Potential Role of PDGFRβ-Associated THBS4 in Colorectal Cancer Development

 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Expression of THBS4 and PDGFRβ in Colorectal Cancer
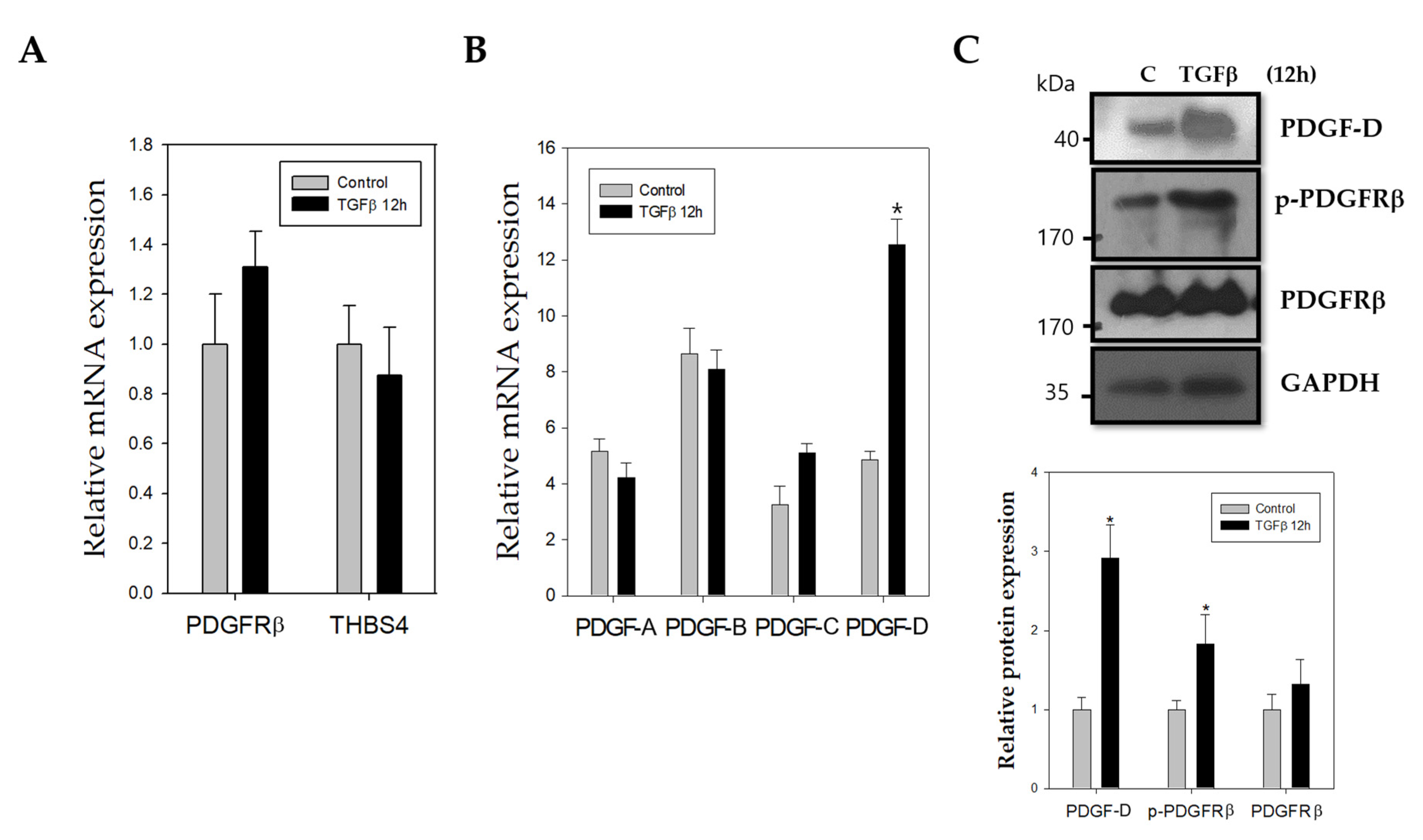
2.2. TGFβ Stimulates Increased mRNA and Protein Expression of PDGF-D
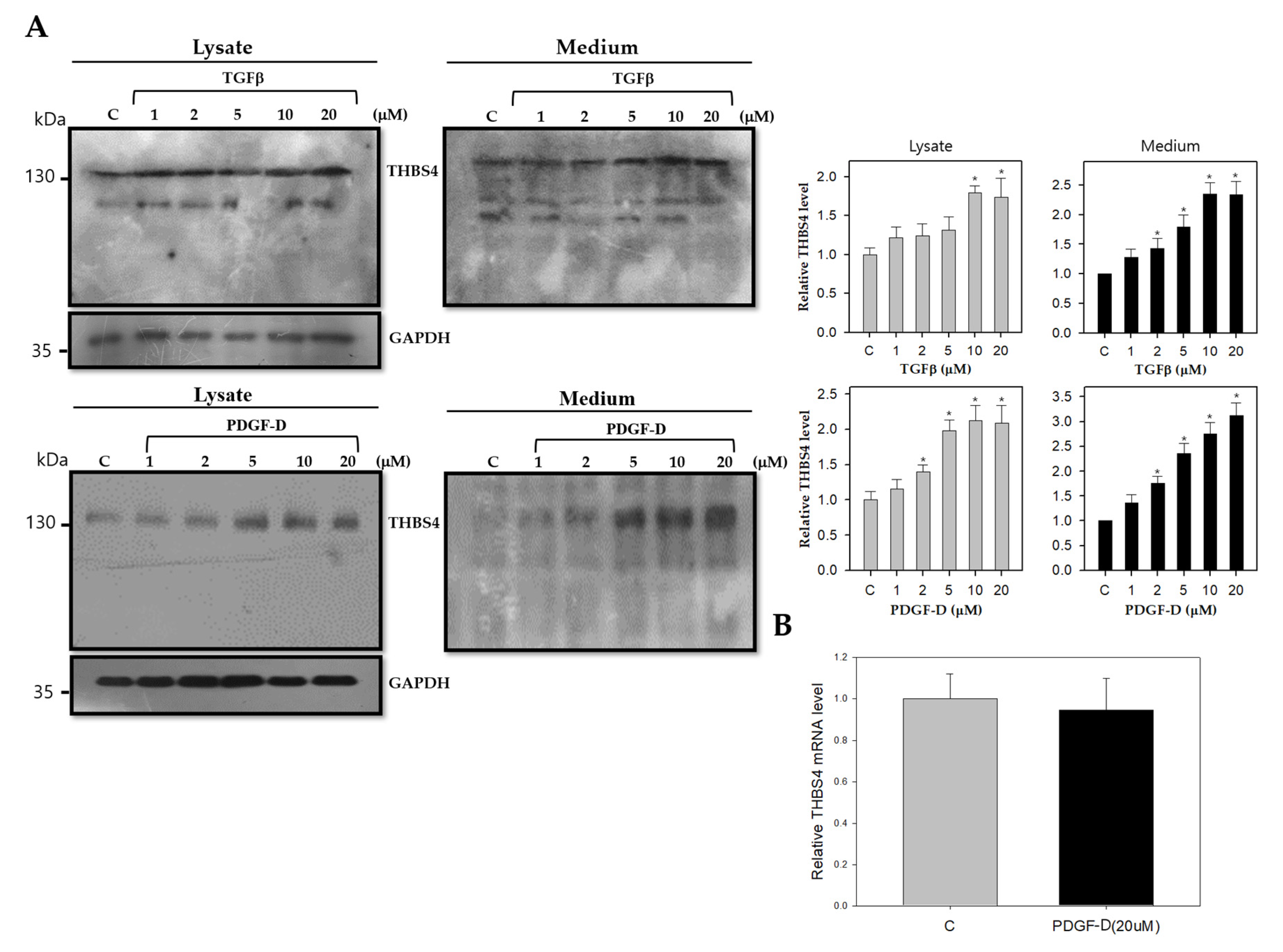
2.3. TGFβ and PDGF-D Influence the Post-Translational Modification and Secretion of THBS4
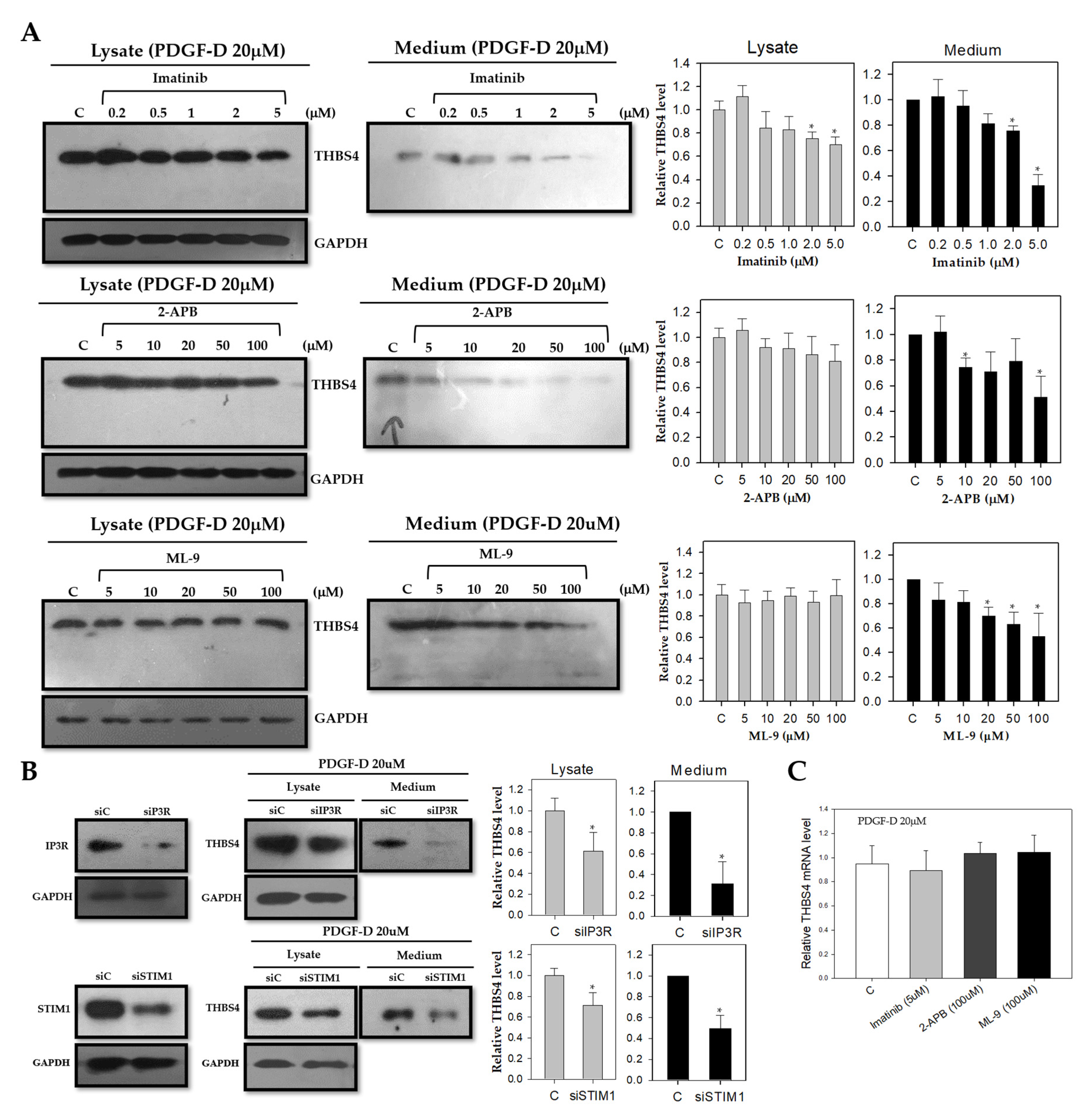
2.4. Knockdown of Ca2+ Signaling Proteins Decreases THBS4 Secretion even on PDGF-D
2.5. PDGF-D Increases THBS4 through PDGFRβ and Ca2+ Signaling Proteins
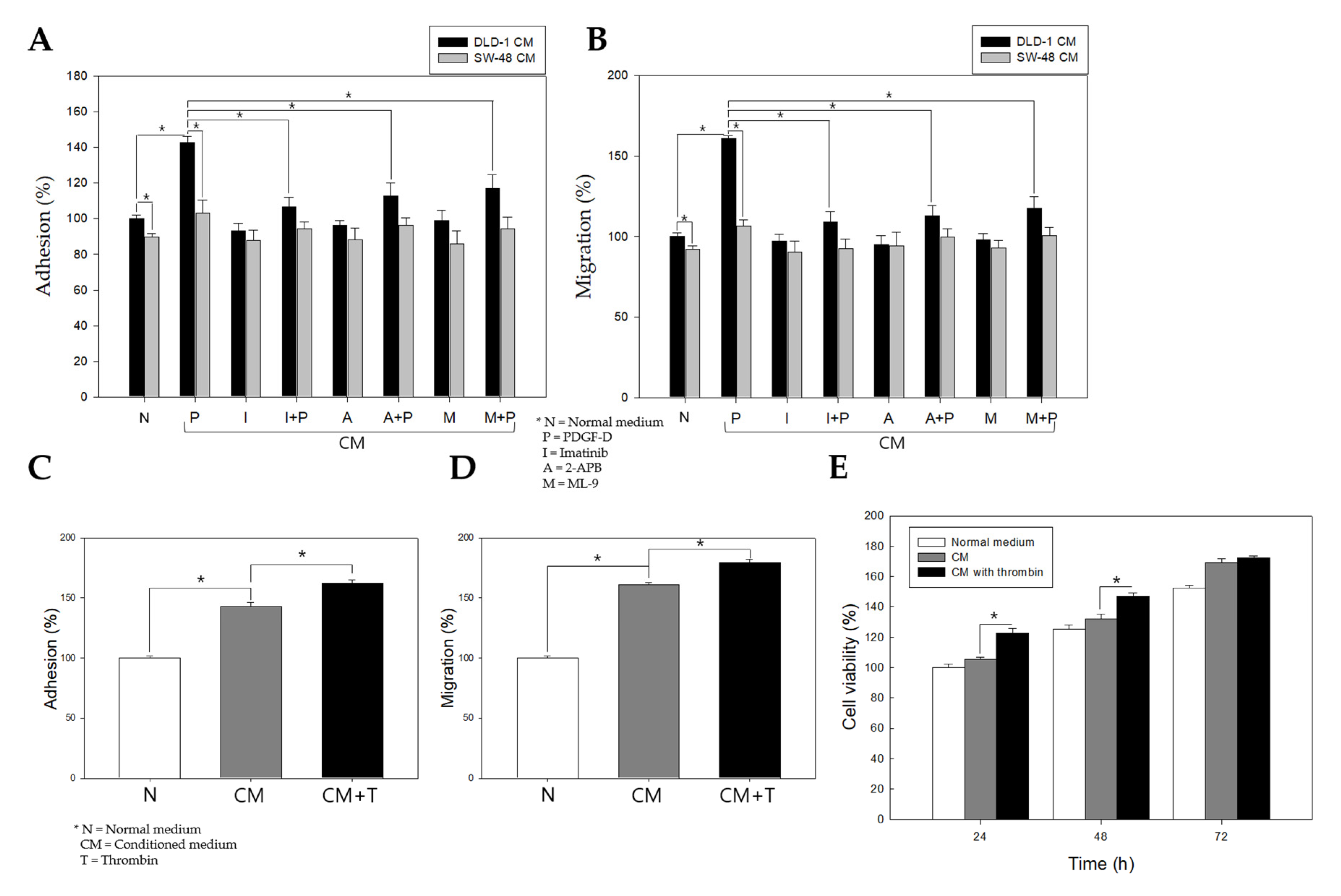
2.6. Stimulation of PDGFRβ in Colon Cancer Cells Promotes Adhesion, Migration, and Proliferation of Colonic Myofibroblasts
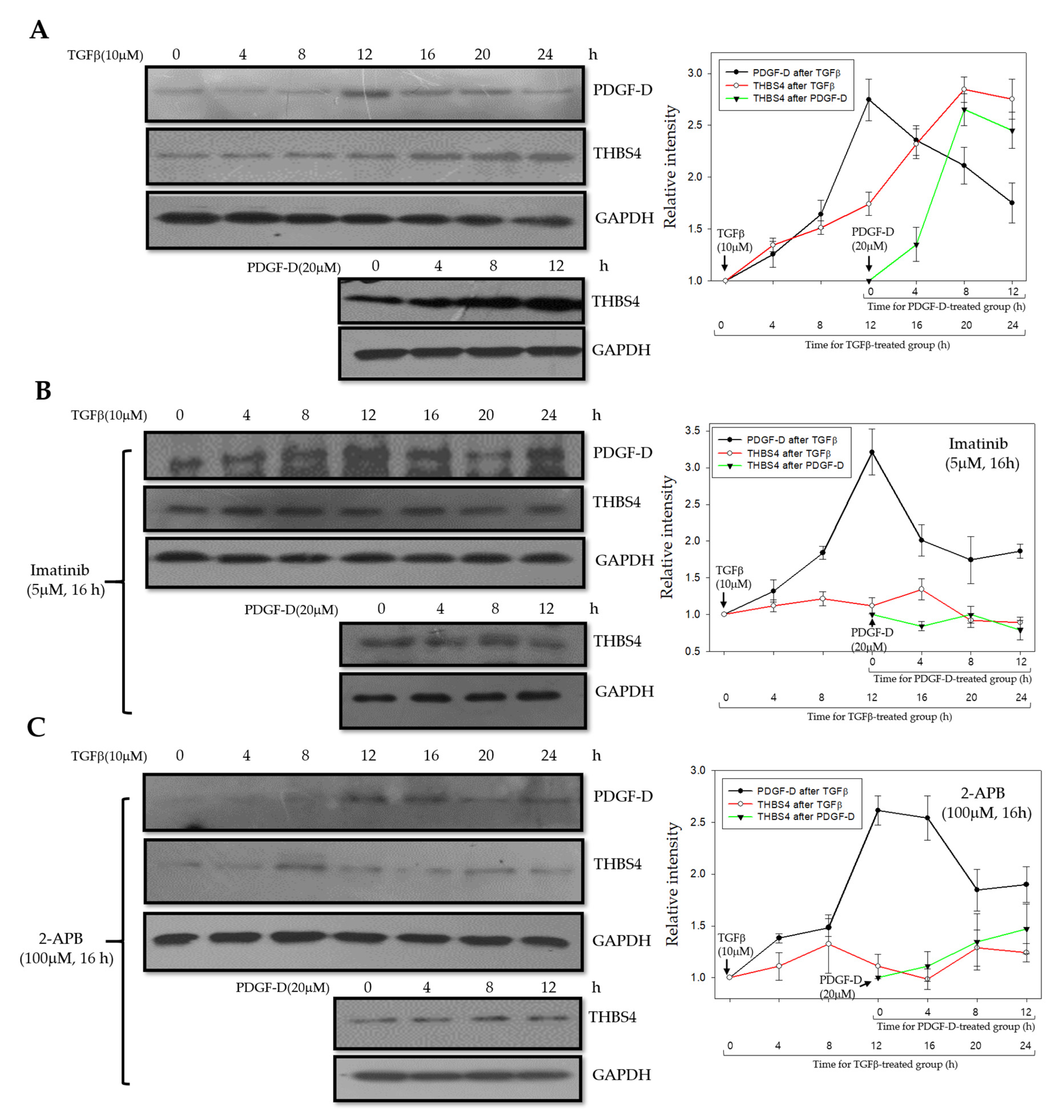
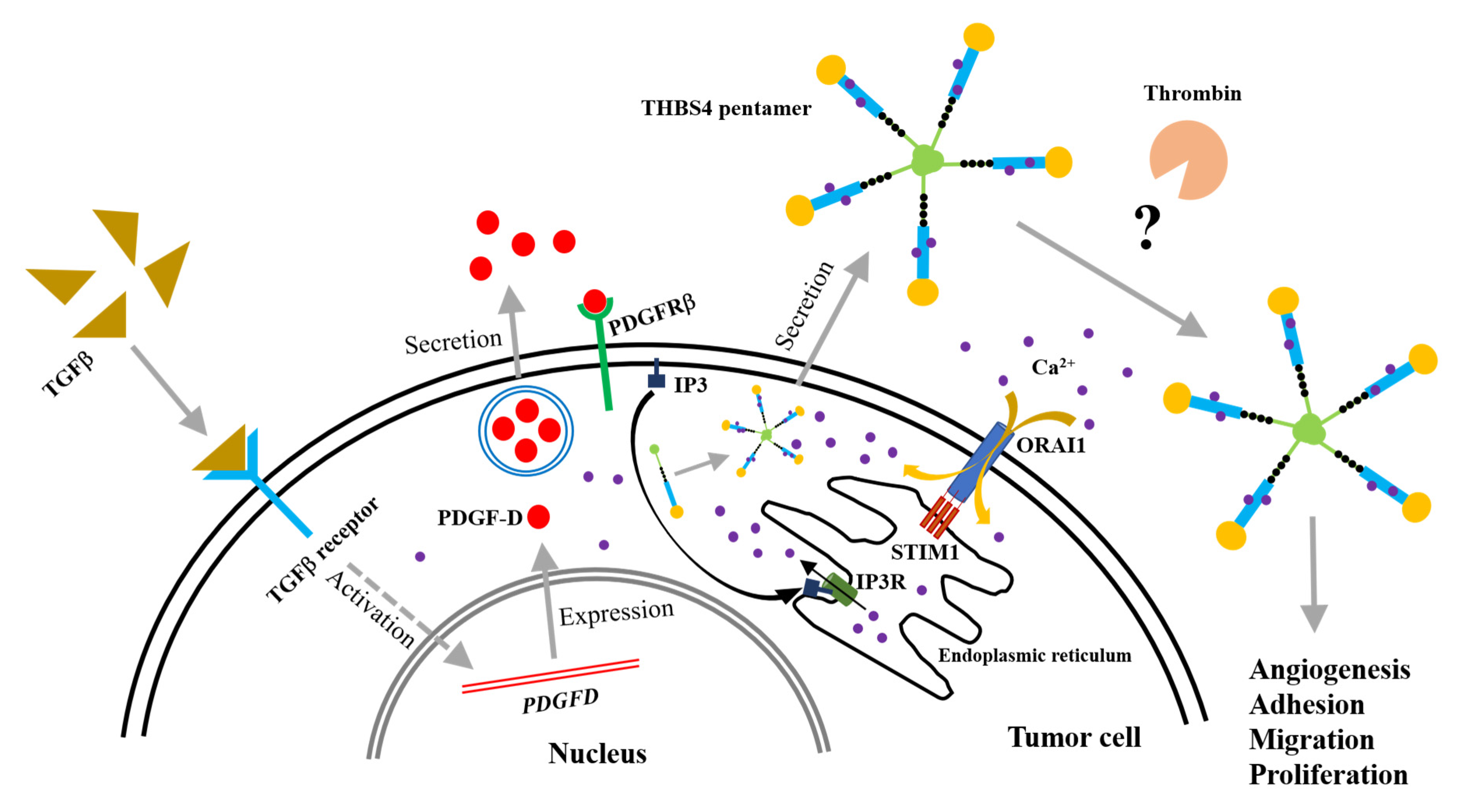
2.7. A Proposed Novel Molecular Pathway for the Development of Colon Cancer
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Human Tissue
4.3. Transfection
4.4. Western Blot Analysis
4.5. Trichloroacetic Acid Precipitation
4.6. Immunofluorescence
4.7. Migration Assay
4.8. Proliferation Assay
4.9. Adhesion Assay
4.10. Quantitative Reverse Transcription-PCR (qRT-PCR)
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- De Rosa, M.; Pace, U.; Rega, D.; Costabile, V.; Duraturo, F.; Izzo, P.; Delrio, P. Genetics, diagnosis and management of colorectal cancer (Review). Oncol. Rep. 2015, 34, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castells, M.; Thibault, B.; Mery, E.; Golzio, M.; Pasquet, M.; Hennebelle, I.; Bourin, P.; Mirshahi, M.; Delord, J.P.; Querleu, D. Ovarian ascites-derived Hospicells promote angiogenesis via activation of macrophages. Cancer Lett. 2012, 326, 59–68. [Google Scholar] [CrossRef]
- Guo, S.; Deng, C.-X. Effect of stromal cells in tumor microenvironment on metastasis initiation. Int. J. Biol. Sci. 2018, 14, 2083. [Google Scholar] [CrossRef] [PubMed]
