Clinicopathological and Molecular Profiles of Sporadic Microsatellite Unstable Colorectal Cancer with or without the CpG Island Methylator Phenotype (CIMP)

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Clinical Data
2.2. Alterations in Molecular Profiles
2.3. Clinicopathological Features and Molecular Signatures of Microsatellite Instability (MSI)-High Colorectal Cancer (CRC) Classified by the 5′-C-phosphate-G-3′ Island Methylator Phenotype (CIMP) Status
2.4. Clinicopathological Features and Molecular Signatures of MSI-High CRC Classified by Somatic Mismatch Repair (MMR) Mutations
2.5. The Mutational Profiles in MSI-High CRC Classified by the Cause of MSI
2.6. Impact of the CIMP and Somatic MMR Statuses on Outcomes of Colorectal Cancer Patients
3. Discussion
4. Materials and Methods
4.1. MSI Analysis
4.2. Targeted NGS
4.3. Sanger Sequencing
4.4. CIMP Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CRC | Colorectal Cancer |
| MSI | Microsatellite Instability |
| MSS | Microsatellite Stable |
| MMR | Mismatch Repair |
| LS | Lynch syndrome |
| CIMP | CpG Island Methylation Phenotype |
| NGS | Next-Generation Sequencing |
| LVI | Lymphovascular Invasion |
| PCR | Polymerase Chain Reaction |
| PMR | Percent Of The Methylated Reference |
| OS | Overall Survival |
| DFS | Disease-Free Survival |
| TNM | Tumor, Node, Metastasis |
| TCGA | The Cancer Genome Atlas |
| ICGC | International Cancer Genome Consortium |
References
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Smits, A.M.; Bos, J.L. Genetic Alterations during Colorectal-Tumor Development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, J.M.; Kim, C.-Y.; Christensen, E.R.; Tester, D.J.; Parc, Y.; Burgart, L.J.; Halling, K.C.; McDonnell, S.K.; Schaid, D.J.; Vockley, C.W.; et al. The Frequency of Hereditary Defective Mismatch Repair in a Prospective Series of Unselected Colorectal Carcinomas. Am. J. Hum. Genet. 2001, 69, 780–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; A Rodriguez-Bigas, M.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar] [PubMed]
- Haraldsdottir, S.; Hampel, H.; Tomsic, J.; Frankel, W.L.; Pearlman, R.; De La Chapelle, A.; Pritchard, C.C. Colon and Endometrial Cancers With Mismatch Repair Deficiency Can Arise From Somatic, Rather Than Germline, Mutations. Gastroenterology 2014, 147, 1308–1316.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensenkamp, A.R.; Vogelaar, I.P.; Van Zelst–Stams, W.A.; Goossens, M.; Ouchene, H.; Hendriks–Cornelissen, S.J.; Kwint, M.P.; Hoogerbrugge, N.; Nagtegaal, I.D.; Ligtenberg, M.J. Somatic Mutations in MLH1 and MSH2 Are a Frequent Cause of Mismatch-Repair Deficiency in Lynch Syndrome-Like Tumors. Gastroenterology 2014, 146, 643–646.e8. [Google Scholar] [CrossRef]
- Provenzale, D.; Gupta, S.; Ahnen, D.J.; Bray, T.; Cannon, J.A.; Cooper, G.; David, D.S.; Early, D.S.; Erwin, D.; Ford, J.M.; et al. Genetic/Familial High-Risk Assessment: Colorectal Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2016, 14, 1010–1030. [Google Scholar] [CrossRef]
- Sekine, S.; Ogawa, R.; Saito, S.; Ushiama, M.; Shida, D.; Nakajima, T.; Taniguchi, H.; Hiraoka, N.; Yoshida, T.; Sugano, K. Cytoplasmic MSH2 immunoreactivity in a patient with Lynch syndrome with anEPCAM-MSH2fusion. Histopathology 2017, 70, 664–669. [Google Scholar] [CrossRef]
