1. Introduction
Pancreatic cancer is the 12th most common cancer and is highly lethal. The overall 5-year survival rate across all stages of pancreatic adenocarcinoma (PAAD) is 9%, although only 3% of patients with metastatic disease are alive after 5 years. PAAD is usually detected at an advanced stage, and most treatment regimens are ineffective, while current interventions to prevent, diagnose, and treat are not satisfactory, leading to the poor overall prognosis [
1,
2,
3,
4,
5].
Pancreatic cancer is characterized by frequent mutations in KRAS, TP53, CDKN2A, and SMAD4, as well as the dysregulation of diverse signaling pathways, including TGF-β, Wnt/Notch, and hedgehog signaling [
3,
6,
7]. Other unique features of pancreatic cancer are the presence of a microenvironment filled with immunosuppressive mediators and a dense stroma. Stromal cells promote tumor growth, invasion, metastasis, and chemoresistance. Acquired immune evasion, which is comprised of an immunosuppressive microenvironment, poor T cell infiltration, and a low mutational burden, is a dynamic entity connected to immune system control. Based on the special pathological traits of pancreatic cancer, novel strategies such as stoma-targeting therapy, immunotherapy, and neoantigen vaccines are emerging as treatments. However, these therapies face the challenge of overcoming the highly immunosuppressive tumor microenvironment in pancreatic cancer [
7,
8,
9].
A novel component in shaping the immune system and affecting disease prognosis for pancreatic cancer is the microbiome. The microbiome is defined as the comprehensive genomic information encoded by the microbiota (bacteria, fungi, protozoa, and viruses) and its ecosystem, products, and host environment. The human microbiota offer protection from disease by maintaining nutrition and hormonal homeostasis, modulating inflammation, and detoxifying compounds [
10,
11]. An intra-tumoral microbiome signature was identified as predictive of the long-term survival of pancreatic cancer [
12]. Recently, the microbiota has begun to be recognized as an important player in cancers. For example, the gut microbiota may act as a mediator of immune system activation, promoting cancer-associated inflammation and affecting tumor responses to therapies in multiple cancers [
13]. For pancreatic cancer, studies demonstrated that the neoantigens of pancreatic cancer showed homology to microbial epitopes, suggesting that microbial factors may determine tumor behavior and patient outcomes [
14]. On the other hand, the gut microbiome from a short-term survivor of pancreatic cancer promoted pancreatic tumor growth in a mouse model via increasing the infiltration of CD4+FOXP3+ and myeloid-derived suppressor cells. These studies suggest that both the intra-pancreatic microbiome and the gut microbiome may be implicated in the immunosuppression and development of PAAD [