- Castells, M.; Thibault, B.; Delord, J.P.; Couderc, B. Implication of tumor microenvironment in chemoresistance: Tumor-associated stromal cells protect tumor cells from cell death. Int. J. Mol. Sci. 2012, 13, 9545–9571. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.C. Thrombospondins: Multifunctional regulators of cell interactions. Annu. Rev. Cell Dev. Biol. 2001, 17, 25–51. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.C.; Lawler, J. The thrombospondins. Int. J. Biochem. Cell Biol. 2004, 36, 961–968. [Google Scholar] [CrossRef]
- Bornstein, P. Thrombospondins as matricellular modulators of cell function. J. Clin. Investig. 2001, 107, 929–934. [Google Scholar] [CrossRef] [Green Version]
- Carlson, C.B.; Lawler, J.; Mosher, D.F. Structures of thrombospondins. Cell. Mol. Life Sci. CMLS 2008, 65, 672–686. [Google Scholar] [CrossRef] [Green Version]
- Narouz-Ott, L.; Maurer, P.; Nitsche, D.P.; Smyth, N.; Paulsson, M. Thrombospondin-4 binds specifically to both collagenous and non-collagenous extracellular matrix proteins via its C-terminal domains. J. Biol. Chem. 2000, 275, 37110–37117. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.C. Functions of the conserved thrombospondin carboxy-terminal cassette in cell–extracellular matrix interactions and signaling. Int. J. Biochem. Cell Biol. 2004, 36, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Caroni, P. Thrombospondin-4, an extracellular matrix protein expressed in the developing and adult nervous system promotes neurite outgrowth. J. Cell Biol. 1995, 131, 1083–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenina, O.I.; Desai, S.Y.; Krukovets, I.; Kight, K.; Janigro, D.; Topol, E.J.; Plow, E.F. Thrombospondin-4 and its variants: Expression and differential effects on endothelial cells. Circulation 2003, 108, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Forster, S.; Gretschel, S.; Jons, T.; Yashiro, M.; Kemmner, W. THBS4, a novel stromal molecule of diffuse-type gastric adenocarcinomas, identified by transcriptome-wide expression profiling. Mod. Pathol. An Off. J. United States Can. Acad. Pathol. Inc. 2011, 24, 1390–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Cheng, G.; Yang, H.; Deng, X.; Qin, C.; Hua, L.; Yin, C. Reciprocal regulation of long noncoding RNAs THBS4003 and THBS4 control migration and invasion in prostate cancer cell lines. Mol. Med. Rep. 2016, 14, 1451–1458. [Google Scholar] [CrossRef] [Green Version]
