The Role of PPARγ Ligands in Breast Cancer: From Basic Research to Clinical Studies

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Search Strategy and Data Extraction
3. Peroxisome Proliferator-Activated Receptor γ: Structure, Distribution, and Mechanism of Action
3.1. Genomic Organization and Tissue Distribution of PPARγ
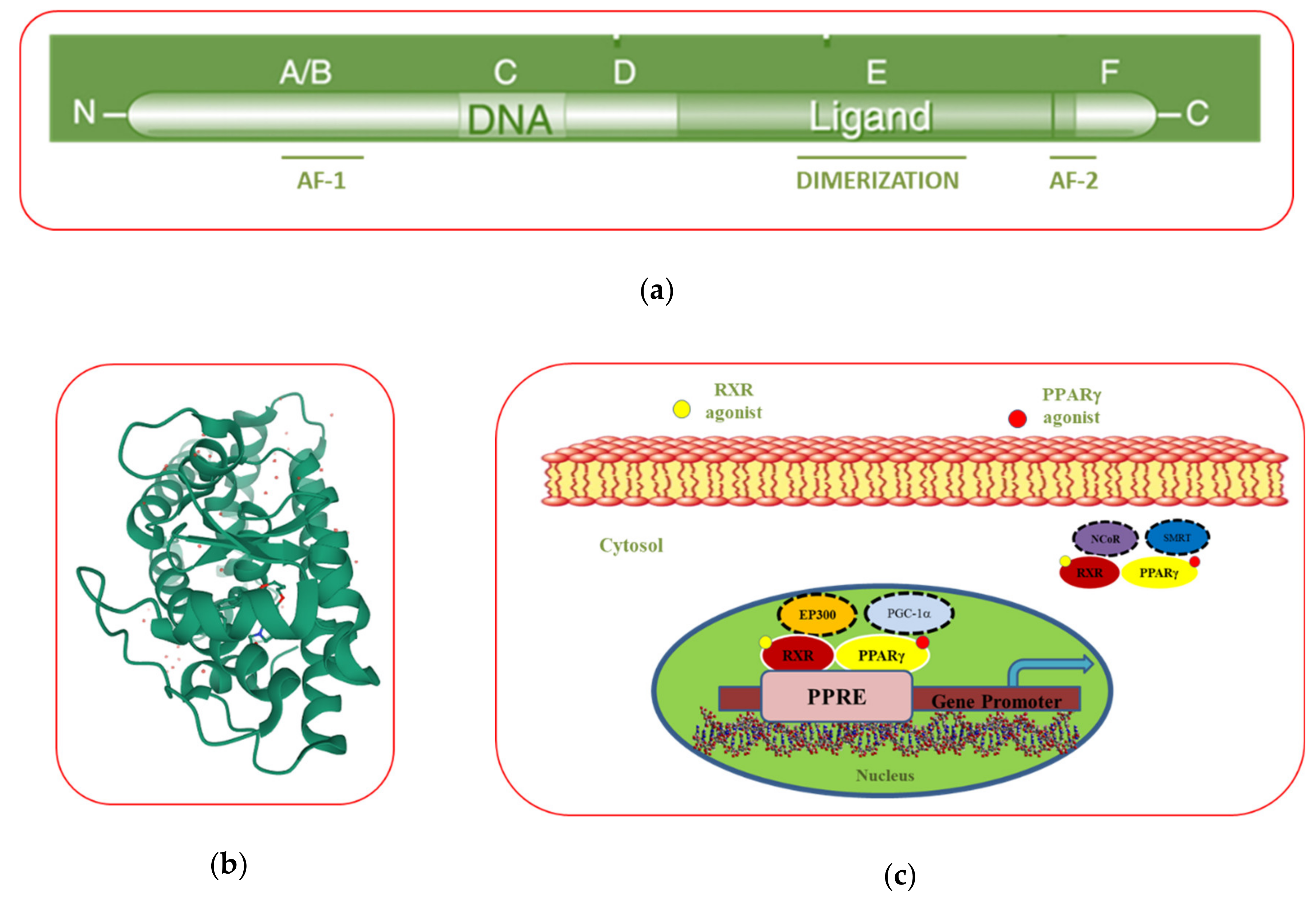
3.2. PPARγ Domain Structure
3.3. The Mechanism of Action of PPARγ
4. PPARγ Agonists
4.1. Natural PPARγ Ligands
4.2. Synthetic PPARγ Ligands
5. The Role of the PPARγ Ligands in Breast Cancer
5.1. In Vitro Studies
5.1.1. Regulation of Cell Growth and Cell Cycle
5.1.2. Regulation of Cell Death
Apoptosis
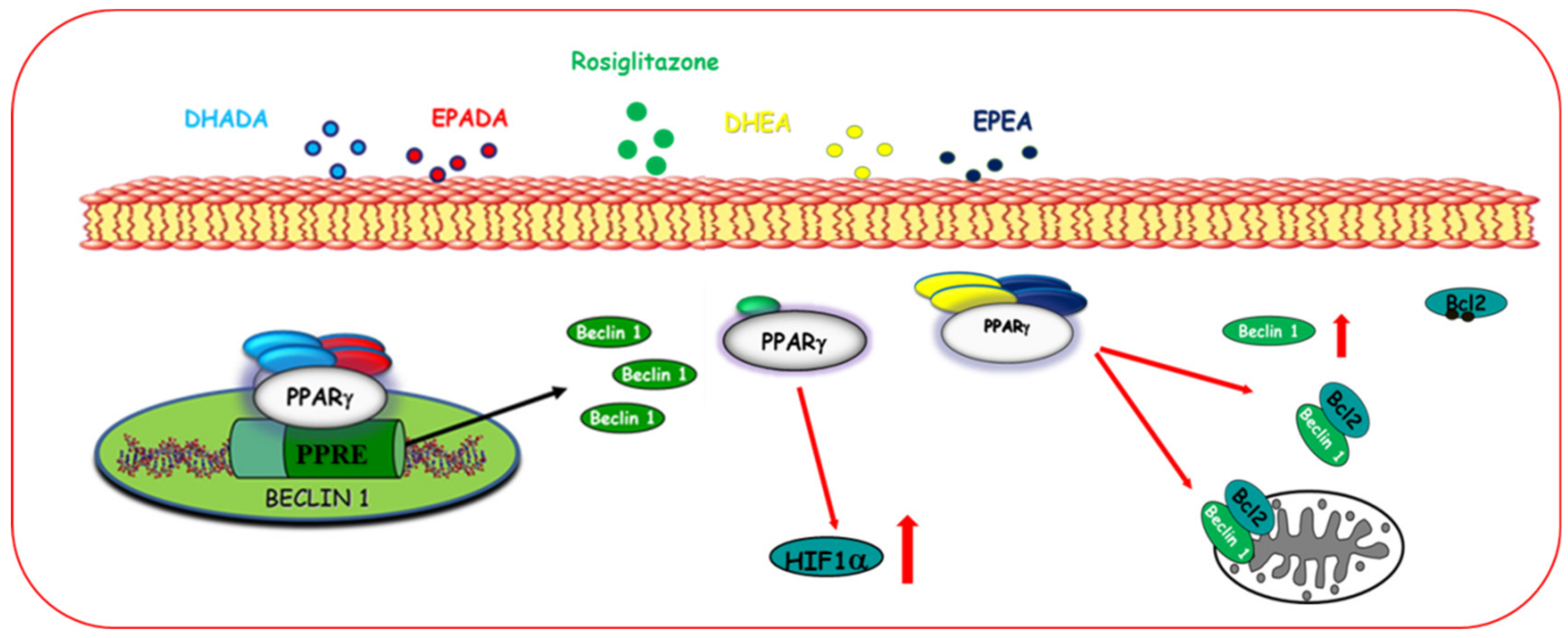
Autophagy
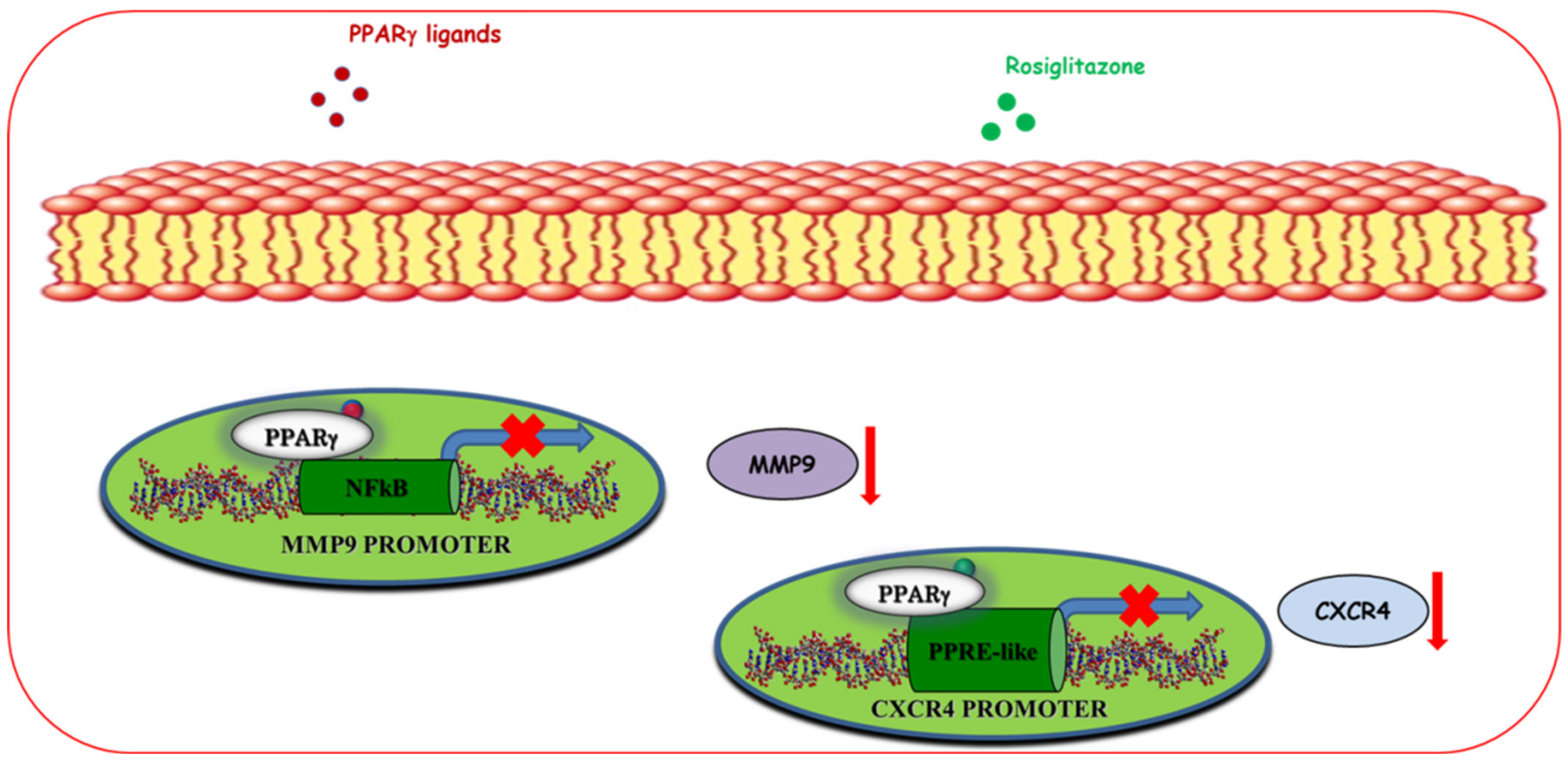
5.1.3. Regulation of Motility and Invasion
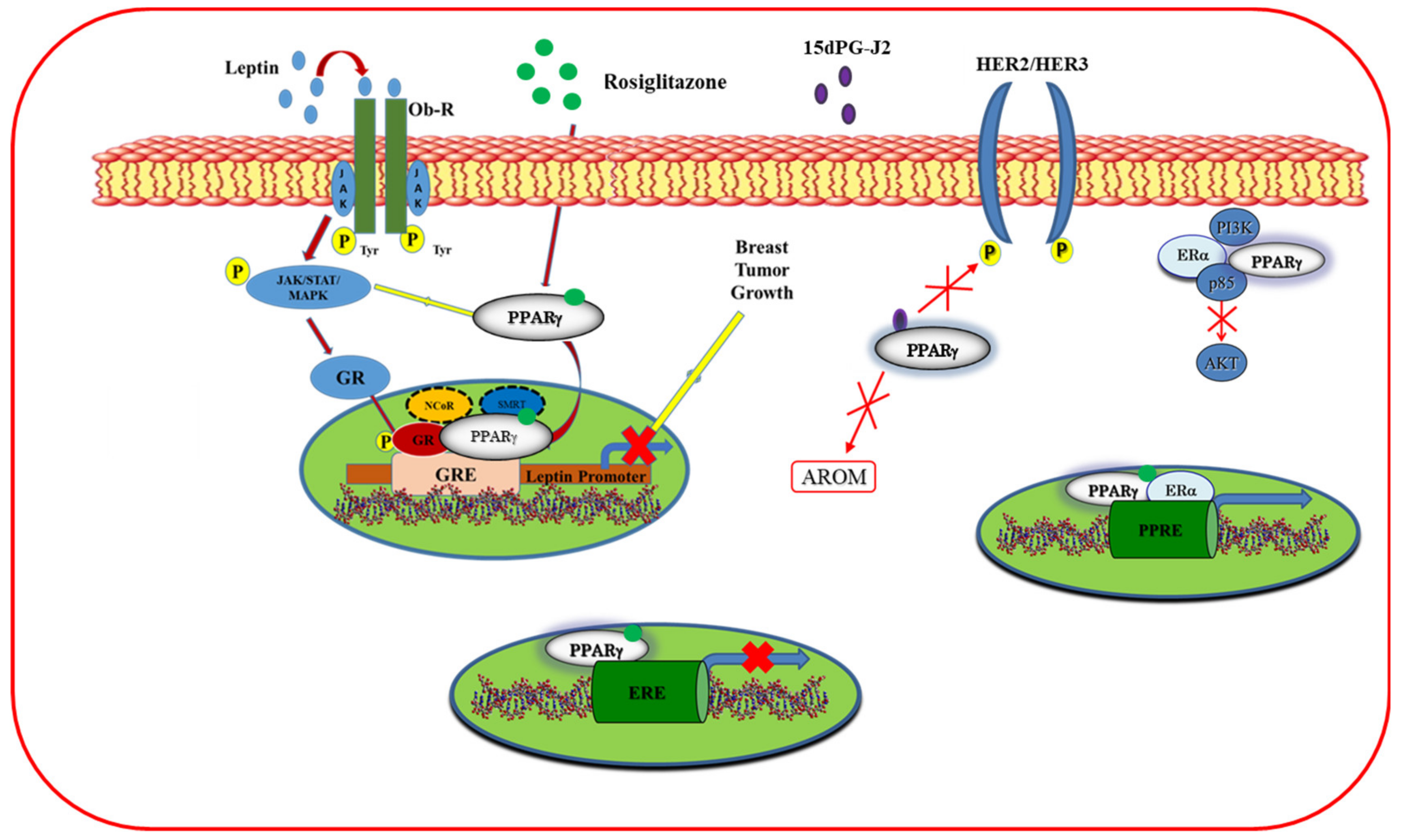
5.1.4. Cross-Talk of PPARγ with Other Signal Transduction Pathways
5.2. In Vivo Studies
5.3. Clinical Studies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Islami, F.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer in women: Burden and trends. Cancer Epidemiol. Biomark. Prev. 2017, 26, 444–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-H.; Jeon, J.; Kim, S.I. Personalized Medicine in Breast Cancer: A Systematic Review. J. Breast Cancer 2012, 15, 265–272. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 5–66. [Google Scholar] [CrossRef]
- Anampa, J.; Makower, D.; Sparano, J.A. Progress in adjuvant chemotherapy for breast cancer: An overview. BMC Med. 2015, 13, 195. [Google Scholar] [CrossRef] [Green Version]
- Weigelt, B.; Peterse, J.L.; Veer, L.J.V. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef]
- Chang-Qing, Y.; Jie, L.; Shi-Qi, Z.; Kun, Z.; Zi-Qian, G.; Ran, X.; Hui-Meng, L.; Ren-Bin, Z.; Gang, Z.; Yin, D.-C.; et al. Recent treatment progress of triple negative breast cancer. Prog. Biophys. Mol. Boil. 2020, 151, 40–53. [Google Scholar] [CrossRef]
- Alasmael, N.; Mohan, R.; Meira, L.; Swales, K.E.; Plant, N. Activation of the Farnesoid X-receptor in breast cancer cell lines results in cytotoxicity but not increased migration potential. Cancer Lett. 2016, 370, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Garattini, E.; Bolis, M.; Garattini, S.K.; Fratelli, M.; Centritto, F.; Paroni, G.; Gianni’, M.; Zanetti, A.; Pagani, A.; Fisher, J.N.; et al. Retinoids and breast cancer: From basic studies to the clinic and back again. Cancer Treat. Rev. 2014, 40, 739–749. [Google Scholar] [CrossRef]
- Murray, A.; Madden, S.F.; Synnott, N.C.; Klinger, R.; O’Connor, D.P.; O’Donovan, N.; Gallagher, W.M.; Crown, J.; Duffy, M.J. Vitamin D receptor as a target for breast cancer therapy. Endocr.-Relat. Cancer 2017, 24, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Catalano, S.; Panza, S.; Vizza, N.; Barone, I.; Bonofiglio, D.; Gelsomino, L.; Rizza, P.; Fuqua, S.A.W.; Andò, S. Farnesoid X receptor inhibits tamoxifen-resistant MCF-7 breast cancer cell growth through downregulation of HER2 expression. Oncogene 2011, 30, 4129–4140. [Google Scholar] [CrossRef] [Green Version]
- Moseti, D.; Regassa, A.; Kim, W.K. Molecular Regulation of Adipogenesis and Potential Anti-Adipogenic Bioactive Molecules. Int. J. Mol. Sci. 2016, 17, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotta-Loizou, I.; Giaginis, C.; Theocharis, S. The role of peroxisome proliferator-activated receptor-γ in breast cancer. Anti-Cancer Agents Med. Chem. 2012, 12, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, I.; Mylona, E.; Giannopoulou, I.; Markaki, S.; Keramopoulos, A.; Nakopoulou, L. PPAR? expression in breast cancer: Clinical value and correlation with ER? Histopathology 2005, 46, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zou, L.; Zhang, C.; He, S.; Cheng, C.; Xu, J.; Lu, W.; Zhang, Y.; Zhang, H.; Wang, D.; et al. PPARγ and Wnt/β-Catenin pathway in human breast cancer: Expression pattern, molecular interaction and clinical/prognostic correlations. J. Cancer Res. Clin. Oncol. 2009, 135, 1551–1559. [Google Scholar] [CrossRef]
- Abduljabbar, R.; Alkaabi, M.; Negm, O.H.; Jerjees, D.; Muftah, A.A.; Mukherjee, A.; Lai, C.F.; Buluwela, L.; Ali, S.; Tighe, P.J.; et al. Prognostic and biological significance of peroxisome proliferator-activated receptor-gamma in luminal breast cancer. Breast Cancer Res. Treat. 2015, 150, 511–522. [Google Scholar] [CrossRef]
- Chou, F.-S.; Wang, P.-S.; Kulp, S.; Pinzone, J.J. Effects of Thiazolidinediones on Differentiation, Proliferation, and Apoptosis. Mol. Cancer Res. 2007, 5, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Burstein, H.J.; Demetri, G.D.; Mueller, E.; Sarraf, P.; Spiegelman, B.M.; Winer, E.P. Use of the Peroxisome Proliferator-Activated Receptor (PPAR) γ Ligand Troglitazone as Treatment for Refractory Breast Cancer: A Phase II Study. Breast Cancer Res. Treat. 2003, 79, 391–397. [Google Scholar] [CrossRef]