12,
15].
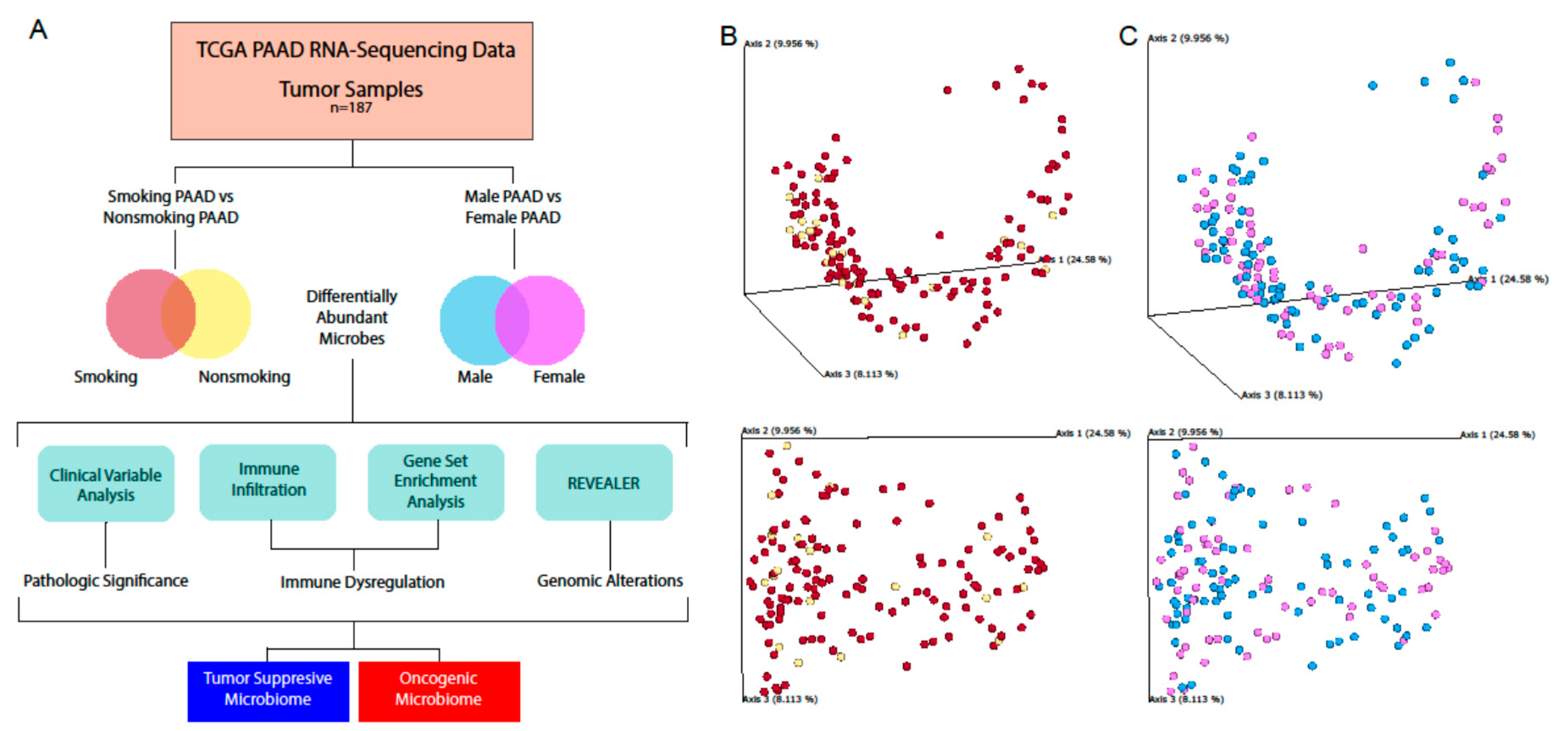
In this study, we aimed to expand our current understanding of the relationship between PAAD and its intratumoral microbiome, using next-generation RNA-sequencing data from 187 PAAD patients. We identified microbes whose abundance was correlated with poor survival and metastasis. We investigated correlations between microbe abundance and the expression of cancer and immune-associated genes. Due to the importance of smoking and gender in PAAD progression, we identified microbes differentially abundant in males versus females and in smokers versus nonsmokers, and analyzed how these microbes correlated with cancer and immune pathways. We observed significant correlations between a high abundance of certain microbes and poor prognosis/immunosuppression. While previous studies have demonstrated connections between PAAD pathogenesis and microbial abundance in mice, our study performed the largest profiling of the intra-pancreatic microbiome in sample size, and we are the first to demonstrate immune and clinical variable associations with microbial abundance in human PAAD samples, to the best of our knowledge.
3. Discussion
Pancreatic cancer has been demonstrated to interact with the intratumor microbiome in multiple recent studies [
12,
15], providing significant evidence of the existence of an intra-pancreatic microbiome that was previously unknown only a few years ago. Since the discovery of this microbiome, several studies in the past two years have attempted to characterize its origins and relationship to diseases. It has been suggested that the microbiota from the stomach, duodenum, biliary tract, and even esophagus could have gained access to the pancreas through the pancreatic duct [
5,
15]. However, studies have not agreed on various issues, including what the most likely route of pancreatic bacterial colonization is, whether the pancreas can be colonized under healthy conditions, and the profile of normal pancreatic flora [
5,
17]. Furthermore, many studies were performed in mice, which may not be relevant to human physiology. In this study, we profiled the intra-tumor pancreatic microbiome through large-scale sequencing data from TCGA, providing the largest comprehensive profiling of microbes in pancreatic tumor samples to date, to the best of our knowledge. Given the known influence of the intra-tumor microbiome on the development of cancers, the microbiota in the pancreas could be a key factor contributing to pancreatic cancer pathogenesis and progression.