- McCart Reed, A.E.; Song, S.; Kutasovic, J.R.; Reid, L.E.; Valle, J.M.; Vargas, A.C.; Smart, C.E.; Simpson, P.T. Thrombospondin-4 expression is activated during the stromal response to invasive breast cancer. Virchows Arch. An Int. J. Pathol. 2013, 463, 535–545. [Google Scholar] [CrossRef]
- Lee, J.; Lee, W.K.; Seol, M.-Y.; Lee, S.G.; Kim, D.; Kim, H.; Park, J.; Jung, S.G.; Chung, W.Y.; Lee, E.J. Coupling of LETM1 up-regulation with oxidative phosphorylation and platelet-derived growth factor receptor signaling via YAP1 transactivation. Oncotarget 2016, 7, 66728. [Google Scholar] [CrossRef]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef]
- Ostman, A.; Heldin, C.H. PDGF receptors as targets in tumor treatment. Adv. Cancer Res. 2007, 97, 247–274. [Google Scholar] [CrossRef]
- Hwang, R.F.; Yokoi, K.; Bucana, C.D.; Tsan, R.; Killion, J.J.; Evans, D.B.; Fidler, I.J. Inhibition of platelet-derived growth factor receptor phosphorylation by STI571 (Gleevec) reduces growth and metastasis of human pancreatic carcinoma in an orthotopic nude mouse model. Clin. Cancer Res. An Off. J. Am. Assoc. Cancer Res. 2003, 9, 6534–6544. [Google Scholar]
- Maass, T.; Thieringer, F.R.; Mann, A.; Longerich, T.; Schirmacher, P.; Strand, D.; Hansen, T.; Galle, P.R.; Teufel, A.; Kanzler, S. Liver specific overexpression of platelet-derived growth factor-B accelerates liver cancer development in chemically induced liver carcinogenesis. Int. J. Cancer 2011, 128, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.; Bezwoda, W.R. Positive immunostaining for platelet derived growth factor (PDGF) is an adverse prognostic factor in patients with advanced breast cancer. Breast Cancer Res. Treat. 1994, 32, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Lindmark, G.; Sundberg, C.; Glimelius, B.; Pahlman, L.; Rubin, K.; Gerdin, B. Stromal expression of platelet-derived growth factor beta-receptor and platelet-derived growth factor B-chain in colorectal cancer. Lab. Investig. A J. Tech. Methods Pathol. 1993, 69, 682–689. [Google Scholar]