- Fabian, C.J.; Kimler, B.F.; Hursting, S.D. Omega-3 fatty acids for breast cancer prevention and survivorship. Breast Cancer Res. 2015, 17, 62. [Google Scholar] [CrossRef] [Green Version]
- Rovito, D.; Giordano, C.; Vizza, D.; Plastina, P.; Barone, I.; Casaburi, I.; Lanzino, M.; De Amicis, F.; Sisci, D.; Mauro, L.; et al. Omega-3 PUFA ethanolamides DHEA and EPEA induce autophagy through PPARγ activation in MCF-7 breast cancer cells. J. Cell. Physiol. 2013, 228, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Berquin, I.M.; Owens, R.T.; O’Flaherty, J.T.; Edwards, I.J. Peroxisome proliferator-activated receptor gamma-mediated up-regulation of syndecan-1 by n-3 fatty acids promotes apoptosis of human breast cancer cells. Cancer Res. 2008, 68, 2912–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, J.P.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajas, L.; Fruchart, J.-C.; Auwerx, J. PPARγ3 mRNA: A distinct PPARγ mRNA subtype transcribed from an independent promoter. FEBS Lett. 1998, 438, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Beamer, B.A.; Negri, C.; Yen, C.-J.; Gavrilova, O.; Rumberger, J.M.; Durcan, M.J.; Yarnall, D.P.; Hawkins, A.L.; Griffin, C.A.; Burns, D.K.; et al. Chromosomal Localization and Partial Genomic Structure of the Human Peroxisome Proliferator Activated Receptor-Gamma (hPPARγ) Gene. Biochem. Biophys. Res. Commun. 1997, 233, 756–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janani, C.; Kumari, B.R. PPAR gamma gene–A review. Diabetes Metab. Syndr. Clin. Res. Rev. 2015, 9, 46–50. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, W.-H. Peroxisome proliferator-activated receptor γ and colorectal cancer. World J. Gastrointest. Oncol. 2010, 2, 159–164. [Google Scholar] [CrossRef]
- Papi, A.; De Carolis, S.; Bertoni, S.; Storci, G.; Sceberras, V.; Santini, N.; Ceccarelli, C.; Taffurelli, M.; Orlandi, M.; Bonafè, M. PPARγ and RXR Ligands Disrupt the Inflammatory Cross-talk in the Hypoxic Breast Cancer Stem Cells Niche. J. Cell. Physiol. 2014, 229, 1595–1606. [Google Scholar] [CrossRef]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.-P.; Verhoeyen, E.; Raggueneau, V.; Manéglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef]
- Gionfriddo, G.; Plastina, P.; Augimeri, G.; Catalano, S.; Giordano, C.; Barone, I.; Morelli, C.; Giordano, F.; Gelsomino, L.; Sisci, D.; et al. Modulating Tumor-Associated Macrophage Polarization by Synthetic and Natural PPARγ Ligands as a Potential Target in Breast Cancer. Cells 2020, 9, 174. [Google Scholar] [CrossRef] [Green Version]
- Quintão, N.L.M.; Santin, J.R.; Stoeberl, L.C.; Corrêa, T.P.; Melato, J.; Costa, R. Pharmacological Treatment of Chemotherapy-Induced Neuropathic Pain: PPARγ Agonists as a Promising Tool. Front. Mol. Neurosci. 2019, 13, 907. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Breyer, M.D. Peroxisome proliferator-activated receptors (PPARs): Novel therapeutic targets in renal disease. Kidney Int. 2001, 60, 14–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houseknecht, K.L.; Cole, B.M.; Steele, P.J. Peroxisome proliferator-activated receptor gamma (PPARγ) and its ligands: A review. Domest. Anim. Endocrinol. 2002, 22, 1–23. [Google Scholar] [CrossRef]
- Tontonoz, P.; Spiegelman, B.M. Fat and Beyond: The Diverse Biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef]
- Kroker, A.J.; Bruning, J.B. Review of the Structural and Dynamic Mechanisms of PPARγPartial Agonism. PPAR Res. 2015, 2015, 816856. [Google Scholar] [CrossRef] [Green Version]
- Powell, E.; Kuhn, P.; Xu, W. Nuclear Receptor Cofactors in PPARγ-Mediated Adipogenesis and Adipocyte Energy Metabolism. PPAR Res. 2006, 2007, 053843. [Google Scholar] [CrossRef] [Green Version]
- Zieleniak, A.; Wójcik, M.; Wozniak, L.A. Structure and physiological functions of the human peroxisome proliferator-activated receptor γ. Arch. Immunol. Ther. Exp. 2008, 56, 331–345. [Google Scholar] [CrossRef]
- Fang, L.; Zhang, M.; Li, Y.; Liu, Y.; Cui, Q.; Wang, N. PPARgene: A Database of Experimentally Verified and Computationally Predicted PPAR Target Genes. PPAR Res. 2016, 2016, 6042162. [Google Scholar] [CrossRef] [Green Version]
- Rovito, D.; Gionfriddo, G.; Barone, I.; Giordano, C.; Grande, F.; De Amicis, F.; Lanzino, M.; Catalano, S.; Andò, S.; Bonofiglio, D. Ligand-activated PPARγ downregulates CXCR4 gene expression through a novel identified PPAR response element and inhibits breast cancer progression. Oncotarget 2016, 7, 65109–65124. [Google Scholar] [CrossRef] [Green Version]