The pancreatic microbiota have been associated with pancreatic ductal adenocarcinoma in only four studies. In Pushalkar et. al., mice experiments provided evidence for the existence of a pancreatic microbiome, and the presence of microbes promoted immunosuppression [
12]. The elimination of microbes protected against invasive pancreatic cancer in mice. However, the results were validated using only 12 human pancreatic cancer samples. In Riquelme et. al., 68 pancreatic tumor samples were profiled, but no normal pancreas samples were examined [
15]. In Thomas et. al., 16 pancreatic cancer samples and 7 normal pancreatic samples were examined [
17]. However, they were not able to find microbiome composition differences between the cancer and normal samples. In Geller et. al., 65 pancreatic tumor samples were profiled [
18]. While some of these studies profiled a large number of samples, none of the four studies investigated correlations between bacterial populations and gene expression dysregulations through mRNA sequencing, and none correlated bacterial abundance to clinical variables and immune phenotypes. These studies also yielded contradictory results. In this study, we extracted microbe reads from 187 pancreatic cancer samples and examined the relationship among microbe abundance, immunological changes, and gene expression signatures in an attempt to utilize this unprecedented large-scale analysis to address the myriad outstanding questions regarding the microbiome’s role in pancreatic cancer.
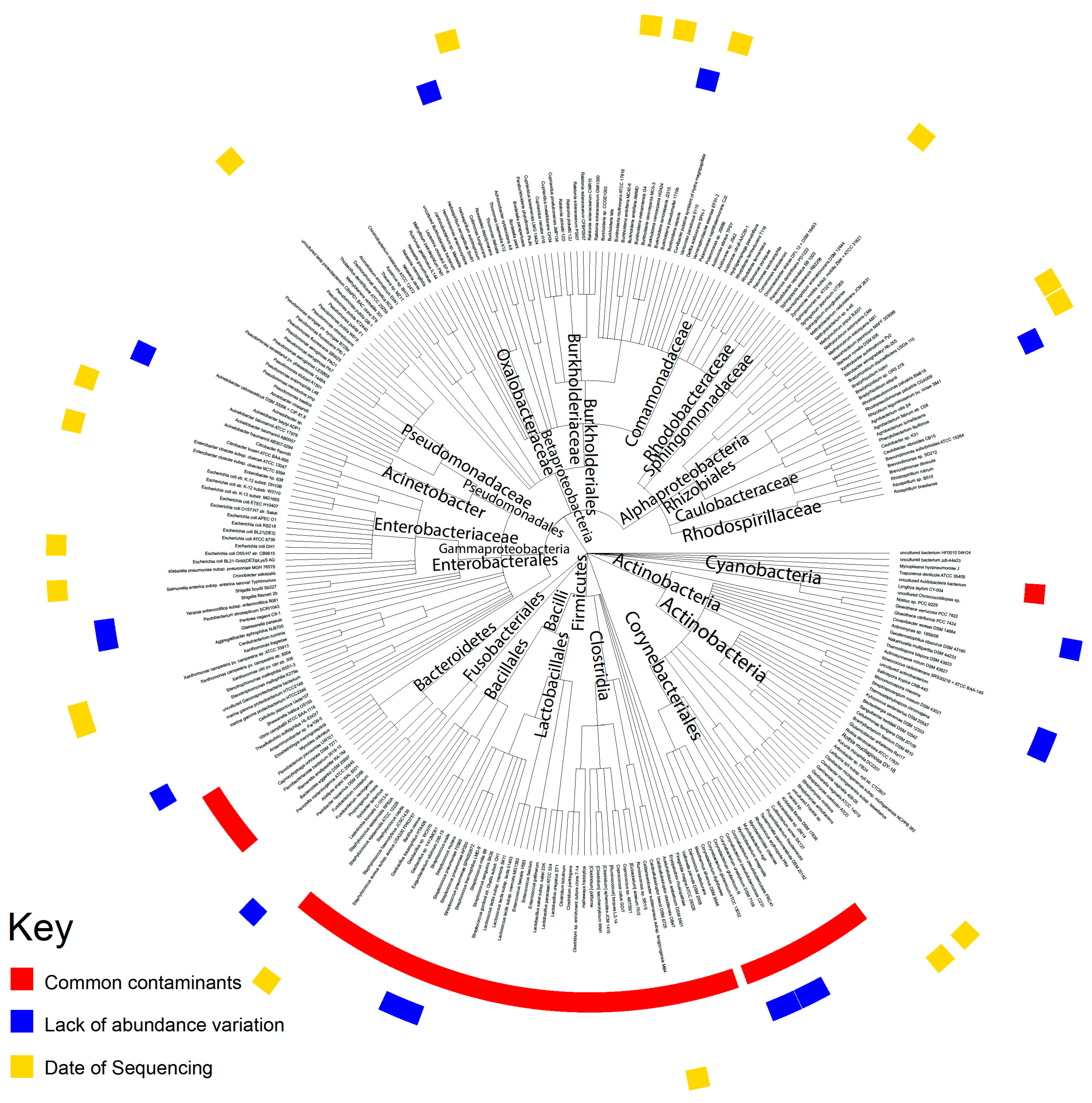
One of the most important questions surrounding the detection of microbes in pancreatic tissue is whether the detected microbes were contaminants from the environment during sample processing. Through three different analyses, we established that most of the microbes we discovered in our data were not contaminants, although we did identify some potential contaminants. While TCGA does not explicitly control for contamination in sample collection procedures, previous studies extracting microbe abundance from TCGA samples generally concluded that tissue-intrinsic and biologically relevant microbes could be extracted from TCGA data given the rigorous identification of contaminants [
19,
20].