- Sundberg, C.; Ljungstrom, M.; Lindmark, G.; Gerdin, B.; Rubin, K. Microvascular pericytes express platelet-derived growth factor-beta receptors in human healing wounds and colorectal adenocarcinoma. Am. J. Pathol. 1993, 143, 1377–1388. [Google Scholar]
- Wehler, T.C.; Frerichs, K.; Graf, C.; Drescher, D.; Schimanski, K.; Biesterfeld, S.; Berger, M.R.; Kanzler, S.; Junginger, T.; Galle, P.R.; et al. PDGFRalpha/beta expression correlates with the metastatic behavior of human colorectal cancer: A possible rationale for a molecular targeting strategy. Oncol. Rep. 2008, 19, 697–704. [Google Scholar]
- Steller, E.J.; Ritsma, L.; Raats, D.A.; Hoogwater, F.J.; Emmink, B.L.; Govaert, K.M.; Laoukili, J.; Rinkes, I.H.; van Rheenen, J.; Kranenburg, O. The death receptor CD95 activates the cofilin pathway to stimulate tumour cell invasion. EMBO Rep. 2011, 12, 931–937. [Google Scholar] [CrossRef] [Green Version]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [Green Version]
- Fantauzzo, K.A.; Soriano, P. PDGFRbeta regulates craniofacial development through homodimers and functional heterodimers with PDGFRalpha. Genes Dev. 2016, 30, 2443–2458. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yuan, W.; Wu, L.; Tang, Q.; Xia, Q.; Ji, J.; Liu, Z.; Ma, Z.; Zhou, Z.; Cheng, Y.; et al. PDGF-D promotes cell growth, aggressiveness, angiogenesis and EMT transformation of colorectal cancer by activation of Notch1/Twist1 pathway. Oncotarget 2017, 8, 9961–9973. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Chen, J.; Yuan, W.; Ji, J.; Liu, Z.; Wu, L.; Tang, Q.; Shu, X. Platelet-derived growth factor-D promotes colorectal cancer cell migration, invasion and proliferation by regulating Notch1 and matrix metalloproteinase-9. Oncol. Lett. 2018, 15, 1573–1579. [Google Scholar] [CrossRef]