- Ponti, G.; Castellsagueé, E.; Ruini, C.; Percesepe, A.; Tomasi, A. Mismatch repair genes founder mutations and cancer susceptibility in Lynch syndrome. Clin. Genet. 2015, 87, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Keijzers, G.; Rasmussen, K.J.R. DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. 2017, 773, 174–187. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.-P.J. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, J.-P. CpG island methylator phenotype in cancer. Nat. Rev. Cancer 2004, 4, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Issa, J.-P.J. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004, 23, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Meyerhardt, J.A.; Loda, M.; Giovannucci, E.L.; Fuchs, C.S. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2009, 58, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Simons, C.C.J.M.; Hughes, L.A.E.; Smits, K.M.; Bakker, C.A.K.-D.; De Bruïne, A.P.; Carvalho, B.; Meijer, G.A.; Schouten, L.J.; Brandt, P.A.V.D.; Weijenberg, M.P.; et al. A novel classification of colorectal tumors based on microsatellite instability, the CpG island methylator phenotype and chromosomal instability: Implications for prognosis. Ann. Oncol. 2013, 24, 2048–2056. [Google Scholar] [CrossRef]
- Nagasaka, T.; Koi, M.; Kloor, M.; Gebert, J.; Vilkin, A.; Nishida, N.; Shin, S.K.; Sasamoto, H.; Tanaka, N.; Matsubara, N.; et al. Mutations in Both KRAS and BRAF May Contribute to the Methylator Phenotype in Colon Cancer. Gastroenterology 2008, 134, 1950–1960.e1. [Google Scholar] [CrossRef] [Green Version]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; I Long, T.; A Faasse, M.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef]
- Boissière-Michot, F.; Frugier, H.; Ho-Pun-Cheung, A.; Lopez-Crapez, E.; Duffour, J.; Bibeau, F. Immunohistochemical staining for p16 and BRAFV600E is useful to distinguish between sporadic and hereditary (Lynch syndrome-related) microsatellite instable colorectal carcinomas. Virchows Archiv 2016, 469, 135–144. [Google Scholar] [CrossRef]
- Capper, D.; Voigt, A.Y.; Bozukova, G.; Ahadova, A.; Kickingereder, P.; Von Deimling, A.; Doeberitz, M.V.K.; Kloor, M. BRAF V600E-specific immunohistochemistry for the exclusion of Lynch syndrome in MSI-H colorectal cancer. Int. J. Cancer 2013, 133, 1624–1630. [Google Scholar] [CrossRef]
- Lagerstedt-Robinson, K.; Liu, T.; Vandrovcova, J.; Halvarsson, B.; Clendenning, M.; Frebourg, T.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Peltomäki, P.; et al. Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer) Diagnostics. J. Natl. Cancer Inst. 2007, 99, 291–299. [Google Scholar] [CrossRef]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. 2000, 16, 168–174. [Google Scholar] [CrossRef]
- Issa, J.P.; Baylin, S.B.; A Belinsky, S. Methylation of the estrogen receptor CpG island in lung tumors is related to the specific type of carcinogen exposure. Cancer Res. 1996, 56, 3655–3658. [Google Scholar] [PubMed]
- Issa, J.-P.J.; Ahuja, N.; Toyota, M.; Bronner, M.P.; A Brentnall, T. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001, 61, 3573–3577. [Google Scholar] [PubMed]