- Malerød, L.; Sporstøl, M.; Juvet, L.K.; Mousavi, A.; Gjøen, T.; Berg, T.O. Hepatic scavenger receptor class B, type I is stimulated by peroxisome proliferator-activated receptor γ and hepatocyte nuclear factor 4α. Biochem. Biophys. Res. Commun. 2003, 305, 557–565. [Google Scholar] [CrossRef]
- Kumar, A.P.; Quake, A.L.; Chang, M.K.X.; Zhou, T.; Lim, K.S.Y.; Singh, R.; Hewitt, R.E.; Salto-Tellez, M.; Pervaiz, S.; Clement, M.-V. Repression of NHE1 Expression by PPAR Activation Is a Potential New Approach for Specific Inhibition of the Growth of Tumor Cells in vitro and in vivo. Cancer Res. 2009, 69, 8636–8644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeng, J.; Byun, J.; Park, J.Y.; Yamamoto, T.; Schesing, K.; Tian, B.; Sadoshima, J.; Oka, S.-I. An Ideal PPAR Response Element Bound to and Activated by PPARα. PLoS ONE 2015, 10, e0134996. [Google Scholar] [CrossRef] [Green Version]
- Schulman, I.G.; Shao, G.; Heyman, R.A. Transactivation by Retinoid X Receptor–Peroxisome Proliferator-Activated Receptor γ (PPARγ) Heterodimers: Intermolecular Synergy Requires Only the PPARγ Hormone-Dependent Activation Function. Mol. Cell. Boil. 1998, 18, 3483–3494. [Google Scholar] [CrossRef] [Green Version]
- Bonofiglio, D.; Cione, E.; Qi, H.; Pingitore, A.; Perri, M.; Catalano, S.; Vizza, N.; Panno, M.L.; Genchi, G.; Fuqua, S.A.; et al. Combined Low Doses of PPARγ and RXR Ligands Trigger an Intrinsic Apoptotic Pathway in Human Breast Cancer Cells. Am. J. Pathol. 2009, 175, 1270–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonofiglio, D.; Cione, E.; Vizza, N.; Perri, M.; Pingitore, A.; Qi, H.; Catalano, S.; Rovito, D.; Genchi, G.; Andò, S. Bid as a potential target of apoptotic effects exerted by low doses of PPARγ and RXR ligands in breast cancer cells. Cell Cycle 2011, 10, 2344–2354. [Google Scholar] [CrossRef] [Green Version]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 1007–1022. [Google Scholar] [CrossRef]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta (BBA)-Mol. Cell Boil. Lipids 2007, 1771, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738. [Google Scholar] [CrossRef] [Green Version]
- Marion-Letellier, R.; Savoye, G.; Ghosh, S. Fatty acids, eicosanoids and PPAR gamma. Eur. J. Pharmacol. 2016, 785, 44–49. [Google Scholar] [CrossRef]
- Shahidi, F.; Ambigaipalan, P. Omega-3 Polyunsaturated Fatty Acids and Their Health Benefits. Annu. Rev. Food Sci. Technol. 2018, 9, 345–381. [Google Scholar] [CrossRef]
- Geelen, A.; Schouten, J.M.; Kamphuis, C.; Stam, B.E.; Burema, J.; Renkema, J.M.S.; Bakker, E.-J.; Veer, P.V.; Kampman, E. Fish Consumption, n-3 Fatty Acids, and Colorectal Cancer: A Meta-Analysis of Prospective Cohort Studies. Am. J. Epidemiol. 2007, 166, 1116–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Shan, K.; Chen, H.; Chen, Y.Q. n-3 Polyunsaturated Fatty Acids and Their Role in Cancer Chemoprevention. Curr. Pharmacol. Rep. 2015, 1, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, B.B.; Derraik, J.G.B.; Brennan, C.M.; Biggs, J.B.; Smith, G.C.; Garg, M.L.; Cameron-Smith, D.; Hofman, P.L.; Cutfield, W.S. Higher omega-3 index is associated with increased insulin sensitivity and more favourable metabolic profile in middle-aged overweight men. Sci. Rep. 2014, 4, 6697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGlory, C.; Calder, P.C.; Nunes, E.A. The Influence of Omega-3 Fatty Acids on Skeletal Muscle Protein Turnover in Health, Disuse, and Disease. Front. Nutr. 2019, 6, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, C.; Afonso, C.; Bandarra, N.M. Dietary DHA and health: Cognitive function ageing. Nutr. Res. Rev. 2016, 29, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, S.; Svahn, S.L.; Johansson, M.E. Effects of Omega-3 Fatty Acids on Immune Cells. Int. J. Mol. Sci. 2019, 20, 5028. [Google Scholar] [CrossRef] [Green Version]
- Mansoori, A.; Sotoudeh, G.; Djalali, M.; Eshraghian, M.-R.; Keramatipour, M.; Nasli-Esfahani, E.; Shidfar, F.; Alvandi, E.; Toupchian, O.; Koohdani, F. Effect of DHA-rich fish oil on PPARγ target genes related to lipid metabolism in type 2 diabetes: A randomized, double-blind, placebo-controlled clinical trial. J. Clin. Lipidol. 2015, 9, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Edwards, I.J.; O’Flaherty, J.T. Omega-3 Fatty Acids and PPARγ in Cancer. PPAR Res. 2008, 2008, 358052. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Plastina, P.; Barone, I.; Catalano, S.; Bonofiglio, D. n–3 Polyunsaturated Fatty Acid Amides: New Avenues in the Prevention and Treatment of Breast Cancer. Int. J. Mol. Sci. 2020, 21, 2279. [Google Scholar] [CrossRef] [Green Version]