- Muppala, S.; Frolova, E.; Xiao, R.; Krukovets, I.; Yoon, S.; Hoppe, G.; Vasanji, A.; Plow, E.; Stenina-Adognravi, O. Proangiogenic properties of Thrombospondin-4. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1975–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muppala, S.; Xiao, R.; Krukovets, I.; Verbovetsky, D.; Yendamuri, R.; Habib, N.; Raman, P.; Plow, E.; Stenina-Adognravi, O. Thrombospondin-4 mediates TGF-beta-induced angiogenesis. Oncogene 2017, 36, 5189–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steller, E.J.; Raats, D.A.; Koster, J.; Rutten, B.; Govaert, K.M.; Emmink, B.L.; Snoeren, N.; van Hooff, S.R.; Holstege, F.C.; Maas, C.; et al. PDGFRB promotes liver metastasis formation of mesenchymal-like colorectal tumor cells. Neoplasia 2013, 15, 204–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anz, D.; Mueller, W.; Golic, M.; Kunz, W.G.; Rapp, M.; Koelzer, V.H.; Ellermeier, J.; Ellwart, J.W.; Schnurr, M.; Bourquin, C.; et al. CD103 is a hallmark of tumor-infiltrating regulatory T cells. Int. J. Cancer 2011, 129, 2417–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrun, J.J. The dual role of TGFbeta in human cancer: From tumor suppression to cancer metastasis. ISRN Mol. Biol. 2012, 2012, 381428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Al-Odaini, A.A.; Wang, Y.; Korah, J.; Dai, M.; Xiao, L.; Ali, S.; Lebrun, J.J. KiSS1 gene as a novel mediator of TGFbeta-mediated cell invasion in triple negative breast cancer. Cell. Signal. 2018, 42, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bornfeldt, K.E.; Raines, E.W.; Graves, L.M.; Skinner, M.P.; Krebs, E.G.; Ross, R. Platelet-derived growth factor. Distinct signal transduction pathways associated with migration versus proliferation. Ann. N. Y. Acad. Sci. 1995, 766, 416–430. [Google Scholar] [CrossRef]
- Heldman, A.W.; Kandzari, D.E.; Tucker, R.W.; Crawford, L.E.; Fearon, E.R.; Koblan, K.S.; Goldschmidt-Clermont, P.J. EJ-Ras inhibits phospholipase C gamma 1 but not actin polymerization induced by platelet-derived growth factor-BB via phosphatidylinositol 3-kinase. Circ. Res. 1996, 78, 312–321. [Google Scholar] [CrossRef]
- Pinzani, M. PDGF and signal transduction in hepatic stellate cells. Front. Biosci. 2002, 7, d1720–d1726. [Google Scholar] [CrossRef]
