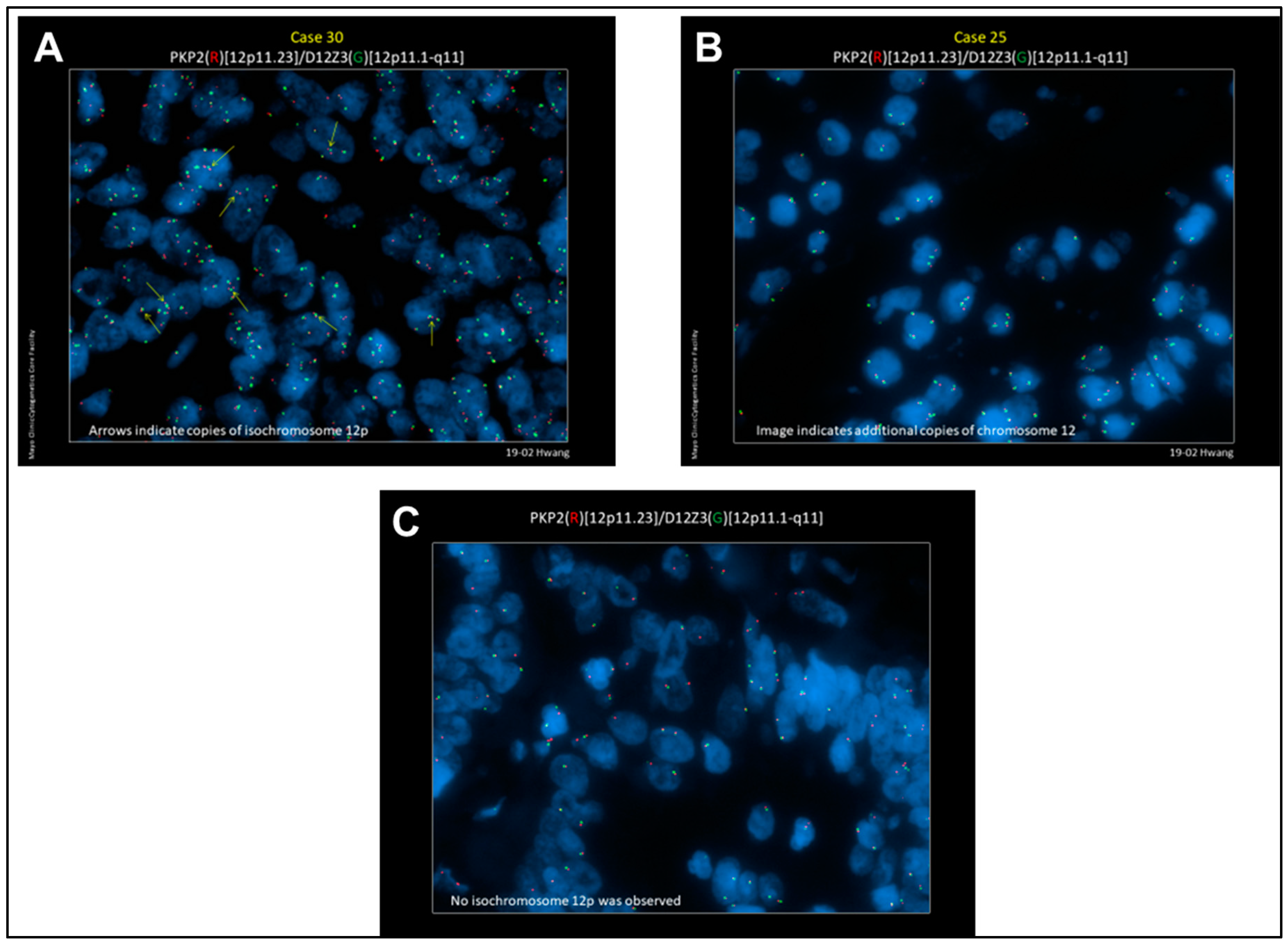
2.1. Detection of i(12p) and Gain of 12p in SMNs
2.2. Evaluation of Genetic Concordance in the Somatic Transformation of TGCTs by Targeted DNA Sequencing
2.3. Evaluation of Differentially Expressed Genes in Matched Teratoma and Somatically Transformed Specimens by RNAseq
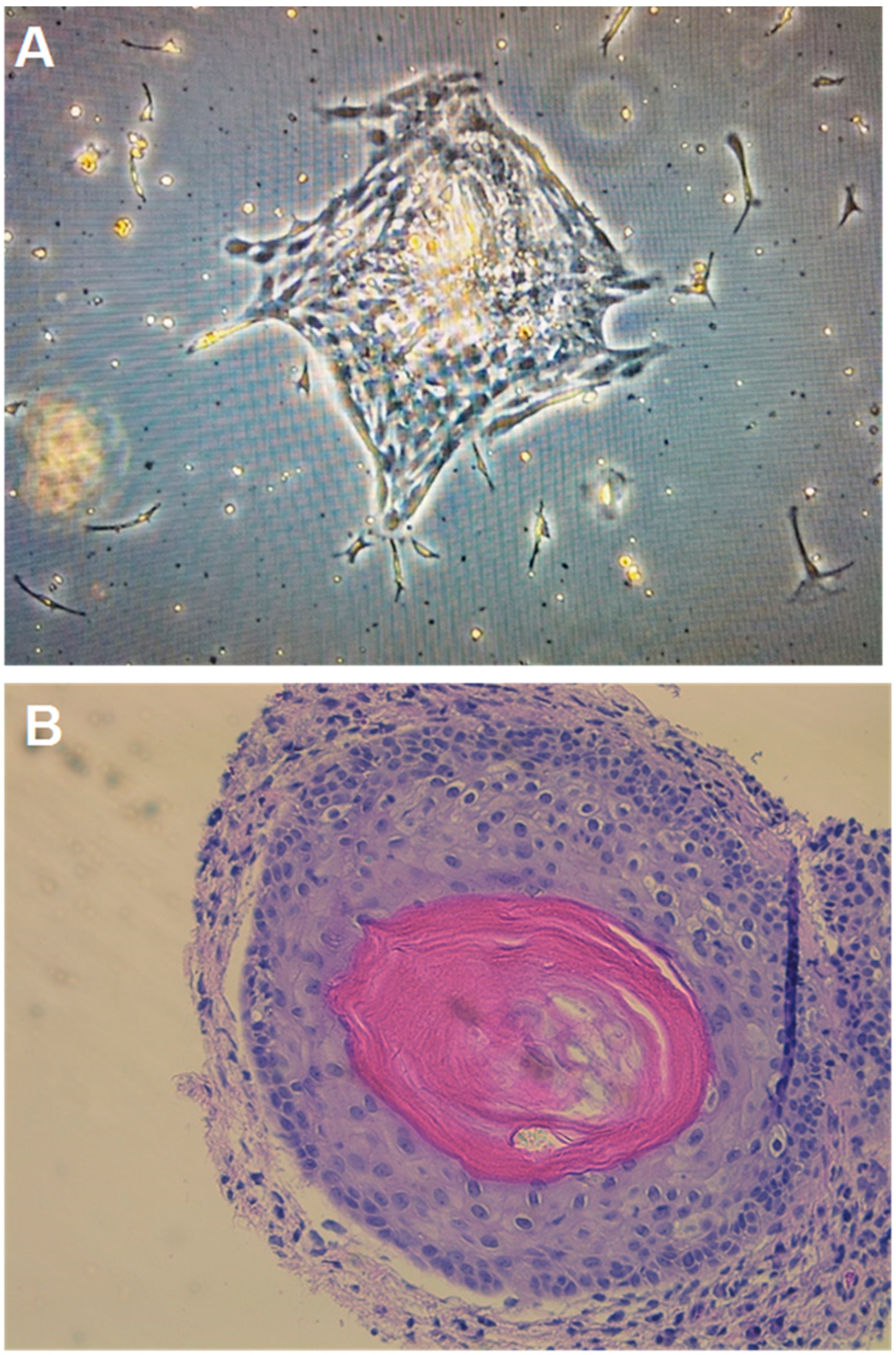
2.4. Growth of Teratoma Cell Cultures and Xenograft
2.5. Stemness Biomarker Expression
3. Discussion
4. Materials and Methods
4.1. Patient Identification
4.2. Fluorescent in Situ Hybridization (FISH)
4.3. DNA and RNA Sequencing
4.4. Primary Culture, Flow Cytometry, and Xenograft
4.5. DNA Sequencing Analysis
4.6. RNA Sequencing Analysis
4.7. Concordance of Mutations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.R.; Steyerberg, E.W.; Habbema, J.D.F. Survival of non-seminomatous germ cell cancer patients according to the IGCC classification: An update based on meta-analysis. Eur. J. Cancer 2006, 42, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Bilen, M.A.; Hess, K.R.; Broaddus, R.R.; Kopetz, S.; Wei, C.; Pagliaro, L.C.; Karam, J.A.; Ward, J.F.; Wood, C.G.; et al. Intratumoral heterogeneity: Role of differentiation in a potentially lethal phenotype of testicular cancer. Cancer 2016, 122, 1836–1843. [Google Scholar] [CrossRef] [PubMed]
- Groot, H.J.; Lubberts, S.; de Wit, R.; Witjes, J.A.; Kerst, J.M.; de Jong, I.J.; Groenewegen, G.; van den Eertwegh, A.J.M.; Poortmans, P.M.; Klümpen, H.-J.; et al. Risk of solid cancer after treatment of testicular germ cell cancer in the platinum era. J. Clin. Oncol. 2018, 36, 2504–2513. [Google Scholar] [CrossRef]
- Kum, J.B.; Ulbright, T.M.; Williamson, S.R.; Wang, M.; Zhang, S.; Foster, R.S.; Grignon, D.J.; Eble, J.N.; Beck, S.D.; Cheng, L. Molecular genetic evidence supporting the origin of somatic-type malignancy and teratoma from the same progenitor cell. Am. J. Surg. Pathol. 2012, 36, 1849–1856. [Google Scholar] [CrossRef]
- Tu, S.M. Origin of Cancers: Clinical Perspectives and Implications of a Stem-Cell Theory of Cancer; Springer: New York, NY, USA, 2010. [Google Scholar]
- Tu, S.M. Story of Hydra: Portrait of Cancer as a Stem-Cell Disease; Nova: New York, NY, USA, 2019. [Google Scholar]
- Nettersheim, D.; Gillis, A.J.M.; Looijenga, L.H.; Schorle, H. TGF-β1, EGF and FGF4 synergistically induce differentiation of the seminoma cell line T Cam-2 into a cell type resembling mixed non-seminoma. Int. J. Androl. 2011, 34, e189–e203. [Google Scholar] [CrossRef]