- Weisenberger, D.J.; Levine, A.J.; Long, T.I.; Buchanan, D.D.; Walters, R.; Clendenning, M.; Rosty, C.; Joshi, A.D.; Stern, M.C.; Le Marchand, L.; et al. Association of the Colorectal CpG Island Methylator Phenotype with Molecular Features, Risk Factors, and Family History. Cancer Epidemiol. Biomark. Prev. 2015, 24, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Deng, G.; Nguyen, A.; Tanaka, H.; Matsuzaki, K.; Bell, I.; Mehta, K.R.; Terdiman, J.P.; Waldman, F.M.; Kakar, S.; Gum, J.; et al. Regional hypermethylation and global hypomethylation are associated with altered chromatin conformation and histone acetylation in colorectal cancer. Int. J. Cancer 2006, 118, 2999–3005. [Google Scholar] [CrossRef]
- Ogino, S.; Brahmandam, M.; Cantor, M.; Namgyal, C.; Kawasaki, T.; Kirkner, G.J.; A Meyerhardt, J.; Loda, M.; Fuchs, C.S. Distinct molecular features of colorectal carcinoma with signet ring cell component and colorectal carcinoma with mucinous component. Mod. Pathol. 2005, 19, 59–68. [Google Scholar] [CrossRef]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Loda, M.; Fuchs, C.S. CpG Island Methylator Phenotype-Low (CIMP-Low) in Colorectal Cancer: Possible Associations with Male Sex and KRAS Mutations. J. Mol. Diagn. 2006, 8, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Toyota, M.; Kondo, Y.; Lin, E.; Zhang, L.; Guo, Y.; Hernandez, N.S.; Chen, X.; Ahmed, S.; Konishi, K.; et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 18654–18659. [Google Scholar] [CrossRef] [Green Version]
- Yagi, K.; Akagi, K.; Hayashi, H.; Nagae, G.; Tsuji, S.; Isagawa, T.; Midorikawa, Y.; Nishimura, Y.; Sakamoto, H.; Seto, Y.; et al. Three DNA Methylation Epigenotypes in Human Colorectal Cancer. Clin. Cancer Res. 2009, 16, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.; Shen, H.; Byun, H.-M.; Berg, D.V.D.; Malik, S.; Pan, F.; Noushmehr, H.; Van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012, 22, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Kraft, P.; Loda, M.; Fuchs, C.S. Evaluation of Markers for CpG Island Methylator Phenotype (CIMP) in Colorectal Cancer by a Large Population-Based Sample. J. Mol. Diagn. 2007, 9, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veigl, M.L.; Kasturi, L.; Olechnowicz, J.; Ma, A.; Lutterbaugh, J.D.; Periyasamy, S.; Li, G.-M.; Drummond, J.; Modrich, P.L.; Sedwick, W.D.; et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc. Natl. Acad. Sci. USA 1998, 95, 8698–8702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, C.N.; Goel, A.; Boland, C.R. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int. J. Cancer 2003, 106, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.; Finn, P.W.; Brown, R.C.; A Plumb, J. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br. J. Cancer 2009, 100, 758–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geurts-Giele, W.R.R.; Leenen, C.H.M.; Dubbink, H.J.; Meijssen, I.C.; Post, E.; Sleddens, H.F.B.M.; Kuipers, E.J.; Goverde, A.; Ouweland, A.M.W.V.D.; Van Lier, M.G.F.; et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J. Pathol. 2014, 234, 548–559. [Google Scholar] [CrossRef]
- Kim, J.H.; Bae, J.M.; Cho, N.-Y.; Kang, G.H. Distinct features between MLH1-methylated and unmethylated colorectal carcinomas with the CpG island methylator phenotype: Implications in the serrated neoplasia pathway. Oncotarget 2016, 7, 14095–14111. [Google Scholar] [CrossRef] [Green Version]
- Richman, S. Deficient mismatch repair: Read all about it (Review). Int. J. Oncol. 2015, 47, 1189–1202. [Google Scholar] [CrossRef] [Green Version]