- Yao, H.; Duan, M.; Yang, L.; Buch, S. Platelet-derived growth factor-BB restores human immunodeficiency virus Tat-cocaine-mediated impairment of neurogenesis: Role of TRPC1 channels. J. Neurosci. 2012, 32, 9835–9847. [Google Scholar] [CrossRef]
- Pérez-Riesgo, E.; Gutiérrez, L.G.; Ubierna, D.; Acedo, A.; Moyer, M.P.; Núñez, L.; Villalobos, C. Transcriptomic analysis of calcium remodeling in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 922. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca2+ transport in cancer: How it contributes to cancer hallmarks? Philos. Trans. Royal Soc. B Biol. Sci. 2014, 369, 20130097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsten, E.; Uutela, M.; Li, X.; Pietras, K.; Ostman, A.; Heldin, C.H.; Alitalo, K.; Eriksson, U. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat. Cell Biol. 2001, 3, 512–516. [Google Scholar] [CrossRef]
- Greco, S.A.; Chia, J.; Inglis, K.J.; Cozzi, S.J.; Ramsnes, I.; Buttenshaw, R.L.; Spring, K.J.; Boyle, G.M.; Worthley, D.L.; Leggett, B.A.; et al. Thrombospondin-4 is a putative tumour-suppressor gene in colorectal cancer that exhibits age-related methylation. BMC Cancer 2010, 10, 494. [Google Scholar] [CrossRef] [Green Version]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Epstein, D.; Haley, J.D. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin. Exp. Metastasis 2008, 25, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Jechlinger, M.; Sommer, A.; Moriggl, R.; Seither, P.; Kraut, N.; Capodiecci, P.; Donovan, M.; Cordon-Cardo, C.; Beug, H.; Grunert, S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Investig. 2006, 116, 1561–1570. [Google Scholar] [CrossRef]
- Katz, L.H.; Li, Y.; Chen, J.S.; Munoz, N.M.; Majumdar, A.; Chen, J.; Mishra, L. Targeting TGF-beta signaling in cancer. Expert Opin. Ther. Targets 2013, 17, 743–760. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Kong, D.; Banerjee, S.; Li, Y.; Adsay, N.V.; Abbruzzese, J.; Sarkar, F.H. Down-regulation of platelet-derived growth factor-D inhibits cell growth and angiogenesis through inactivation of Notch-1 and nuclear factor-kappaB signaling. Cancer Res. 2007, 67, 11377–11385. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Csordas, G.; Varnai, P.; Golenar, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnoczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Varnai, P.; Balla, A.; Hunyady, L.; Balla, T. Targeted expression of the inositol 1,4,5-triphosphate receptor (IP3R) ligand-binding domain releases Ca2+ via endogenous IP3R channels. Proc. Natl. Acad. Sci. USA 2005, 102, 7859–7864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondratskyi, A.; Yassine, M.; Kondratska, K.; Skryma, R.; Slomianny, C.; Prevarskaya, N. Calcium-permeable ion channels in control of autophagy and cancer. Front. Physiol. 2013, 4, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandewalle, B.; Hornez, L.; Wattez, N.; Revillion, F.; Lefebvre, J. Vitamin-D3 derivatives and breast-tumor cell growth: Effect on intracellular calcium and apoptosis. Int. J. Cancer 1995, 61, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.H.; Boivin, G.P.; Prasad, V.; Periasamy, M.; Shull, G.E. Squamous cell tumors in mice heterozygous for a null allele of Atp2a2, encoding the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 Ca2+ pump. J. Biol. Chem. 2001, 276, 26737–26740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [Green Version]
- Karacosta, L.G.; Foster, B.A.; Azabdaftari, G.; Feliciano, D.M.; Edelman, A.M. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J. Biol. Chem. 2012, 287, 24832–24843. [Google Scholar] [CrossRef] [Green Version]
- Monteith, G.R.; Davis, F.M.; Roberts-Thomson, S.J. Calcium channels and pumps in cancer: Changes and consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef] [Green Version]
- Lawler, J.; Connolly, J.E.; Ferro, P.; Derick, L.H. Thrombin and chymotrypsin interactions with thrombospondin. Ann. New York Acad. Sci. 1986, 485, 273–287. [Google Scholar] [CrossRef]
- McLaughlin, J.N.; Mazzoni, M.R.; Cleator, J.H.; Earls, L.; Perdigoto, A.L.; Brooks, J.D.; Muldowney, J.A., 3rd; Vaughan, D.E.; Hamm, H.E. Thrombin modulates the expression of a set of genes including thrombospondin-1 in human microvascular endothelial cells. J. Biol. Chem. 2005, 280, 22172–22180. [Google Scholar] [CrossRef] [Green Version]