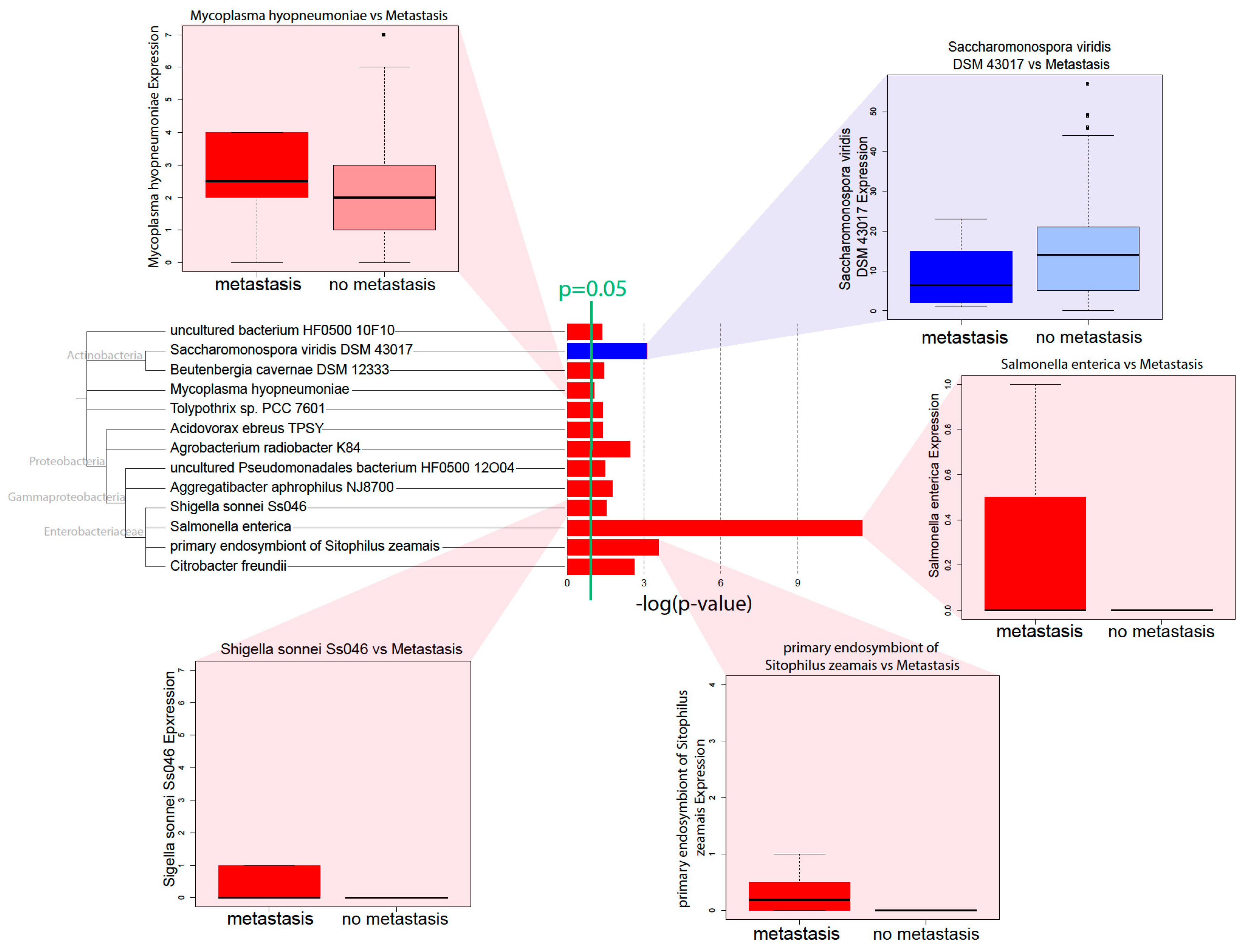
Through correlations with clinical variables, we identified that a high microbial abundance of a number of bacterial species is correlated with poor prognosis, in terms of metastasis and survival. Microbe abundance is also generally associated with the high expression of methylation-related gene expression signatures, suggesting that microbe abundance may be associated with increased methylation. In accordance with Pushalkar et al., we found that a high microbial abundance was associated with immunosuppression, which includes the low infiltration of M2 macrophages and T-cells. High microbe abundance was also correlated with the activation of oncogenic pathways and downregulation of tumor suppressive pathways, possibly creating a pro-tumor microenvironment.