- Advani, S.M.; Advani, P.; DeSantis, S.M.; Brown, D.; VonVille, H.M.; Lam, M.; Loree, J.M.; Sarshekeh, A.M.; Bressler, J.; Lopez, D.S.; et al. Clinical, Pathological, and Molecular Characteristics of CpG Island Methylator Phenotype in Colorectal Cancer: A Systematic Review and Meta-analysis. Transl. Oncol. 2018, 11, 1188–1201. [Google Scholar] [CrossRef]
- Donehower, L.A.; Creighton, C.J.; Schultz, N.; Shinbrot, E.; Chang, K.; Gunaratne, P.H.; Muzny, N.; Sander, C.; Hamilton, S.R.; Gibbs, R.A.; et al. MLH1-Silenced and Non-Silenced Subgroups of Hypermutated Colorectal Carcinomas Have Distinct Mutational Landscapes. J. Pathol. 2013, 229, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Network, T.C.G.A. Comprehensive molecular characterization of human colon and rectal cancer. Nat. Cell Biol. 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Tahara, T.; Yamamoto, E.; Madireddi, P.; Suzuki, H.; Maruyama, R.; Chung, W.; Garriga, J.; Jelinek, J.; Yamano, H.-O.; Sugai, T.; et al. Colorectal Carcinomas With CpG Island Methylator Phenotype 1 Frequently Contain Mutations in Chromatin Regulators. Gastroenterology 2014, 146, 530–538.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettington, M.; Walker, N.; Rosty, C.; Brown, I.; Clouston, A.; Wockner, L.; Whitehall, V.; Leggett, B. Critical Appraisal of the Diagnosis of the Sessile Serrated Adenoma. Am. J. Surg. Pathol. 2014, 38, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Rosty, C.; Young, J.P.; Walsh, M.D.; Clendenning, M.; Sanderson, K.; Walters, R.J.; Parry, S.; A Jenkins, M.; Win, A.K.; Southey, M.C.; et al. PIK3CA Activating Mutation in Colorectal Carcinoma: Associations with Molecular Features and Survival. PLoS ONE 2013, 8, e65479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, M.; Norris, K.F.; Wang, R.Y.; Kuo, K.C.; Gehrke, C.W. DNA cytosine methylation and heat-induced deamination. Biosci. Rep. 1986, 6, 387–393. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Initiative, A.P.C.G.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Poulos, R.C.; Olivier, J.; Wong, J.W. The interaction between cytosine methylation and processes of DNA replication and repair shape the mutational landscape of cancer genomes. Nucleic Acids Res. 2017, 45, 7786–7795. [Google Scholar] [CrossRef]
- Bardeesy, N.; Cheng, K.-H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.; Vickers, S.M.; Adsay, N.V.; Jhala, N.C.; Kim, H.-G.; Schoeb, T.R.; Grizzle, W.E.; Klug, C.A. Inactivation of Smad4 Accelerates KrasG12D-Mediated Pancreatic Neoplasia. Cancer Res. 2007, 67, 8121–8130. [Google Scholar] [CrossRef] [Green Version]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.X.; Hruban, R.H.; Adsay, N.V.; Tuveson, D.A.; et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007, 11, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-K.; Chang, S.-C.; Yang, Y.-C.; Li, A.F.-Y. Loss of heterozygosity and DNA aneuploidy in colorectal adenocarcinoma. Ann. Surg. Oncol. 2003, 10, 1086–1094. [Google Scholar] [CrossRef]
- Chang, S.-C.; Lin, J.-K.; Yang, S.H.; Wang, H.-S.; Li, A.F.-Y.; Chi, C.-W. Relationship between genetic alterations and prognosis in sporadic colorectal cancer. Int. J. Cancer 2006, 118, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Catalogue of Somatic Mutations in Cancer. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 28 January 2019).