- Schopfer, F.J.; Lin, Y.; Baker, P.R.S.; Cui, T.; Garcia-Barrio, M.; Zhang, J.; Chen, K.; Chen, Y.E.; Freeman, B.A. Nitrolinoleic acid: An endogenous peroxisome proliferator-activated receptor ligand. Proc. Natl. Acad. Sci. USA 2005, 102, 2340–2345. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.T.; Lakshmi, S.P.; Reddy, R.C. The Nitrated Fatty Acid 10-Nitro-oleate Diminishes Severity of LPS-Induced Acute Lung Injury in Mice. PPAR Res. 2012, 2012, 617063. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Waltenberger, B.; Pferschy-Wenzig, E.-M.; Blunder, M.; Liu, X.; Malainer, C.; Blažević, T.; Schwaiger, S.; Rollinger, J.M.; Heiss, E.H.; et al. Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARγ): A review. Biochem. Pharmacol. 2014, 92, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonofiglio, D.; Giordano, C.; De Amicis, F.; Lanzino, M.; Andò, S. Natural Products as Promising Antitumoral Agents in Breast Cancer: Mechanisms of Action and Molecular Targets. Mini-Rev. Med. Chem. 2016, 15, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Magee, P.; Pearson, S.J.; Whittingham-Dowd, J.K.; Allen, J. PPARγ as a molecular target of EPA anti-inflammatory activity during TNF-α-impaired skeletal muscle cell differentiation. J. Nutr. Biochem. 2012, 23, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Chambrier, C.; Bastard, J.-P.; Rieusset, J.; Chevillotte, E.; Bonnefont-Rousselot, M.; Thérond, P.; Hainque, B.; Riou, J.-P.; Laville, M.; Vidal, H. Eicosapentaenoic Acid Induces mRNA Expression of Peroxisome Proliferator-Activated Receptor γ. Obes. Res. 2002, 10, 518–525. [Google Scholar] [CrossRef]
- Marion-Letellier, R.; Butler, M.; Déchelotte, P.; Playford, R.J.; Ghosh, S. Comparison of cytokine modulation by natural peroxisome proliferator-activated receptor gamma ligands with synthetic ligands in intestinal-like Caco-2 cells and human dendritic cells—Potential for dietary modulation of peroxisome proliferator-activated receptor gamma in intestinal inflammation. Am. J. Clin. Nutr. 2008, 87, 939–948. [Google Scholar]
- Rovito, D.; Giordano, C.; Plastina, P.; Barone, I.; De Amicis, F.; Mauro, L.; Rizza, P.; Lanzino, M.; Catalano, S.; Bonofiglio, D.; et al. Omega-3 DHA- and EPA–dopamine conjugates induce PPARγ-dependent breast cancer cell death through autophagy and apoptosis. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 2185–2195. [Google Scholar] [CrossRef]
- Yu, C.; Chen, L.-L.; Luo, H.; Chen, J.; Cheng, F.; Gui, C.; Zhang, R.; Shen, J.; Chen, K.; Jiang, H.; et al. Binding analyses between Human PPARgamma-LBD and ligands. Surface plasmon resonance biosensor assay correlating with circular dichroic spectroscopy determination and molecular docking. J. Boil. Inorg. Chem. 2004, 271, 386–397. [Google Scholar] [CrossRef]
- Bull, A.W.; Steffensen, K.R.; Leers, J.; Rafter, J.J. Activation of PPAR in colon tumor cell lines by oxidized metabolites of linoleic acid, endogenous ligands for PPAR. Carcinogenesis 2003, 24, 1717–1722. [Google Scholar] [CrossRef]
- Itoh, T.; Fairall, L.; Amin, K.; Inaba, Y.; Szanto, A.; Balint, B.L.; Nagy, L.; Yamamoto, K.; Schwabe, J. Structural basis for the activation of PPARγ by oxidized fatty acids. Nat. Struct. Mol. Boil. 2008, 15, 924–931. [Google Scholar] [CrossRef]
- Wang, C.; Fu, M.; D’Amico, M.; Albanese, C.; Zhou, J.-N.; Brownlee, M.; Lisanti, M.P.; Chatterjee, V.K.K.; Lazar, M.A.; Pestell, R.G. Inhibition of Cellular Proliferation through IκB Kinase-Independent and Peroxisome Proliferator-Activated Receptor γ-Dependent Repression of Cyclin D1. Mol. Cell. Boil. 2001, 21, 3057–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Correll, P.; Heuvel, J.V. Conjugated linoleic acid decreases production of pro-inflammatory products in macrophages: Evidence for a PPARγ-dependent mechanism. Biochim. Biophys. Acta (BBA)-Mol. Cell Boil. Lipids 2002, 1581, 89–99. [Google Scholar] [CrossRef]
- Mueller, M.; Jungbauer, A. Red clover extract. Menopause 2008, 15, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.-K.; Gao, J.; Zhu, D.-N. Kaempferol and quercetin isolated from Euonymus alatus improve glucose uptake of 3T3-L1 cells without adipogenesis activity. Life Sci. 2008, 82, 615–622. [Google Scholar] [CrossRef]
- Puhl, A.C.; Bernardes, A.; Silveira, R.L.; Yuan, J.; Campos, J.L.D.O.; Saidemberg, D.M.; Palma, M.S.; Cvoro, A.; Ayers, S.D.; Webb, P.; et al. Mode of Peroxisome Proliferator-Activated Receptor γ Activation by Luteolin. Mol. Pharmacol. 2012, 81, 788–799. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, A.; Yanover, C.; Cahan, A.; Goldschmidt, Y. Estimating the effects of second-line therapy for type 2 diabetes mellitus: Retrospective cohort study. BMJ Open Diabetes Res. Care 2017, 5, e000435. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency. Available online: https://www.ema.europa.eu/en/news/european-medicines-agency-recommends-suspension-avandia-avandamet-avaglim (accessed on 10 July 2020).