- Bouskine, A.; Vega, A.; Nebout, M.; Benahmed, M.; Fenichel, P. Expression of embryonic stem cell markers in cultured JKT?1, a cell line derived from a human seminoma. Int. J. Androl. 2010, 33, 54–63. [Google Scholar] [CrossRef]
- Som, A.; Xiao, L.; Zhu, R.; Guo, C.C.; Xiao, L.; Rao, P.; Efstathiou, E.; Matin, A.; Tu, S. Clinically atypical seminomas with yolk sac tumor features. Can. J. Urol. 2013, 20, 6860–6867. [Google Scholar]
- Looijenga, L.H.J.; Oosterhuis, J.W. Pathogenesis of testicular germ cell tumors. Rev. Reprod. 1999, 4, 90–100. [Google Scholar] [CrossRef]
- Van Echten, J.; Oosterhuis, J.W.; Looijenga, L.H.J.; Van De Pol, M.; Wiersema, J.; Meerman, G.J.T.; Koops, H.S.; Sleijfer, D.T.; De Jong, B. No recurrent structural abnormalities apart from i(12p) in primary germ cell tumors of the adult testis. Genes Chromosom. Cancer 1995, 14, 133–144. [Google Scholar] [CrossRef]
- Suijkerbuijk, R.F.; Sinke, R.J.; Meloni, A.M.; Parrington, J.M.; van Echten, J.; de Jong, B.; Oosterhuis, J.W.; Sandberg, A.A.; Geurts van Kessel, A. Overepresentation of chromosome 12p sequences and karyotypic evolution in i(12p)-negative testicular germ cell tumors revealed by fluorescence in situ hybridization. Cancer Genet. Cytogenet. 1993, 70, 85–93. [Google Scholar] [CrossRef]
- Reuter, V.E. Origins and molecular biology of testicular germ cell tumors. Mod. Pathol. 2005, 18, S51–S60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahrami, A.; Ro, J.Y.; Ayala, A.G. An overview of testicular germ cell tumors. Arch. Pathol. Lab. Med. 2007, 131, 1267–1280. [Google Scholar] [PubMed]
- Ottesen, A.M.; Skakkebaek, N.E.; Lundsteen, C.; Leffers, H.; Larsen, J.; Meyts, E.R.-D. High-resolution comparative genomic hybridization detects extra chromosome arm 12p material in most cases of carcinoma in situ adjacent to overt germ cell tumors, but not before the invasive tumor development. Genes Chromosom. Cancer 2003, 38, 117–125. [Google Scholar] [CrossRef]
- Heinonen, K.; Rao, P.N.; Slack, J.L.; Cruz, J.; Bloomfield, C.D.; Mrózek, K. Isochromosome 12p in two cases of acute myeloid leukemia without evidence of germ cell tumor. Br. J. Haematol. 1996, 93, 677–680. [Google Scholar] [CrossRef]