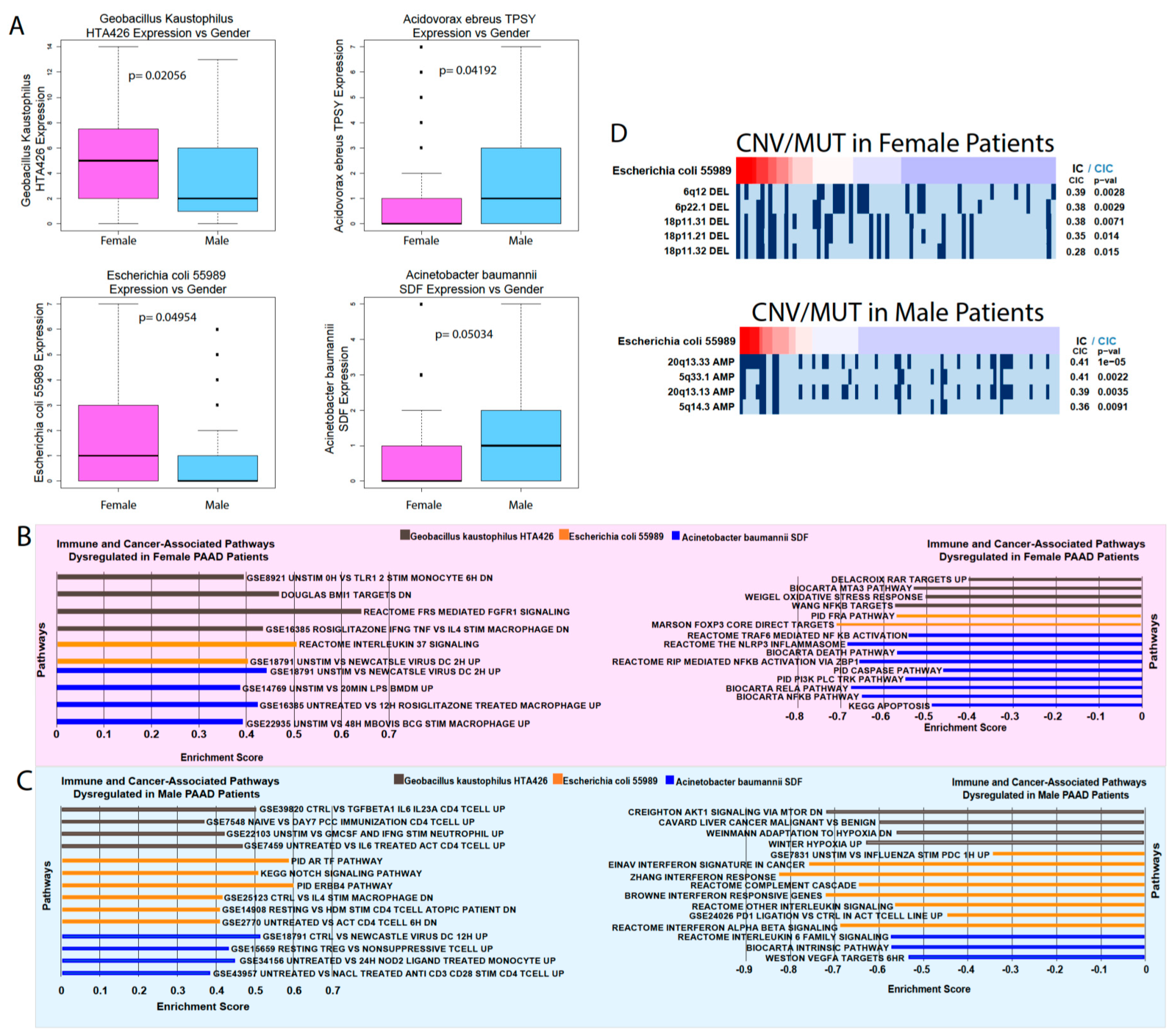
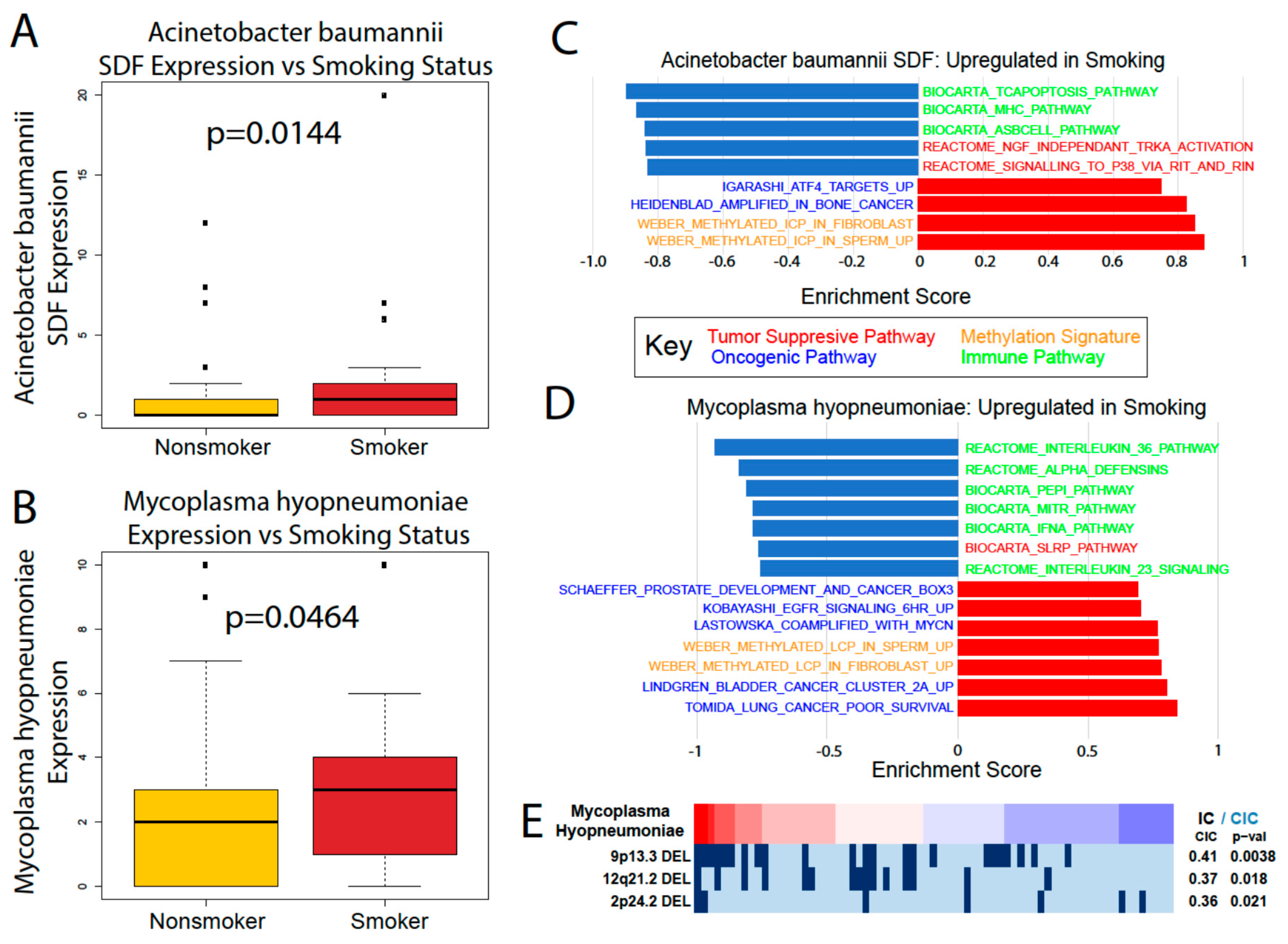
We were also able to find significant differences in microbe abundance between smokers and nonsmokers and between male and female patients. Smoking is a significant risk factor for pancreatic cancer that increases its risk by 75%, with a persistent increased risk even 10 years after quitting [
21]. Gender is also a risk factor, with males being slightly more likely to develop pancreatic cancer than females, although it is unclear if this trend is due to the greater prevalence of smoking among men [
21]. While we found provocative correlations between smoking/gender-associated microbes’ abundance and immune and cancer-associated pathways, more in vitro and in vivo experiments are required to elucidate the role of these microbes and their relationship to gender and smoking.