- Shang, J.; Brust, R.; Mosure, S.A.; Bass, J.; Munoz-Tello, P.; Lin, H.; Hughes, T.S.; Tang, M.; Ge, Q.; Kamenekca, T.M.; et al. Cooperative cobinding of synthetic and natural ligands to the nuclear receptor PPARγ. eLife 2018, 7. [Google Scholar] [CrossRef]
- Koffarnus, R.L.; A Wargo, K.; Phillippe, H.M. Rivoglitazone: A New Thiazolidinedione for the Treatment of Type 2 Diabetes Mellitus. Ann. Pharmacother. 2013, 47, 877–885. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Marshall, J.L.; Wagner, A.J.; Hwang, J.J.; Malik, S.; Cotarla, I.; Deeken, J.F.; He, A.R.; Daniel, H.; Halim, A.-B.; et al. A phase 1 study of efatutazone, an oral peroxisome proliferator-activated receptor gamma agonist, administered to patients with advanced malignancies. Cancer 2012, 118, 5403–5413. [Google Scholar] [CrossRef]
- Hauner, H. The mode of action of thiazolidinediones. Diabetes/Metab. Res. Rev. 2002, 18, S10–S15. [Google Scholar] [CrossRef]
- Nanjan, M.; Mohammed, M.; Kumar, B.R.P.; Chandrasekar, M. Thiazolidinediones as antidiabetic agents: A critical review. Bioorganic Chem. 2018, 77, 548–567. [Google Scholar] [CrossRef] [PubMed]
- Rizos, C.; Elisaf, M.; Mikhailidis, D.; Liberopoulos, E.; Liberopoulos, E. How safe is the use of thiazolidinediones in clinical practice? Expert Opin. Drug Saf. 2008, 8, 15–32. [Google Scholar] [CrossRef]
- Zhang, F.; LaVan, B.E.; Gregoire, F.M. Selective Modulators of PPAR-γActivity: Molecular Aspects Related to Obesity and Side-Effects. PPAR Res. 2007, 2007, 32696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindarajulu, M.; Pinky, P.D.; Bloemer, J.; Ghanei, N.; Suppiramaniam, V.; Amin, R.H. Signaling Mechanisms of Selective PPARγ Modulators in Alzheimer’s Disease. PPAR Res. 2018, 2018, 2010675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, J.M.; Lenhard, J.M.; Oliver, B.B.; Ringold, G.M.; Kliewer, S.A. Peroxisome Proliferator-activated Receptors a and g Are Activated by Indomethacin and Other Non-steroidal Anti-inflammatory Drugs. J. Biol. Chem. 1997, 272, 3406–3410. [Google Scholar] [CrossRef] [Green Version]
- Puhl, A.C.; Milton, F.A.; Cvoro, A.; Sieglaff, D.H.; Campos, J.L.D.O.; Bernardes, A.; Filgueira, C.S.; Lindemann, J.L.; Deng, T.; Neves, F.A.; et al. Mechanisms of Peroxisome Proliferator Activated Receptor γ Regulation by Non-steroidal Anti-inflammatory Drugs. Nucl. Recept. Signal. 2015, 13. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhang, W.; Liang, B.; Casimiro, M.C.; Whitaker-Menezes, D.; Wang, M.; Lisanti, M.P.; Lanza-Jacoby, S.; Pestell, R.G.; Wang, C. PPARγ activation induces autophagy in breast cancer cells. Int. J. Biochem. Cell Boil. 2009, 41, 2334–2342. [Google Scholar] [CrossRef] [Green Version]
- Hulin, B.; McCarthy, P.; Gibbs, E. The glitazone family of antidiabetic agents. In Current Pharmaceutical Design; Bentham Science Publishers: Bussum, The Netherlands, 1996; Volume 2, pp. 85–102. [Google Scholar]
- Liu, H.; Zang, C.; Fenner, M.; Possinger, K.; Elstner, E. PPARγ Ligands and ATRA Inhibit the Invasion of Human Breast Cancer Cells in vitro. Breast Cancer Res. Treat. 2003, 79, 63–74. [Google Scholar] [CrossRef]
- Bonofiglio, D.; Aquila, S.; Catalano, S.; Gabriele, S.; Belmonte, M.; Middea, E.; Qi, H.; Morelli, C.; Gentile, M.; Maggiolini, M.; et al. Peroxisome Proliferator-Activated Receptor-γ Activates p53 Gene Promoter Binding to the Nuclear Factor-κB Sequence in Human MCF7 Breast Cancer Cells. Mol. Endocrinol. 2006, 20, 3083–3092. [Google Scholar] [CrossRef]
- Oberfield, J.L.; Collins, J.L.; Holmes, C.P.; Goreham, D.M.; Cooper, J.P.; Cobb, J.E.; Lenhard, J.M.; Hull-Ryde, E.A.; Mohr, C.P.; Blanchard, S.G.; et al. A peroxisome proliferator-activated receptor ligand inhibits adipocyte differentiation. Proc. Natl. Acad. Sci. USA 1999, 96, 6102–6106. [Google Scholar] [CrossRef] [Green Version]
- Rocchi, S.; Picard, F.; Vamecq, J.; Gelman, L.; Potier, N.; Zeyer, D.; Dubuquoy, L.; Bac, P.; Champy, M.F.; Plunket, K.D.; et al. A unique PPARgamma ligand with potent insulin-sensitizing yet weak adipogenic activity. Mol. Cell 2001, 8, 737–747. [Google Scholar] [CrossRef]
- Henriksen, K.; Byrjalsen, I.; Qvist, P.; Beck-Nielsen, H.; Hansen, G.; Riis, B.J.; Perrild, H.; Svendsen, O.L.; Gram, J.; Karsdal, M.A.; et al. Efficacy and safety of the PPARγ partial agonist balaglitazone compared with pioglitazone and placebo: A phase III, randomized, parallel-group study in patients with type 2 diabetes on stable insulin therapy. Diabetes/Metab. Res. Rev. 2011, 27, 392–401. [Google Scholar] [CrossRef]
- Lazarenko, O.P.; Rzonca, S.O.; Suva, L.; Lecka-Czernik, B. Netoglitazone is a PPAR-gamma ligand with selective effects on bone and fat. Bone 2006, 38, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonofiglio, D.; Qi, H.; Gabriele, S.; Catalano, S.; Aquila, S.; Belmonte, M.; Ando, S. Peroxisome proliferator-activated receptor g inhibits follicular and anaplastic thyroid carcinoma cells growth by upregulating p21Cip1/WAF1 gene in a Sp1-dependent manner. Endocr.-Relat. Cancer 2008, 15, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Nicolosi, M.L.; Giuliano, S.; Bellomo, M.; Belfiore, A.; Malaguarnera, R. PPAR-γ Agonists as Antineoplastic Agents in Cancers with Dysregulated IGF Axis. Front. Endocrinol. 2017, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thu, K.; Bretones, I.S.; Mak, T.; Cescon, D.W. Targeting the cell cycle in breast cancer: Towards the next phase. Cell Cycle 2018, 17, 1871–1885. [Google Scholar] [CrossRef] [Green Version]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Chantar, M.L.M.; Nawroth, R.; Sánchez-García, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Boil. 2015, 35, S25–S54. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Jingwen, B.; Yaochen, L.; Guojun, Z.; Bai, J.; Li, Y.; Zhang, G.-J. Cell cycle regulation and anticancer drug discovery. Cancer Boil. Med. 2017, 14, 348–362. [Google Scholar] [CrossRef]
- Williams, G.H.; Stoeber, K. The cell cycle and cancer. J. Pathol. 2011, 226, 352–364. [Google Scholar] [CrossRef]
- Tachibana, K.; Yamasaki, D.; Ishimoto, K.; Doi, T. The Role of PPARs in Cancer. PPAR Res. 2008, 2008, 102737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, F.; Wakino, S.; Liu, Z.; Kim, S.; Hsueh, W.A.; Collins, A.R.; Van Herle, A.J.; Law, R.E. Troglitazone Inhibits Growth of MCF-7 Breast Carcinoma Cells by Targeting G1 Cell Cycle Regulators. Biochem. Biophys. Res. Commun. 2001, 286, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-N.; Noh, E.-M.; Lee, Y.-R.; Roh, S.-G.; Song, E.-K.; Han, M.-K.; Lee, Y.-C.; Shim, I.K.; Lee, S.J.; Jung, S.H.; et al. Troglitazone enhances tamoxifen-induced growth inhibitory activity of MCF-7 cells. Biochem. Biophys. Res. Commun. 2008, 377, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Triana-Martínez, F.; Loza, M.I.; Dominguez, E. Beyond Tumor Suppression: Senescence in Cancer Stemness and Tumor Dormancy. Cells 2020, 9, 346. [Google Scholar] [CrossRef] [Green Version]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S.A. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Pignatelli, M.; Sanchez-Rodriguez, J.; Santos, A.; Pérez-Castillo, A. 15-Deoxy-Δ-12,14-prostaglandin J2 induces programmed cell death of breast cancer cells by a pleiotropic mechanism. Carcinogenesis 2004, 26, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Colin-Cassin, C.; Meyer, M.; Cerella, C.; Kleinclauss, A.; Monard, G.; Boisbrun, M.; Han, B.W.; Flament, S.; Grillier-Vuissoz, I.; Kuntz, S. Biotinylation enhances the anticancer effects of 15d-PGJ2 against breast cancer cells. Int. J. Oncol. 2018, 52, 1991–2000. [Google Scholar]
- Nikhil, K.; Sharan, S.; Singh, A.K.; Chakraborty, A.; Roy, P. Anticancer Activities of Pterostilbene-Isothiocyanate Conjugate in Breast Cancer Cells: Involvement of PPARγ. PLoS ONE 2014, 9, e104592. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Huang, Y.; Cheng, C.; Lu, W.; Zhang, Y.; Liu, X.; Zou, L.; Ben, Q.; Shen, A. Combination of thiazolidinedione and hydralazine suppresses proliferation and induces apoptosis by PPARγ up-expression in MDA-MB-231 cells. Exp. Mol. Pathol. 2011, 91, 768–774. [Google Scholar] [CrossRef]