- Cormier-Daire, V.; Le Merrer, M.; Gigarel, N.; Morichon, N.; Prieur, M.; Lyonnet, S.; Vekemans, M.; Munnich, A. Prezygotic origin of the isochromosome 12p in Pallister-Killian syndrome. Am. J. Med. Genet. 1997, 69, 166–168. [Google Scholar] [CrossRef]
- Chen, K.; Meric-Bernstam, F.; Zhao, H.; Zhang, Q.; Ezzeddine, N.; Tang, L.Y.; Qi, Y.; Mao, Y.; Chen, T.; Chong, Z.; et al. Clinical actionability enhanced through deep targeted sequencing of solid tumors. Clin. Chem. 2015, 61, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.R.; Evans, M.J. The morphology and growth of a pluripotent teratocarcinoma cell line and its derivatives in tissue culture. Cell 1974, 2, 163–172. [Google Scholar] [CrossRef]
- Kim, W.T.; Ryu, A.C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Finõnes, R.R.; Yeargin, J.; Lee, M.; Kaur, A.P.; Cheng, C.; Sun, P.; Wu, C.; Nguyen, C.; Wang-Rodriguez, J.; Meyer, A.N.; et al. Early human prostate adenocarcinomas harbor androgen-independent cancer cells. PLoS ONE 2013, 8, e74438. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Lee, A.S.; Volkmer, J.P.; Sahoo, D.; Nag, D.; Mosley, A.R.; Inlay, M.A.; Ardehali, R.; Chavez, S.L.; Pera, R.R.; et al. An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat. Biotechnol. 2011, 29, 829–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, L.B.; Fosså, S.D.; Schonfeld, S.J.; McMaster, M.L.; Lynch, C.F.; Storm, H.H.; Hall, P.; Holowaty, E.J.; Andersen, A.; Pukkala, E.; et al. Second cancers among 40 576 testicular cancer patients: Focus on long-term survivors. J. Natl. Cancer Inst. 2005, 97, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Moller, H.; Horwich, A. Mortality and incidence of second cancers following treatment for testicular cancer. Br. J. Cancer 2007, 96, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvammen, Ø.; Myklebust, T.A.; Solberg, A.; Møller, B.; Klepp, O.H.; Fosså, S.D.; Tandstad, T. Causes of inferior relative survival after testicular germ cell tumor diagnosed 1953–2015: A population-based prospective cohort study. PLoS ONE 2019, 14, e0225942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, D.W.; Zickwolf, G.K.; Weil, G.J.; Sylvester, S.; DeMaria, M.A. Male fetal progenitor cells persist in maternal blood for as long as 27 years postpartum. Proc. Natl. Acad. Sci. USA 1996, 93, 705–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.C.; Lambert, N.; Maloney, S.; Furst, D.E.; Moore, J.M.; Nelson, J.L. Long-term fetal microchimerism in peripheral blood mononuclear cell subsets in healthy women and women with scleroderma. Blood 1999, 93, 2033–2037. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, S.; Wick, H.C.; Slonim, D.K.; Johnson, K.L.; Bianchi, D.W. Comprehensive analysis of genes expressed by rare microchimeric fetal cells in the maternal mouse lung. Biol. Reprod. 2012, 87, 1–6. [Google Scholar] [CrossRef]
- Chan, W.F.N.; Gurnot, C.; Montine, T.J.; Sonnen, J.A.; Guthrie, K.A.; Nelson, J.L. Male Microchimerism in the human female brain. PLoS ONE 2012, 7, e45592. [Google Scholar] [CrossRef] [Green Version]
- Barsky, S.H.; Ye, Y.; Xiao, Y.; Yearsley, K. Insights into the stem cell origin of human cancers by studying a registry of bone marrow and other organ transplant recipients who later developed solid cancers. J. Clin. Oncol. 2008, 26, 11010. [Google Scholar] [CrossRef]
- Fung, C.; Fossa, S.D.; Milano, M.T.; Oldenburg, J.; Travis, L.B. Solid tumors after chemotherapy or surgery for testicular nonseminoma: A population-based study. J. Clin. Oncol. 2013, 31, 3807–3814. [Google Scholar] [CrossRef] [Green Version]
- Jones, W.G.; Fossa, S.D.; Mead, G.M.; Roberts, J.T.; Sokal, M.; Horwich, A.; Stenning, S.P. Randomized trial of 30 versus 20 Gy in the adjuvant treatment of stage I testicular seminoma: A report on Medical Research Council Trial TE18, European Organization for the Research and Treatment of cancer trial 30942. J. Clin. Oncol. 2005, 23, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.R.; Bosl, G.J.; Sheinfeld, J.; Motzer, R.J. Medical treatment of advanced testicular cancer. JAMA 2008, 299, 672–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollmannsberger, C.; Tyldesley, S.; Moore, C.; Chi, K.N.; Murray, N.; Daneshmand, S.; Black, P.; Duncan, G.; Hayes-Lattin, B.; Nichols, C. Evolution in management of testicular seminoma: Population-based outcomes with selective utilization of active therapies. Ann. Oncol. 2011, 22, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.H.; Einhorn, L.H. Testicular cancer: A reflection on 50 years of discovery. J. Clin. Oncol. 2014, 32, 3085–3092. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Jones, T.D.; Wang, M.; Sung, M.-T.; Zhang, S.; Ulbright, T.M.; Eble, J.N.; Beck, S.D.; Foster, R.S.; Anagnostou, J.J.; Conner, C.; et al. Clonal origin of metastatic testicular teratomas. Clin. Cancer Res. 2006, 12, 5377–5383. [Google Scholar] [CrossRef] [Green Version]