- Baenziger, N.L.; Brodie, G.N.; Majerus, P.W. A thrombin-sensitive protein of human platelet membranes. Proc. Natl. Acad. of Sci. USA 1971, 68, 240–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex | Age | Location |
|---|---|---|
| Male | 79 | Rectum |
| Male | 57 | Rectum |
| Male | 88 | Sigmoid |
| Male | 65 | Sigmoid |
| Male | 35 | Rectum |
| Male | 82 | Rectosigmoid |
| Male | 46 | Descending colon |
| Female | 77 | Sigmoid |
| Female | 41 | Sigmoid |
| Female | 76 | Rectum |
| Female | 59 | Rectosigmoid |
| Female | 79 | Rectum |
| Gene | Sequence (5’ → 3’) |
|---|---|
| PDGF-A | Forward: CGTAGGGAGTGAGGATTCTTTG Reverse: AAATGACCGTCCTGGTCTTG |
| PDGF-B | Forward: CTCGATCCGCTCCTTTGATG Reverse: AGGAAGTTGGCGTTGGTG |
| PDGF-C | Forward: GTCAATGTGTCCCAAGCAAAG Reverse: CCACGTCGGTGAGTGATTT |
| PDGF-D | Forward: GAAATTGTGGCTGTGGAACTG Reverse: GGCCAGGCTCAAACTGTAATA |
| PDGFR | Forward: GTGACAGACTACCTCTTTGG Reverse: CTACATCTCCCAGTGTCTCC |
| THBS4 | Forward: GTTCAGCCACCATCTTCGGTC Reverse: GCACCTTCCCATCGTTCTTCAG |
| GAPDH | Forward: CCACATCGCTCAGACACCATG Reverse: GTCAATGAAGGGGTCATTGATGGC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.S.; Choi, H.S.; Wu, M.; Myung, J.; Kim, E.J.; Kim, Y.S.; Ro, S.; Ha, S.E.; Bartlett, A.; Wei, L.; et al. Potential Role of PDGFRβ-Associated THBS4 in Colorectal Cancer Development. Cancers 2020, 12, 2533. https://doi.org/10.3390/cancers12092533
Kim MS, Choi HS, Wu M, Myung J, Kim EJ, Kim YS, Ro S, Ha SE, Bartlett A, Wei L, et al. Potential Role of PDGFRβ-Associated THBS4 in Colorectal Cancer Development. Cancers. 2020; 12(9):2533. https://doi.org/10.3390/cancers12092533
Chicago/Turabian StyleKim, Min Seob, Hyun Seok Choi, Moxin Wu, JiYeon Myung, Eui Joong Kim, Yong Sung Kim, Seungil Ro, Se Eun Ha, Allison Bartlett, Lai Wei, and et al. 2020. "Potential Role of PDGFRβ-Associated THBS4 in Colorectal Cancer Development" Cancers 12, no. 9: 2533. https://doi.org/10.3390/cancers12092533