- Michael, M.S.; Badr, M.Z.; Badawi, A.F. Inhibition of cyclooxygenase-2 and activation of peroxisome proliferator-activated receptor-y synergistically induces apoptosis and inhibits growth of human breast cancer cells. Int. J. Mol. Med. 2003, 11, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Elstner, E.; Williamson, E.; Zang, C.; Fritz, J.; Heber, D.; Fenner, M.; Possinger, K.; Koeffler, H. Novel Therapeutic Approach: Ligands for PPARγ and Retinoid Receptors Induce Apoptosis in bcl-2-positive Human Breast Cancer Cells. Breast Cancer Res. Treat. 2002, 74, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Bonofiglio, D.; Gabriele, S.; Aquila, S.; Qi, H.; Belmonte, M.; Catalano, S.; Andò, S. Peroxisome proliferator-activated receptor gamma activates fas ligand gene promoter inducing apoptosis in human breast cancer cells. Breast Cancer Res. Treat. 2008, 113, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Khandia, R.; Dadar, M.; Munjhal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2017, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.Y.; Hong, O.Y.; Youn, H.J.; Kim, M.G.; Kim, C.H.; Jung, S.H.; Kim, J.S. 15d-PGJ2 inhibits NF-κB and AP-1-mediated MMP-9 expression and invasion of breast cancer cell by means of a heme oxygenase-1-dependent mechanism. BMB Rep. 2020, 53, 212–217. [Google Scholar] [CrossRef]
- Hong, O.-Y.; Youn, H.J.; Jang, H.-Y.; Jung, S.H.; Noh, E.-M.; Chae, H.S.; Jeong, Y.-J.; Kim, W.; Kim, C.-H.; Kim, J.-S. Troglitazone Inhibits Matrix Metalloproteinase-9 Expression and Invasion of Breast Cancer Cell through a Peroxisome Proliferator-Activated Receptor γ-Dependent Mechanism. J. Breast Cancer 2018, 21, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.-K.; Yu, H.-N.; Noh, E.-M.; Kim, J.-M.; Hong, O.-Y.; Youn, H.J.; Jung, S.H.; Kwon, K.-B.; Kim, J.-S.; Lee, Y.-R. DHA blocks TPA-induced cell invasion by inhibiting MMP-9 expression via suppression of the PPAR-γ/NF-κB pathway in MCF-7 cells. Oncol. Lett. 2016, 13, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Keller, H.; Givel, F.; Perroud, M.; Wahli, W. Signaling cross-talk between peroxisome proliferator-activated receptor/retinoid X receptor and estrogen receptor through estrogen response elements. Mol. Endocrinol. 1995, 9, 794–804. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kilgore, M.W. Signal cross-talk between estrogen receptor alpha and beta and the peroxisome proliferator-activated receptor gamma1 in MDA-MB-231 and MCF-7 breast cancer cells. Mol. Cell. Endocrinol. 2002, 194, 123–133. [Google Scholar] [CrossRef]
- Bonofiglio, D. Estrogen Receptor Binds to Peroxisome Proliferator-Activated Receptor Response Element and Negatively Interferes with Peroxisome Proliferator-Activated Receptor Signaling in Breast Cancer Cells. Clin. Cancer Res. 2005, 11, 6139–6147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, G.L.; Zhao, Y.; Kalus, A.M.; Simpson, E.R. Peroxisome Proliferator-activated Receptor γ Ligands Inhibit Estrogen Biosynthesis in Human Breast Adipose Tissue: Possible Implications for Breast Cancer Therapy. Cancer Res. 2000, 60, 1604–1608. [Google Scholar] [PubMed]
- Pignatelli, M.; Cortés-Canteli, M.; Lai, C.; Santos, A.; Perez-Castillo, A. The peroxisome proliferator-activated receptor gamma is an inhibitor of ErbBs activity in human breast cancer cells. J. Cell Sci. 2001, 114, 4117–4126. [Google Scholar] [PubMed]
- Catalano, S.; Mauro, L.; Bonofiglio, D.; Pellegrino, M.; Qi, H.; Rizza, P.; Vizza, D.; Bossi, G.; Andò, S. In Vivo and in Vitro Evidence That PPARγ Ligands Are Antagonists of Leptin Signaling in Breast Cancer. Am. J. Pathol. 2011, 179, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Miyoshi, K.; Claudio, E.; Siebenlist, U.; Gonzalez, F.J.; A Flaws, J.; Wagner, K.-U.; Hennighausen, L. Loss of the Peroxisome Proliferation-activated Receptor gamma (PPARγ) Does Not Affect Mammary Development and Propensity for Tumor Formation but Leads to Reduced Fertility. J. Boil. Chem. 2002, 277, 17830–17835. [Google Scholar] [CrossRef] [Green Version]
- Elstner, E.; Müller, C.; Koshizuka, K.; Williamson, E.A.; Park, D.; Asou, H.; Shintaku, P.; Said, J.W.; Heber, D.; Koeffler, H.P. Ligands for peroxisome proliferator-activated receptor and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc. Natl. Acad. Sci. USA 1998, 95, 8806–8811. [Google Scholar] [CrossRef] [Green Version]
- E Clay, C.; Namen, A.M.; Atsumi, G.-I.; Willingham, M.C.; High, K.P.; Kute, T.E.; Trimboli, A.J.; Fonteh, A.N.; Dawson, P.A.; Chilton, F.H. Influence of J series prostaglandins on apoptosis and tumorigenesis of breast cancer cells. Carcinogenesis 1999, 20, 1905–1911. [Google Scholar] [CrossRef] [Green Version]
- Lapillonne, H.; Konopleva, M.; Tsao, T.; Gold, D.; McQueen, T.; Sutherland, R.L.; Madden, T.; Andreeff, M. Activation of Peroxisome Proliferator-activated Receptor γ by a Novel Synthetic Triterpenoid 2-Cyano-3,12-dioxooleana-1,9-dien-28-oic Acid Induces Growth Arrest and Apoptosis in Breast Cancer Cells. Cancer Res. 2003, 63, 5926–5939. [Google Scholar]
- Xue, M.; Wang, Q.; Zhao, J.; Dong, L.; Ge, Y.; Hou, L.; Liu, Y.; Zheng, Z. Docosahexaenoic acid inhibited the Wnt/β-Catenin pathway and suppressed breast cancer cells in vitro and in vivo. J. Nutr. Biochem. 2014, 25, 104–110. [Google Scholar] [CrossRef]
- Ghosh-Choudhury, T.; Mandal, C.C.; Woodruff, K.; Clair, P.S.; Fernandes, G.; Choudhury, G.G.; Ghosh-Choudhury, N. Fish oil targets PTEN to regulate NFκB for downregulation of anti-apoptotic genes in breast tumor growth. Breast Cancer Res. Treat. 2008, 118, 213–228. [Google Scholar] [CrossRef] [Green Version]
- Vibet, S.; Goupille, C.; Bougnoux, P.; Steghens, J.-P.; Goré, J.; Mahéo, K. Sensitization by docosahexaenoic acid (DHA) of breast cancer cells to anthracyclines through loss of glutathione peroxidase (GPx1) response. Free. Radic. Boil. Med. 2008, 44, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.S.; Wang, P.; Yamabe, N.; Fukui, M.; Jay, T.R.; Zhu, B.T. Docosahexaenoic Acid Induces Apoptosis in MCF-7 Cells in Vitro and in Vivo via Reactive Oxygen Species Formation and Caspase 8 Activation. PLoS ONE 2010, 5, e10296. [Google Scholar] [CrossRef] [Green Version]
- Rose, D.P.; Connolly, J.M. Dietary fat and breast cancer metastasis by human tumor xenografts. Breast Cancer Res. Treat. 1997, 46, 225–237. [Google Scholar] [CrossRef] [PubMed]
- A Leslie, M.; A Abdelmagid, S.; Perez, K.H.; Muller, W.J.; Ma, D.W. Mammary tumour development is dose-dependently inhibited by n-3 polyunsaturated fatty acids in the MMTV-neu(ndl)-YD5 transgenic mouse model. Lipids Health Dis. 2014, 13, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, L.D.; Agarwal, D.; Rosol, T.J.; Lehman, A.; Tian, M.; Hatton, J.; Heestand, J.; Belury, M.A.; Clinton, S.K.; Cook, J. The inhibition of early stages of HER-2/neu-mediated mammary carcinogenesis by dietary n -3 PUFAs. Mol. Nutr. Food Res. 2012, 57, 320–327. [Google Scholar] [CrossRef] [Green Version]
- MacLennan, M.B.; Clarke, S.E.; Perez, K.; Wood, G.A.; Muller, W.J.; Kang, J.X.; Ma, D.W. Mammary tumor development is directly inhibited by lifelong n-3 polyunsaturated fatty acids. J. Nutr. Biochem. 2013, 24, 388–395. [Google Scholar] [CrossRef]
- Jiang, W.; Zhu, Z.; McGinley, J.N.; El Bayoumy, K.; Manni, A.; Thompson, H.J. Identification of a Molecular Signature Underlying Inhibition of Mammary Carcinoma Growth by Dietary N-3 Fatty Acids. Cancer Res. 2012, 72, 3795–3806. [Google Scholar] [CrossRef] [Green Version]
- Yee, L.D.; Williams, N.; Wen, P.; Young, N.C.; Lester, J.; Johnson, M.V.; Farrar, W.B.; Walker, M.J.; Povoski, S.P.; Suster, S.; et al. Pilot Study of Rosiglitazone Therapy in Women with Breast Cancer: Effects of Short-term Therapy on Tumor Tissue and Serum Markers. Clin. Cancer Res. 2007, 13, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Straka, S.; Lester, J.L.; Cole, R.M.; Andridge, R.R.; Puchala, S.; Rose, A.M.; Clinton, S.K.; Belury, M.A.; Yee, L.D. Incorporation of eicosapentaenioic and docosahexaenoic acids into breast adipose tissue of women at high risk of breast cancer: A randomized clinical trial of dietary fish and n-3 fatty acid capsules. Mol. Nutr. Food Res. 2015, 59, 1780–1790. [Google Scholar] [CrossRef]
- Gomes, M.A.; Jia, X.; Kolenski, I.; Duncan, A.M.; A Meckling, K. The role of background diet on the effects of eicosapentaenoic acid and docosahexaenoic acid supplementation in healthy pre-menopausal women: A randomized, cross-over, controlled study. Lipids Health Dis. 2016, 15, 168. [Google Scholar] [CrossRef] [Green Version]