- Kernek, K.M.; Ulbright, T.M.; Zhang, S.; Billings, S.D.; Cummings, O.W.; Henley, J.D.; Michael, H.; Brunelli, M.; Martignoni, G.; Foster, R.S.; et al. Identical allelic losses in mature teratoma and other histologic components of malignant mixed germ cell tumors of the testis. Am. J. Pathol. 2003, 163, 2477–2484. [Google Scholar] [CrossRef] [Green Version]
- Funt, S.A.; Patil, S.; Feldman, D.R.; Motzer, R.J.; Bajorin, D.F.; Sheinfeld, J.; Tickoo, S.K.; Reuter, V.E.; Bosl, G.J. Impact of teratoma on the cumulative incidence of disease-related death in patients with advanced germ cell tumors. J. Clin. Oncol. 2019, 37, 2329–2337. [Google Scholar] [CrossRef]
- Bilen, M.A.; Hess, K.R.; Campbell, M.T.; Wang, J.; Broaddus, R.R.; Karam, J.A.; Ward, J.F.; Wood, C.G.; Choi, S.L.; Rao, P.; et al. Intratumoral heterogeneity and chemoresistance in nonseminomatous germ cell tumor of the testis. Oncotarget 2016, 7, 86280–86289. [Google Scholar] [CrossRef] [Green Version]
- Graham, R.P.; Jin, L.; Knutson, D.L.; Kloft-Nelson, S.M.; Greipp, P.T.; Waldburger, N.; Roessler, S.; Longerich, T.; Roberts, L.R.; Oliveira, A.M.; et al. DNAJB1-PRKACA is specific for fibrolamellar carcinoma. Mod. Pathol. 2015, 28, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Shugay, M.; De Mendíbil, I.O.; Vizmanos, J.L.; Novo, F.J. Oncofuse: A computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics 2013, 29, 2539–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Clark, A.T.; Bodnar, M.S.; Fox, M.; Rodriquez, R.T.; Abeyta, M.J.; Firpo, M.T.; Pera, R.A.R. Spontaneous differentiation of germ cells from human embryonic stem cells in vitro. Hum. Mol. Genet. 2004, 13, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Gordeeva, O.F.; Zinovieva, R.; Smirnova, Y.; Payushina, O.; Nikonova, T.; Khrushchov, N. Differentiation of embryonic stem cells after transplantation into peritoneal cavity of irradiated mice and expression of specific germ cell genes in pluripotent cells. Transplant. Proc. 2005, 37, 295–298. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Second Malignancy | Number of Cases |
|---|---|
| Total | 43 |
| Gastrointestinal | 9 |
| Colorectal | 6 |
| Pancreas | 2 |
| Adenocarcinoma | 1 |
| Neuroendocrine | 1 |
| Esophageal | 1 |
| Lung | 7 |
| Squamous | 3 |
| Adenocarcinoma | 3 |