The most prominent microbes associated with clinical variables and immune pathways in pancreatic cancer are mostly from the phylum
Proteobacteria.
Acidovorax ebreus, a
Betaproteobacteria, was correlated with poor prognosis and decreased immune infiltration. Members of the class
Gammaproteobacteria are especially featured in our correlations. Their abundance correlates positively with increasing metastasis more than the abundance of any other class of microbes.
C. freundii is associated with poor prognosis and the dysregulation of multiple immune and cancer-associated pathways.
Pseudomonadales are associated with inflammasome activation and poor prognosis.
S. sonnei abundance is associated with poor prognosis and the upregulation of cancer-associated pathways. Both
C. feundii and
S. sonnei are in the
Enterobacteriaceae family and are known to be present in the human gut, sometimes as pathogens [
22], suggesting that pancreatic bacteria are possibly translocated from the gut. Our results corroborate previous findings that show that
Proteobacteria is the dominant bacterial species in pancreatic cancer, comprising almost half of all pancreatic bacteria [
12]. It has been hypothesized that
Gammaproteobacteria in the pancreas could promote chemotherapy resistance to the drug gemcitabine by metabolizing it [
18], and our study is the first to demonstrate a concrete correlation between
Gammaproteobacteria levels and pancreatic cancer prognosis.
5. Materials and Methods
5.1. Data Acquisition from TCGA
5.2. Extraction of Microbial Reads and Calculation of Microbial Abundance
Using the Pathoscope 2.0 program [
23], the RNA-sequencing data were filtered for bacterial reads via direct alignment through a wrapper for Bowtie2. This framework utilizes a reference library to select for reads unique to organisms of interest. For this analysis, bacterial sequences deposited at the NCBI nucleotide database (
https://www.ncbi.nlm.nih.gov/nucleotide/) were used as a reference library. Pathoscope generates two output measures quantifying the amount of bacterial species present in samples. One measure, best guess, quantifies the relative abundance of each species, expressed as a percentage. The other measure, best hit read numbers, signifies the absolute integer count of each species in the sequencing data.
5.3. Evaluation of Contamination Using Date of Sequencing
We applied a heuristic algorithm to extract the sequencing dates where this overexpression occurs, which allowed us to determine potential contaminants’ relationship with the sequencing date. We visualized the microbial abundance of cancer patients in the form of a heat map and removed any microbe where stretches of dates with high microbial abundance exist, which we identified as contamination. In other words, contaminants are marked by a non-uniform abundance across sequencing dates. For all the following analyses, we removed all the microbes that were identified as contaminants.
5.4. Evaluation of Contamination Based on Plates
The TCGA sequencing protocol includes the collection of tissue samples from multiple sites at a common sequencing center. Tissue samples are then sequenced on the same plates at such common centers. The information about which patient samples was sequenced on which plate is publicly available via the Broad Institute’s GDAC Firehose. We used this resource to group patients based on common sequencing plate IDs. The abundance values of microbes were associated with plates on which the samples were stored prior to sequencing using the Kruskal–Wallis test (p < 0.05) and the visual examination of abundance differences between different plates using a boxplot.
5.5. Evaluation of Contamination Using Microbial Abundance Counts
The abundance of individual microbes in each patient is plotted against the total microbe reads in the same patient to determine whether any microbe is likely a contaminant. The best hit results from Pathoscope are used for this analysis because absolute counts are required. In the resulting scatterplots, if a positive slope exists it is likely that the microbe was biologically relevant and physically present in the sample, since the counts per microbe increased with the number of microbes sequenced. If the scatterplot has a slope of close to zero and the counts of all the microbes are substantially above zero, it is likely that the microbe was a contaminant. This reasoning follows from the assumption that similar amounts of microbes will be present regardless of how many microbes are present in the tissue sample if the microbe is an environmental contaminant. The Spearman correlation test and the correlation coefficient (R2) were used to calculate the significance of a linear trendline and the slope of that trendline, respectively.