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Aaltonen, L.A.; Salovaara, R.; Kristo, P.; Canzian, F.; Hemminki, A.; Peltomäki, P.; Chadwick, R.B.; Kääriäinen, H.; Eskelinen, M.; Järvinen, H.; et al. Incidence of Hereditary Nonpolyposis Colorectal Cancer and the Feasibility of Molecular Screening for the Disease. N. Engl. J. Med. 1998, 338, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, R.B.; Pyatt, R.E.; Niemann, T.H.; Richards, S.K.; Johnson, C.K.; Stevens, M.W.; Meek, J.E.; Hampel, H.; Prior, T.W.; de la Chapelle, A. Hereditary and somatic DNA mismatch repair gene mutations in sporadic endometrial carcinoma. J. Med. Genet. 2001, 38, 461–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumilla, K.; Schowalter, K.V.; Lindor, N.M.; Thomas, B.C.; Mensink, K.A.; Gallinger, S.; Holter, S.; Newcomb, P.A.; Potter, J.D.; Jenkins, M.A.; et al. Frequency of Deletions of EPCAM (TACSTD1) in MSH2-Associated Lynch Syndrome Cases. J. Mol. Diagn. 2011, 13, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Cotton, R.G.; Scriver, C.R. Proof of “disease causing” mutation. Hum. Mutat. 1998, 12, 1–3. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Curtin, K.; Lin, H.H.; Robertson, M.A.; Schaffer, D.; Nichols, M.; Gruenthal, K.; Leppert, M.F.; Slattery, M.L. The colon cancer burden of genetically defined hereditary nonpolyposis colon cancer. Gastroenterology 2001, 121, 830–838. [Google Scholar] [CrossRef]
- Kim, J.H.; Rhee, Y.-Y.; Bae, J.-M.; Kwon, H.-J.; Cho, N.-Y.; Kim, M.J.; Kang, G.H. Subsets of microsatellite-unstable colorectal cancers exhibit discordance between the CpG island methylator phenotype and MLH1 methylation status. Mod. Pathol. 2013, 26, 1013–1022. [Google Scholar] [CrossRef] [Green Version]



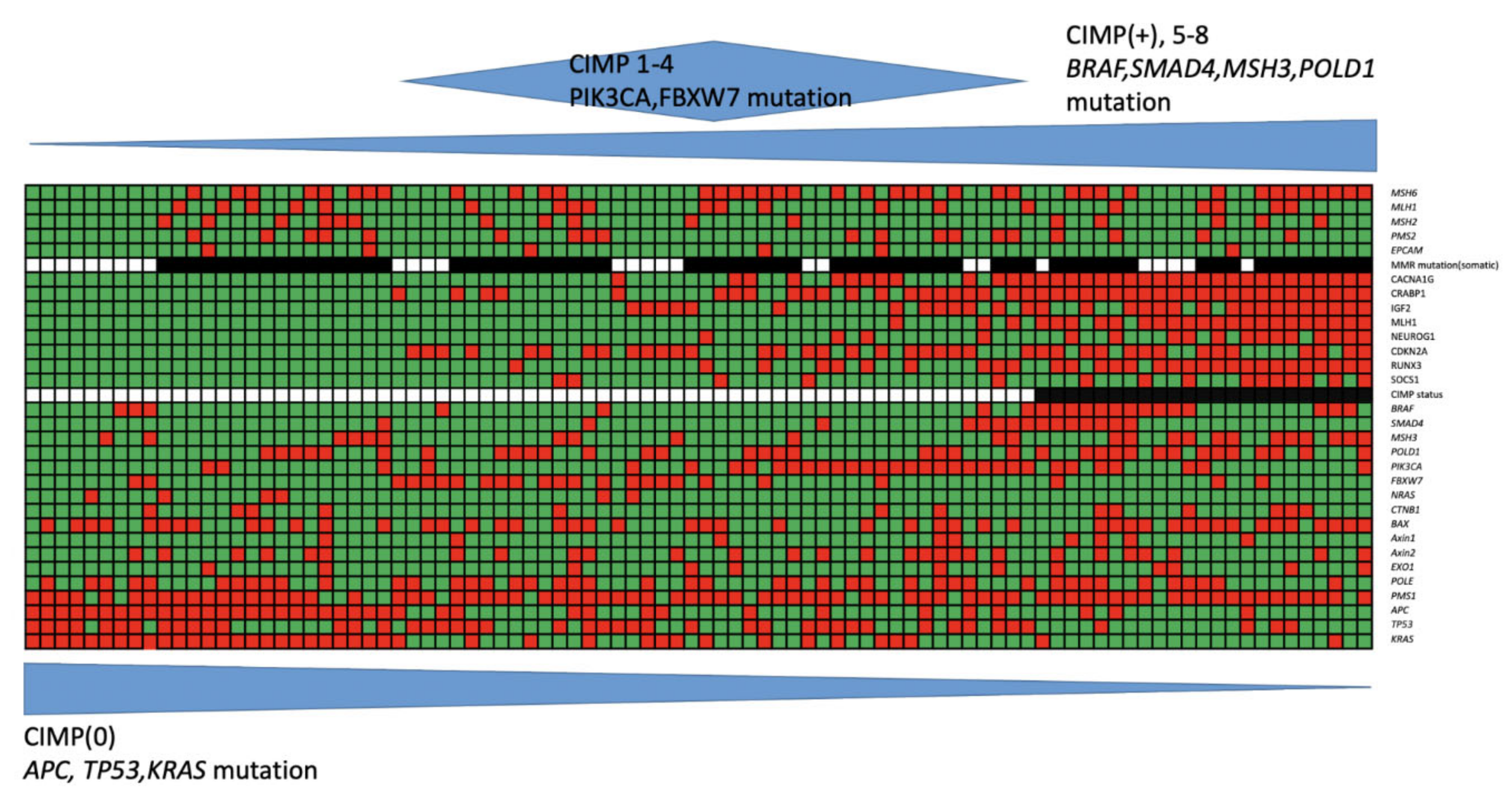{kind=link}
{kind=link}
| Variables | Total Patients n = 92 n (%) | CIMP-0 n = 25 n (%) | CIMP-Low n = 44 n (%) | CIMP-High n = 23 n (%) | p Value |