- Manni, A.; Richie, J.P.; E Schetter, S.; Calcagnotto, A.; Trushin, N.; Aliaga, C.; El-Bayoumy, K. Stearoyl-CoA desaturase-1, a novel target of omega-3 fatty acids for reducing breast cancer risk in obese postmenopausal women. Eur. J. Clin. Nutr. 2017, 71, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Inglis, J.E.; Kleckner, A.S.; Lin, P.-J.; Gilmore, N.J.; Culakova, E.; VanDerWoude, A.C.; Mustian, K.M.; Fernandez, I.D.; Dunne, R.F.; Deutsch, J.; et al. Excess Body Weight and Cancer-Related Fatigue, Systemic Inflammation, and Serum Lipids in Breast Cancer Survivors. Nutr. Cancer 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hershman, D.L.; Unger, J.M.; Crew, K.D.; Awad, D.; Dakhil, S.R.; Gralow, J.; Greenlee, H.; Lew, D.L.; Minasian, L.M.; Till, C.; et al. Randomized Multicenter Placebo-Controlled Trial of Omega-3 Fatty Acids for the Control of Aromatase Inhibitor–Induced Musculoskeletal Pain: SWOG S0927. J. Clin. Oncol. 2015, 33, 1910–1917. [Google Scholar] [CrossRef]
- Fabian, C.J.; Kimler, B.F.; Phillips, T.A.; Box, J.A.; Kreutzjans, A.L.; Carlson, S.E.; Hidaka, B.H.; Metheny, T.; Zalles, C.M.; Mills, G.B.; et al. Modulation of Breast Cancer Risk Biomarkers by High-Dose Omega-3 Fatty Acids: Phase II Pilot Study in Premenopausal Women. Cancer Prev. Res. 2015, 8, 912–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, C.J.; Kimler, B.F.; Phillips, T.A.; Nydegger, J.L.; Kreutzjans, A.L.; Carlson, S.E.; Hidaka, B.H.; Metheny, T.; Zalles, C.M.; Mills, G.B.; et al. Modulation of Breast Cancer Risk Biomarkers by High-Dose Omega-3 Fatty Acids: Phase II Pilot Study in Postmenopausal Women. Cancer Prev. Res. 2015, 8, 922–931. [Google Scholar] [CrossRef] [Green Version]
- Young, L.R.; Kurzer, M.S.; Thomas, W.; Redmon, J.; Raatz, S.K. Effect of Dietary Fat and Omega-3 Fatty Acids on Urinary Eicosanoids and Sex Hormone Concentrations in Postmenopausal Women: A Randomized Controlled Feeding Trial. Nutr. Cancer 2011, 63, 930–939. [Google Scholar] [CrossRef] [PubMed]
- McColley, S.P.; Georgopoulos, A.; Young, L.R.; Kurzer, M.S.; Redmon, J.; Raatz, S.K. A high-fat diet and the threonine-encoding allele (Thr54) polymorphism of fatty acid–binding protein 2 reduce plasma triglyceride–rich lipoproteins. Nutr. Res. 2011, 31, 503–508. [Google Scholar] [CrossRef] [Green Version]
- Raatz, S.K.; Young, L.R.; Picklo, M.J.; Sauter, E.R.; Qin, W.; Kurzer, M.S. Total dietary fat and fatty acid content modifies plasma phospholipid fatty acids, desaturase activity indices, and urinary prostaglandin E in women. Nutr. Res. 2012, 32, 1–7. [Google Scholar] [CrossRef]
- Young, L.R.; Raatz, S.K.; Thomas, W.; Redmon, J.; Kurzer, M.S. Total dietary fat and omega-3 fatty acids have modest effects on urinary sex hormones in postmenopausal women. Nutr. Metab. 2013, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Holt, P.R.; Aleman, J.O.; Walker, J.M.; Jiang, C.S.; Liang, Y.; De Rosa, J.C.; Giri, D.D.; Iyengar, N.M.; Milne, G.L.; A Hudis, C.; et al. Docosahexaenoic acid supplementation is not anti-inflammatory in adipose tissue of healthy obese postmenopausal women. Int. J. Nutr. 2017, 1, 31–49. [Google Scholar] [CrossRef] [Green Version]
- Zick, S.M.; Colacino, J.A.; Cornellier, M.; Khabir, T.; Surnow, K.; Djuric, Z. Fatigue reduction diet in breast cancer survivors: A pilot randomized clinical trial. Breast Cancer Res. Treat. 2016, 161, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Esteva, F.J.; Moulder, S.L.; Gonzalez-Angulo, A.M.; Ensor, J.; Murray, J.L.; Green, M.C.; Koenig, K.B.; Lee, M.-H.; Hortobagyi, G.N.; Yeung, S.-C. Phase I trial of exemestane in combination with metformin and rosiglitazone in nondiabetic obese postmenopausal women with hormone receptor–positive metastatic breast cancer. Cancer Chemother. Pharmacol. 2012, 71, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Common Name | IUPAC NAME | Chemical Structure | References |
|---|---|---|---|---|
| ω-3 PUFAs 1 | Eicosapentaenoic acid (EPA) 20:5(ω-3) | (5Z,8Z,11Z,14Z,17)-Eicosa-5,8,11,14,17-pentaenoic acid |  | [64,65] |
| Docosahexaenoic acid (DHA) 22:6(ω-3) | (4Z,7Z,10Z,13Z,16Z,19Z)-Docosa-4,7,10,13,16,19-hexaenoic acid |  | [57,66] | |
| ω-3 PUFA conjugates | Eicosapentaenoyl ethanolamine (EPEA) | (5Z,8Z,11Z,14Z,17)-N-(2-Hydroxyethyl)-eicosa-5,8,11,14,17-pentaenamide |  | [21] |
| Docosahexaenoyl ethanolamine (DHEA) | (4Z,7Z,10Z,13Z,16Z,19Z)-N-(2-Hydroxyethyl)-docosa-4,7,10,13,16,19-hexaenamide |  | [21] | |
| Eicosapentaenoyl dopamine (EPADA) | (5Z,8Z,11Z,14Z,17)-N-(3,4-Dihydroxyphenethyl)eicosa-5,8,11,14,17-pentaenamide |  | [67] | |
| Docosahexaenoyl dopamine (DHADA) | (4Z,7Z,10Z,13Z,16Z,19Z)-N-(3,4-Dihydroxyphenethyl)docosa-4,7,10,13,16,19-hexaenamide |  | [67] | |
| Docosahexaenoyl serotonin (DHA-5-HT) | (4Z,7Z,10Z,13Z,16Z,19Z)-N-[2-(5-hydroxy-1H-indol-3-yl)ethyl]-docosa-4,7,10,13,16,19-hexaenamide |  | [30] | |
| ω-6 PUFAs | Linoleic acid (LA) 18:2(ω-6) | (9Z,12Z)-Octadeca-9,12-dienoic acid |  | [68] |
| 9-Hydroxyoctadecadienoic acid(9-HODE) | (9S,10E,12E)-9-Hydroxyoctadeca-10,12-dienoic acid |  | [69,70] | |
| 13-Hydroxyoctadecadienoic acid (13-HODE) | (9Z,11E,13S)-13-Hydroxyoctadeca-9,11-dienoic acid |  | [69,70] | |
| 15-Deoxy-Δ-12,14-prostaglandin J2 (15d-PGJ2) | (Z)-7-((S,E)-5-((E)-oct-2-en-1-ylidene)-4-oxocyclopent-2-en-1-yl)hept-5-enoic acid |  | [71] | |
| Conjugated linoleic acid (CLA) | (9Z,11E)-CLA | (9Z,11E)-Octadeca-9,11-dienoic acid |  | [72] |
| (9E,11E)-CLA | (9E,11E)-Octadeca-9,11-dienoic acid |  | [72] | |
| (9Z,11Z)-CLA | (9Z,11Z)-Octadeca-9,11-dienoic acid |  | [72] | |
| (10E,12E)-CLA | (10E,12E)-Octadeca-9,11-dienoic acid |  | [72] | |
| Nitrated fatty acids | Nitrolinolenic acid | (9Z,12Z)-2-Nitrooctadeca-9,12-dienoic acid |  | [60] |
| 10-Nitrooleic acid | (9Z)-10-Nitrooctadec-9-enoic acid |  | [61] | |
| Bioactive compounds | Genistein | 5,7-Dihydroxy-3-(4-hydroxyphenyl)-4H-chromen-4-one |  | [73] |
| Quercetin | 2-(3,4-Dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one |  | [74] | |
| Luteolin | 2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-4H-chromen-4-one |  | [75] |
| Class | Common Name | IUPAC Name | Chemical Structure | References |
|---|---|---|---|---|
| TDZs 1 | Troglitazone | 5-[[4-[(6-Hydroxy-2,5,7,8-tetramethyl-3,4-dihydrochromen-2-yl)methoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione |  | [88] |
| Ciglitazone | 5-{4-[(1-Methylcyclohexyl)methoxy]benzyl}-1,3-thiazolidine-2,4-dione |  | [89] | |
| Pioglitazone | 5-[[4-[2-(5-Ethylpyridin-2-yl)ethoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione |  | [90] | |
| Rosiglitazone | 5-(4-(2-(Methyl(pyridin-2-yl)amino)ethoxy)benzyl)thiazolidine-2,4-dione |  | [91] | |
| Edaglitazone | 5-[[4-[2-(5-Methyl-2-phenyl-1,3-oxazol-4-yl)ethoxy]-1-benzothiophen-7-yl]methyl]-1,3-thiazolidine-2,4-dione |  | [78] | |
| Efatutazone | 5-[(4-{[6-(4-Amino-3,5-dimethylphenoxy)-1-methyl-1H-1,3-benzodiazol-2-yl]methoxy}phenyl)methyl]-1,3-thiazolidine-2,4-dione |  | [80] | |
| Rivoglitazone | 5-({4-[(6-Methoxy-1-methyl-1H-benzimidazol-2-Yl)methoxy]phenyl}methyl)-1,3-thiazolidine-2,4-dione |  | [79] | |
| SPPARMs 2 | GW0072 | 3-(4-(4-Carboxyphenyl)butyl)-2-heptyl-4-oxo-5-thiazolidine |  | [92] |
| Fmoc-l-leucine | 2-(9H-Fluoren-9-ylmethoxycarbonylamino)-4-methylpentanoic acid |  | [93] | |
| Balaglitazone | 5-(4-((3-Methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methoxy)benzyl)thiazolidine-2,4-dione |  | [94] | |
| Netoglitazone | 5-[[6-[(2-Fluorophenyl)methoxy]naphthalen-2-yl]methyl]-1,3-thiazolidine-2,4-dione |  | [95] | |
| NSAIDs 3 | Sulindac sulfide | 5-Fluoro-2-methyl-1-[p-(methylthio)benzylidene]indene-3-acetic acid |  | [87] |
| Diclofenac | 2-[2-(2,6-Dichloroanilino)phenyl]acetic acid |  | [87] | |
| Indomethacin | 2-[1-(4-Chlorobenzoyl)-5-methoxy-2-methylindol-3-yl]acetic acid |  | [68,84,87] | |
| Ibuprofen | 2-(4-Isobutylphenyl)propanoic acid |  | [87] |
| NCT | Intervention/Treatment | Phase | Eligibility Criteria, Primary Outcome and Purpose | References |
|---|---|---|---|---|
| NCT01282580 | -Lovaza -ω-3 fatty acid capsules -Dietary fish (canned salmon, albacore) | I | Eligibility criteria: female (≥18 years) having increased risk for breast cancer based on family and personal history with a normal mammogram in the past 12 years and >1 years from pregnancy, lactation, or chemotherapy. Primary outcome: fatty acid profiles of breast adipose tissue. Purpose: to determine the effects of increased fish consumption on serum and breast fat tissue fatty acids and to assess adherence and tolerability of increased dietary intake of fish relative to an ω-3 fatty acid supplement. | [141] |