5.6. Evaluation of Contamination Using Previously Identified Contaminants
A list of phyla that have been previously determined to be present in DNA sequencing kits was obtained from a study by Glassing et al. [
24]. A list of phyla that are common in the hospital setting was obtained from a study by Rampelotto et al. [
25]. These two lists per used to identify bacteria as common contaminants.
5.7. Determination of Microbiome Diversity in Patient Samples
Using the Qiime2 framework, the best guess data output from Pathoscope were used to calculate the alpha diversity and beta diversity using the qiime diversity alpha and qimme diversity beta modules, respectively. Principle component analysis of the beta diversity results was performed via the qiime diversity pcoa module and visualized using the qiime emperor plot module, the latter of which uses the EMPeror tool.
5.8. Differential Microbial Abundance between Cancer Patients of Different Smoking Status and Gender
A differential abundance analysis was performed to compare the microbe abundance (percent abundance) in cancer tissues based on male vs. female and smoking vs. nonsmoking comparisons. Microbes that are present in less than 10 percent of the patients in a cancer cohort were excluded. The Kruskal–Walls analysis test was then applied to determine the differential abundance (p < 0.05).
5.9. Correlation of Microbial Abundance to Survival and Clinical Variables
Survival analyses were performed while using the Kaplan–Meier Model, with microbe expression being designated as a binary variable based on the presence or absence of microbes in tumor samples. Univariate Cox regression analysis was used to identify candidates that were significantly associated with patient survival (p < 0.05). Clinical variable analysis was performed using the Kruskal–Wallis test, as described above.
5.10. Correlation of Microbial Abundance to Immune Infiltration
The estimated relative immune cell infiltration levels for 22 cell types were computed using the software CibersortX [
26]. The microbe abundance was then correlated with the immune cell infiltration levels for each microbe using the Kruskal–Wallis test (
p < 0.05). Microbe abundance was modeled as a binary variable of presence and absence. The immune cell types examined include naïve B-cells, memory B-cells, plasma cells, CD8 T-cells, CD4 naïve T-cells, CD4 memory resting T-cells, CD4 memory activated T-cells, follicular helper T-cells, regulatory T-cells, gamma-delta T-cells, resting NK cells, activated NK cells, monocytes, M0-M2 macrophages, resting dendritic cells, activated dendritic cells, resting mast cells, activated mast cells, eosinophils, and neutrophils.
5.11. Correlation of Microbial Abundance to Cancer and Immune-associates Signatures
The signature enrichment corresponding to microbial abundance was measured using the Geneset Enrichment Analysis (GSEA). Cancer and stem cell-associated signatures were chosen from the C6 set of signatures from the Molecular Signatures Database (MSigDB) [
27]. Immune-associated signatures were chosen from the C7 set of signatures. Significantly enriched signatures were identified by a nominal enrichment score > 1 and a nominal
p-value < 0.05. Canonical (CP) pathways were also included from the C2 set of signatures. The direction of pathway enrichment was filtered to match the direction of clinical variable correlations per microbe.
5.12. Correlation of Microbial Abundance to Copy Number Variations and Mutations
The copy number variation (CNV) and mutation data were obtained from annotation files generated by the BROAD Institute GDAC Firehose on March 31, 2018. The surface-level trends of mutation presence were analyzed by calculating the percentage of patients with each mutation, indicated by a binary value per mutation. The GDAC files were compiled into input files for the repeated evaluation of variables conditional entropy and redundancy (REVEALER) algorithm, which identifies sets of specific CNVs and mutations that are most likely implicated in changes to the target expression profile. The target profile was identified as the expression of a single immune-associated gene. The REVEALER algorithm runs in multiple iterations in order to identify the most prominent genomic alterations. For our study, we set the maximum number of iterations to three. The algorithm also allows the use of a seed, or a particular mutation of a CNV gain or loss event that may account for target activity. However, because we did not know the individual genetic alterations that were responsible for each IA gene dysregulation, the seed was set to null. Significant correlations were indicated by p < 0.05 and CIC > 0.03.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