|---|---|---|---|---|---|
| Age | 69.0 ± 14.6 | 68.0 ± 15.2 | 68.4 ± 15.7 | 71.3 ± 12.1 | 0.690 |
| Gender | |||||
| Male | 49 (53.2) | 18 (72.0) | 25 (56.8) | 6 (26.1) | 0.005 |
| Female | 43 (57.6) | 7 (28.0) | 19 (43.2) | 17 (73.9) | |
| Location | |||||
| Proximal colon | 45 (48.9) | 9 (36.0) | 17 (38.6) | 19 (82.6) | 0.002 |
| Distal colon | 20 (21.7) | 7 (28.0) | 9 (20.5) | 4 (17.4) | |
| Rectum | 27 (29.3) | 9 (36.0) | 8 (40.9) | 0 | |
| TNM stage | |||||
| I | 11 (12.0) | 4 (16.0) | 4 (9.1) | 3 (13.0) | 0.415 |
| II | 48 (52.2) | 15 (60.0) | 20 (45.5) | 13 (56.5) | |
| III | 20 (21.7) | 5 (20.0) | 10 (22.7) | 5 (21.7) | |
| IV | 13 (14.1) | 1 (4.0) | 10 (22.7) | 2 (8.7) | |
| Lymphovascular invasion | |||||
| Positive | 14 (15.2) | 5 (20.0) | 6 (13.6) | 3 (13.0) | 0.736 |
| Negative | 78 (8.7) | 20 (80.0) | 38 (86.4) | 20 (87.0) | |
| Cell Differentiation | |||||
| Well/moderate | 77 (83.7) | 23 (92.0) | 39 (88.6) | 15 (65.2) | 0.020 |
| Poor | 15 (16.3) | 2 (8.0) | 5 (11.4) | 8 (34.8) | |
| Mucinous component | |||||
| ≥50% | 13 (14.1) | 3 (12.0) | 5 (11.4) | 5 (21.7) | 0.480 |
| <50% | 79 (85.9) | 22 (88.0) | 39 (88.6) | 18 (78.3) |
| Genetic Mutation | Total Patients n = 92 n (%) | CIMP-0 n = 25 n (%) | CIMP-Low n = 44 n (%) | CIMP-High n = 23 n (%) | p Value |
|---|---|---|---|---|---|
| APC | 40 (43.4) | 25 (100) | 12 (27.3) | 3 (13.0) | <0.001 |
| TP53 | 41 (44.6) | 25 (100) | 14 (31.8) | 2 (8.7) | <0.001 |
| KRAS | 45 (48.9) | 15 (60.0) | 26 (59.1) | 4 (17.4) | 0.001 |
| PIK3CA | 33 (35.9) | 3 (12.0) | 23 (52.3) | 7 (30.4) | 0.003 |
| FBXW7 | 24 (26.1) | 2 (8.0) | 19 (43.2) | 3 (13.0) | 0.002 |
| NRAS | 6 (7.5) | 4 (16.0) | 2 (4.5) | 0 | 0.062 |
| CTNB1 | 13 (14.1) | 4 (16.0) | 3 (6.8) | 6 (26.1) | 0.094 |
| BAX | 48 (52.2) | 13 (52.0) | 19 (43.2) | 16 (69.6) | 0.122 |
| AXIN1 | 10 (10.9) | 1 (4.0) | 4 (9.1) | 5 (21.7) | 0.125 |
| AXIN2 | 28 (30.4) | 6 (24.0) | 15 (34.1) | 7 (30.4) | 0.692 |