| Neuroendocrine | 1 |
| Genitourinary | 15 |
| Renal cell | 8 |
| Clear cell | 7 |
| Papillary | 1 |
| Urothelial | 5 |
| Bladder | 4 |
| Upper tract | 1 |
| Prostate | 2 |
| Sarcoma | 7 |
| Leiomyosarcoma | 4 |
| Retroperitoneal | 3 |
| Gluteal | 1 |
| Rhabdomyosarcoma | 1 |
| Angiosarcoma | 1 |
| Unclassified | 1 |
| Melanoma | 3 |
| Glioblastoma multiforme | 1 |
| Adrenal cortical carcinoma | 1 |
| Case No. | Primary Testicular Tumor | Age at Initial Diagnosis | Primary Treatment | Age at SMN | Time to SMN (Yrs.) | SMN Histology | Metastatic Tumor | % i(12p) FISH Positive | # i(12p) Copies |
| 1 | Seminoma * | 51 | BEP | 57 | 6.5 | Unclassified sarcoma | RP | 56% | 1–2 |
| 2 | Seminoma | 31 | XRT | 54 | 22.0 | Colorectal | Liver | 68% | 1 |
| 3 | Seminoma | 28 | XRT, BEP, HDC + SCT | 34 | 5.9 | LMS | Gluteus | 64% | 1–2 |
| 4 | Seminoma | 53 | BEP | 56 | 3.1 | Urothelial | Bladder | 39% | 1 |
| 5 | Nonseminoma | 39 | BEP | 39 | 0.4 | Pancreatic | Pancreas | 63% | 1–3 |
| Case No. | Primary Histology | Age at Initial Diagnosis | Primary Treatment | Age at SMN | Time to SMN (Yrs.) | SMN Histology | Site of SMN | % Extra Copies Chromosome 12 | # i(12p) Copies |
| 6 | Seminoma | 33 | XRT | 51 | 18.0 | LMS | RP | 73% | 0 |
| 7 | Seminoma | 51 | XRT | 59 | 7.6 | Rectal (adenocarcinoma) | Rectum | 55% | 0 |
| 8 | Seminoma | 58 | XRT | 71 | 13.3 | Colorectal | Cecum | 36% | 0 |
| 9 | Seminoma | 57 | XRT | 65 | 8.5 | Lung (adenocarcinoma) | Lung | 72% | 0 |
| 10 | Seminoma | 29 | XRT | 59 | 24.6 | Melanoma | Axillary lymph node | 83% | 0 |
| Nonseminoma | 44 | BEP | |||||||
| 11 | Seminoma | 22 | XRT | 58 | 9.6 | LMS | RP | 72% | 0 |
| 12 | Nonseminoma | 28 | CISCA/VB | 45 | 36.3 | LMS | RP | 74% | 0 |
| 13 | Seminoma | 53 | XRT | 58 | 44.8 | Colorectal | Lung | 83% | 0 |
| 14 | Seminoma | 62 | 65 | 5.2 | Lung (squamous) | Lung | 61% | 0 | |
| 15 | Seminoma | 27 | XRT | 81 | 3.0 | Urothelial | Bladder | 66% | 0 |
| 16 | NA | 20 | XRT + VB | 57 | 53.6 | Adrenal Cortical Carcinoma | Adrenal | 89% | 0 |
| 17 | Seminoma | 31 | XRT | 61 | 36.6 | Bladder | Bladder | 82% | 0 |
| 18 | Seminoma | 29 | CTX/carboplatin | 49 | 29.9 | Urothelial | Renal pelvis | 51% | 0 |
| 19 | Seminoma | 47 | XRT | 65 | 20.3 | Renal cell carcinoma | Kidney | 57% | 0 |
| 20 | Seminoma | 29 | PVB | 54 | 19.4 | Urothelial | Bladder | 91% | 0 |