| NCT02150525 | -ω-3 fatty acids —Placebo | II | Eligibility criteria: postmenopausal women (45–65 years) with a history of breast cancer after 12 months from surgery and 3 months from completion of chemotherapy. Subjects must present at least one symptom of atrophic vaginitis. Primary outcome: improvement of vaginitis’ symptoms by oral ω-3 fatty acids. Purpose: to study the effectiveness of ω-3 fatty acid on atrophic vaginitis in postmenopausal breast cancer patients. | NA |
| NCT02101970 | -Weight Loss + ω-3 fatty acids -Weight Loss + Placebo | NA | Eligibility criteria: female having evidence of hyperplasia with Masood score of 13 or higher, 500 or more epithelial cells on cytology slide of screening random periareolar fine-needle aspiration, and reasonable hematopoietic, kidney, and liver function. Primary outcome: dropout rate. Purpose: to determine if high dose supplementation with the ω-3 fatty acids EPA and DHA, when added to a weight loss program, is well tolerated and if there is an increase in the favorable change in blood and tissue breast cancer risk factors when compared to weight loss alone. | NA |
| NCT02816125 | -Diet supplemented with EPA + DHA -Dietary fat at less than 20% energy + EPA + DHA | III | Eligibility criteria: menstruating and premenopausal women (≥18 years). Primary outcome: fatty acid incorporation into red blood cells and in cell material from nipple aspirate fluid. Purpose: to examine the effect of the combined treatment with EPA and DHA on breast cancer risk factors in healthy premenopausal women. | [142] |
| NCT00723398 | -Raloxifene -Lovaza -Lovaza + Raloxifene | NA | Eligibility criteria: no smoker postmenopausal women (35–70 years) without hormone-replaced therapy for at least 6 months. Primary outcome: absolute breast density. Purpose: to evaluate the effects of Raloxifene alone or in combination with omega-3 fatty acids on breast cancer development markers in postmenopausal women. | [143] |
| NCT01823991 | -Lovaza -VitaBlue -Placebo | Early I | Eligibility criteria: women (40–70 years) having stage II-IIIA of breast cancer after completion of adjuvant treatment with chemo- and/or radiotherapy in the past 6 months. Primary outcome: cognitive function scores with intervention. Purpose: to evaluate the safety of nutritional intervention with n-3 fatty acids and blueberry anthocyanins on cognitive performance. | NA |
| NCT01869764 | -ω-3 fatty acids -Placebo | II | Eligibility criteria: newly diagnosed women (≥18 years) having in situ carcinoma and stage I to III of breast cancer that will receive breast surgery at least after 7 days from the day of enrollment. Tumor size of at least 1 cm. Primary outcome: ω-3 fatty acid levels in breast tissues in plasma and in erythrocytes before and after surgery. Purpose: to study ω-3 fatty acid effects in patients with breast cancer. | NA |
| NCT02352779 | -Low doses of ω-3 fatty acid -High dose of ω-3 fatty acid -Placebo | NA | Eligibility criteria: women having a confirmed diagnosis of breast cancer and undergone some type or combination of standard adjuvant treatment. Patients must have cancer-related fatigue. Primary outcome: mean change and standard deviation in cancer-related fatigue. Purpose: to study ω-3 fatty acid in reducing cancer-related fatigue. | [144] |
| NCT01385137 | -ω-3-fatty acids -Placebo | III | Eligibility criteria: postmenopausal women with estrogen-receptor positive and/or progesterone-receptor positive invasive breast adenocarcinoma (I-IIIA stage) currently taking a third-generation aromatase inhibitor; patients must be subjected to modified radical mastectomy or breast-sparing surgery. Primary outcome: Brief Pain Inventory (BPI) Worst Pain/Stiffness Score. Purpose: to study ω-3 fatty acid effects in muscle, bone pain, and stiffness in breast cancer patients receiving hormone therapy. | [145] |
| NCT01252277 | -Lovaza | II | Eligibility criteria: premenopausal female (18–54 years) having a breast cancer risk evaluated on the basis of several criteria. Primary outcome: proportion of subjects that complete an intervention. Purpose: to evaluate the effects of Lovaza on breast cancer biomarkers in premenopausal patients. | [146] |
| NCT01252290 | -Lovaza | II | Eligibility criteria: postmenopausal women (25–69 years) having a breast cancer risk evaluated on the basis of several criteria. Primary outcome: proportion of subjects that complete the intervention. Purpose: to evaluate the effects of Lovaza on breast cancer biomarkers in postmenopausal patients. | [147] |
| NCT00627276 | -ω-3 fatty acids-Placebo | NA | Eligibility criteria: breast cancer patients having confirmed diagnosis of ductal carcinoma in situ and/or atypical ductal hyperplasia. Primary outcome: genetic evaluation of markers for breast cancer risk and progression, fatty acids profile, occurrence of ductal carcinoma in situ and/or atypical ductal hyperplasia or invasive cancer in tissue samples. Purpose: to study the effects of ω-3 fatty acids in patients with ductal carcinoma in situ and/or atypical ductal hyperplasia. | NA |
| NCT01824498 | -Low-fat diet -Low fat with high ω -3 diet-High-fat diet | NA | Eligibility criteria: postmenopausal women (45–70 years) having BMI between 19 and 29. Consumption of a “typical” American diet. Primary outcome: plasma sex hormone levels, Estradiol. Purpose: to determine whether diets designed to increase plasma ω-3 concentrations will favorably affect sex hormone distribution in women in a direction associated with reduced risk of sex hormone-mediated cancer development. | [148,149,150,151] |
| NCT02062255 | -Aspirin -ω-3 EPA and DHA -Aspirin + ω-3 EPA and DHA | Early phase I | Eligibility criteria: postmenopausal women (≥18 years). Primary outcome: prostaglandin E2, aromatase, pro-inflammatory cytokines, steroids, and lipids. Purpose: to study the impact of COX2 on sera biomarkers from obese subjects. | NA |
| NCT01821833 | -ω-3 fatty acids + paclitaxel -Placebo + paclitaxel | NA | Eligibility criteria women (≥18 years) with breast cancer or ovarian cancer who will receive paclitaxel treatment at least for 2 months. Primary outcome: mean severity of pain. Purpose: to study the effects of ω-3 fatty acids on pain of cancer patients. | NA |
| NCT01127867 | -DHA | NA | Eligibility criteria: postmenopausal women (40–70 years) having low serum estradiol level (<40 ng/mL) and BMI 35–50. Primary outcome: evaluation of monocyte aggregations and macrophage markers in fat biopsies. Purpose: to study the effects of DHA in decreasing inflammation in fat tissue and reducing breast cancer risk. | [152] |
| NCT01902745 | -Fatigue-reduction diet (typical caloric intake and replacement of some calories with the following foods: whole grains, vegetables, fruit, fatty fish, and nuts and/or seeds) | NA | Eligibility criteria: breast cancer patients (≥18 years) having completed the cancer-related treatments and showing persistent, moderate, or severe fatigue despite standard treatment. Primary outcome: fatigue in breast cancer survivors. Purpose: to expand the data on fatigue-reduction diet in breast cancer survivors | [153] |
| NCT00933309 | -Exemestane -Exemestane + Avandamet (rosiglitazone and metformin) | I | Eligibility criteria: postmenopausal women with BMI ≥ 25 kg/m2 having estrogen receptor positive and/or progesterone receptor positive breast cancer along with clinical evidence of metastasis. Primary outcome: Dose-limiting toxicity (DLT). Purpose: to define the highest tolerable dose of Avandamet in combination with exemestane in postmenopausal obese breast cancer patients. | [154] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Augimeri, G.; Giordano, C.; Gelsomino, L.; Plastina, P.; Barone, I.; Catalano, S.; Andò, S.; Bonofiglio, D. The Role of PPARγ Ligands in Breast Cancer: From Basic Research to Clinical Studies. Cancers 2020, 12, 2623. https://doi.org/10.3390/cancers12092623
Augimeri G, Giordano C, Gelsomino L, Plastina P, Barone I, Catalano S, Andò S, Bonofiglio D. The Role of PPARγ Ligands in Breast Cancer: From Basic Research to Clinical Studies. Cancers. 2020; 12(9):2623. https://doi.org/10.3390/cancers12092623
Chicago/Turabian StyleAugimeri, Giuseppina, Cinzia Giordano, Luca Gelsomino, Pierluigi Plastina, Ines Barone, Stefania Catalano, Sebastiano Andò, and Daniela Bonofiglio. 2020. "The Role of PPARγ Ligands in Breast Cancer: From Basic Research to Clinical Studies" Cancers 12, no. 9: 2623. https://doi.org/10.3390/cancers12092623