| EXO1 | 12 (13.0) | 2 (8.0) | 5 (11.4) | 5 (21.7) | 0.332 |
| POLE | 44 (47.8) | 11 (44.0) | 23 (52.3) | 10 (43.5) | 0.715 |
| PMS1 | 76 (82.7) | 19 (76.0) | 36 (81.8) | 21 (91.3) | 0.370 |
| BRAF | 21 (22.8) | 3 (12.0) | 4 (9.1) | 14 (60.9) | <0.001 |
| SMAD4 | 15 (16.3) | 1 (4.0) | 7 (15.9) | 7 (30.4) | 0.046 |
| MSH3 | 24 (26.1) | 6 (24.0) | 6 (13.6) | 12 (52.2) | 0.003 |
| POLD1 | 36 (39.1) | 6 (24.0) | 16 (36.4) | 14 (60.9) | 0.029 |
| Variables | MMR Mutation (+) n = 64 n (%) | MMR Mutation (−) n = 28 n (%) | p Value |
|---|---|---|---|
| Age (year) | 69.3 ± 14.9 | 68.4 ± 14.2 | 0.804 |
| Gender | |||
| Male | 36 (56.3) | 13 (46.4) | 0.496 |
| Female | 28 (43.8) | 15 (53.6) | |
| Location | |||
| Proximal colon | 33 (51.6) | 12 (42.4) | 0.739 |
| Distal colon | 13 (20.3) | 7 (20.3) | |
| Rectum | 18 (28.1) | 9 (32.1) | |
| TNM stage | |||
| I | 7 (10.9) | 4 (14.3) | 0.365 |
| II | 37 (57.8) | 11 (39.3) | |
| III | 13 (20.3) | 7 (25.0) | |
| IV | 7 (10.9) | 6 (21.4) | |
| Lymphovascular invasion | |||
| Positive | 7 (10.9) | 7 (25.0) | 0.115 |
| Negative | 57 (89.1) | 21 (75.0) | |
| Cell Differentiation | |||
| Well/moderate | 54 (84.4) | 23 (82.1) | 0.768 |
| Poor | 10 (15.6) | 5 (17.9) | |
| Mucinous component | |||
| ≥50% | 13 (20.3) | 0 | 0.008 |
| <50% | 51 (79.7) | 28 (100) |
| Genetic Mutation | MMR Mutation (+) n = 64 n (%) | MMR Mutation (−) n = 28 n (%) | p Value |
|---|---|---|---|
| APC | 24 (37.5) | 16 (57.1) | 0.110 |
| TP53 | 28 (43.8) | 17 (60.7) | 0.265 |
| KRAS | 41 (59.3) | 4 (17.4) | 0.175 |
| BRAF | 11 (17.2) | 10 (35.7) | 0.063 |
| SMAD4 | 11 (17.2) | 4 (14.3) | 1.000 |
| MSH3 | 19 (29.7) | 5 (17.9) | 0.306 |
| POLD1 | 30 (46.9) | 6 (21.4) | 0.036 |
| CTNB1 | 11 (1.2) | 2 (7.1) | 0.330 |
| BAX | 36 (56.3) | 12 (42.9) | 0.254 |
| AXIN1 | 9 (14.2) | 1 (3.6) | 0.273 |
| AXIN2 | 22 (34.4) | 6 (21.4) | 0.325 |
| FBXW7 | 14 (21.9) | 10 (35.7) | 0.200 |
| EXO1 | 10 (15.6) | 2 (7.1) | 0.333 |