| CSC Surface Marker | Function | Adult Stem Cell Expression | Normal Tissue Expression | CSC Expression | % Cells Positive in Cell Line tera13 | % Cells Positive in Cell Line tera22 |
|---|---|---|---|---|---|---|
| SSEA3 | hESC marker | Mesenchymal | Rare | Embryonal, Breast | <1% | <1% |
| SSEA4 | hESC marker | Mesenchymal, Cardiac, Gonad | Rare | Embryonal, Breast | 55% | 55% |
| SSEA5 | hESC marker | NA | Unknown | Embryonal, Pancreatic, Colorectal, Urothelial | <1% | <1% |
| TRA-1-60 | hESC marker | NA | Rare | Embryonal, Breast, Prostate | -- | -- |
| TRA-1-81 | hESC marker | NA | Rare | Embryonal, Breast | -- | -- |
| CD133 | Marker of hematopoietic stem cell | Hematopoietic, Prostate, Neural | Rare | Many * | 5% | <1% |
| CD90 | Cell adhesion | Mesenchymal, Cardiac | Rare | Brain, Liver | >99% | >99% |
| CD326 | Cell adhesion | No | Rare | Colon, Pancreas, Liver | -- | -- |
| Cripto-1 (TDGF1) | Self-renewal | NA | Rare | Breast, Colon, Lung | 4% | 5% |
| CD24 | B cell proliferation | Intestinal | Rare | Breast, Gastric, Pancreas | 25% | 2% |
| CD117 (c-kit) | Receptor for stem cell factor | Mesenchymal, Cardiac | Rare | Ovary | 2% | -- |
| CD26 | Dipeptidyl peptidase iv | Hematopoietic | Rare | Colorectal, Leukemia | 80% | >99% |
| CD34 | Cell adhesion | Hematopoietic | Rare | Leukemia, SCC | 3% | 2% |
| CD44 | Hyaluronic acid receptor | Hematopoietic, Adipose | Rare | Many ** | >99% | >99% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umbreit, E.C.; Siddiqui, B.A.; Hwang, M.J.; Joon, A.Y.; Maity, T.; Westerman, M.E.; Merriman, K.W.; Alhasson, H.; Uthup, J.; Guo, T.; et al. Origin of Subsequent Malignant Neoplasms in Patients with History of Testicular Germ Cell Tumor. Cancers 2020, 12, 3755. https://doi.org/10.3390/cancers12123755
Umbreit EC, Siddiqui BA, Hwang MJ, Joon AY, Maity T, Westerman ME, Merriman KW, Alhasson H, Uthup J, Guo T, et al. Origin of Subsequent Malignant Neoplasms in Patients with History of Testicular Germ Cell Tumor. Cancers. 2020; 12(12):3755. https://doi.org/10.3390/cancers12123755
Chicago/Turabian StyleUmbreit, Eric C., Bilal A. Siddiqui, Michael J. Hwang, Aron Y. Joon, Tapati Maity, Mary E. Westerman, Kelly W. Merriman, Hussam Alhasson, Joma Uthup, Tao Guo, and et al. 2020. "Origin of Subsequent Malignant Neoplasms in Patients with History of Testicular Germ Cell Tumor" Cancers 12, no. 12: 3755. https://doi.org/10.3390/cancers12123755