| NRAS | 4 (6.3) | 2 (7.1) | 1.000 |
| POLE | 31 (48.4) | 13 (46.4) | 1.000 |
| PIK3CA | 26 (40.6) | 7 (25.0) | 0.166 |
| TGFBR2 | 64 (100) | 28 (100) | 1.000 |
| PMS1 | 53 (82.8) | 23 (82.1) | 1.000 |
| Factors | Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|---|
| Hazard Ratio | 95% CI | p Value | Hazard Ratio | 95% CI | p Value | |
| TNM stage | 3.39 | 2.17–5.30 | <0.001 | 4.22 | 2.245–7.923 | <0.001 |
| LVI | 3.49 | 1.54–7.91 | 0.003 | 2.30 | 0.927–5.691 | 0.073 |
| Mucinous histology | 0.44 | 0.13–1.47 | 0.182 | |||
| Poor differentiation | 1.98 | 0.85–4.61 | 0.115 | |||
| CIMP status | 0.99 | 0.44–2.22 | 0.982 | |||
| Somatic MMR mutation | 0.69 | 0.33–1.44 | 0.320 | |||
| Adjuvant chemotherapy | 2.80 | 1.36–5.76 | 0.005 | 0.49 | 0.166–1.445 | 0.196 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, S.-C.; Li, A.F.-Y.; Lin, P.-C.; Lin, C.-C.; Lin, H.-H.; Huang, S.-C.; Lin, C.-H.; Liang, W.-Y.; Chen, W.-S.; Jiang, J.-K.; et al. Clinicopathological and Molecular Profiles of Sporadic Microsatellite Unstable Colorectal Cancer with or without the CpG Island Methylator Phenotype (CIMP). Cancers 2020, 12, 3487. https://doi.org/10.3390/cancers12113487
Chang S-C, Li AF-Y, Lin P-C, Lin C-C, Lin H-H, Huang S-C, Lin C-H, Liang W-Y, Chen W-S, Jiang J-K, et al. Clinicopathological and Molecular Profiles of Sporadic Microsatellite Unstable Colorectal Cancer with or without the CpG Island Methylator Phenotype (CIMP). Cancers. 2020; 12(11):3487. https://doi.org/10.3390/cancers12113487
Chicago/Turabian StyleChang, Shih-Ching, Anna Fen-Yau Li, Pei-Ching Lin, Chun-Chi Lin, Hung-Hsin Lin, Shen-Chieh Huang, Chien-Hsing Lin, Wen-Yi Liang, Wei-Shone Chen, Jeng-Kai Jiang, and et al. 2020. "Clinicopathological and Molecular Profiles of Sporadic Microsatellite Unstable Colorectal Cancer with or without the CpG Island Methylator Phenotype (CIMP)" Cancers 12, no. 11: 3487. https://doi.org/10.3390/cancers12113487