Translational Considerations to Improve Response and Overcome Therapy Resistance in Immunotherapy for Hepatocellular Carcinoma

Abstract
:Simple Summary
Abstract
1. Introduction
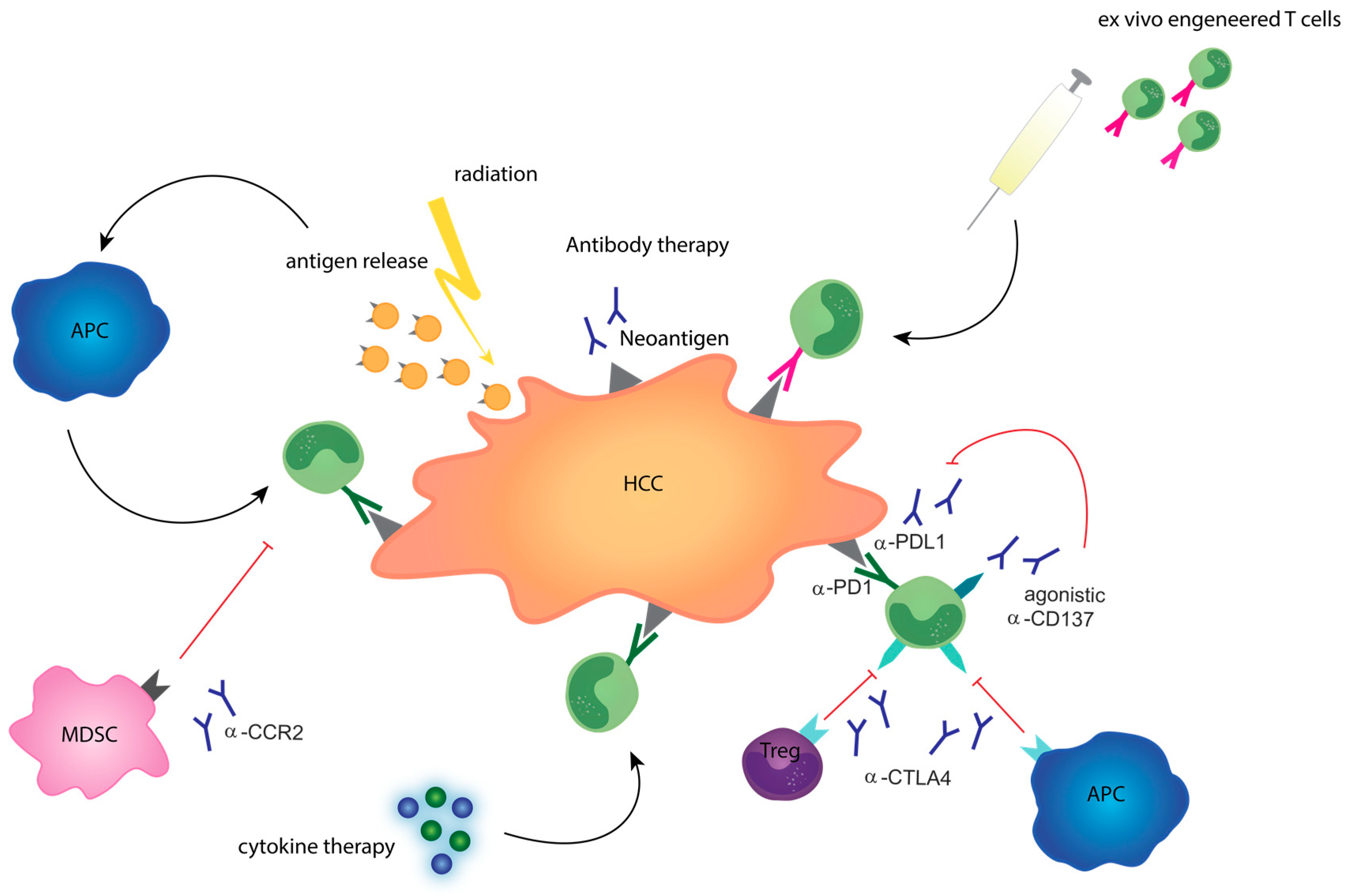
2. General Strategies for Immunomodulatory Treatments in Primary Liver Cancer
3. Predictors for Response or Resistance to Current Immune-Modulatory Therapies
3.1. Tumor Characteristics and Tumor Infiltrating Lymphocytes as Predictors for Response
3.2. Molecular Subtyping of HCC
4. Combination Treatments to Improve Therapy Response in HCC
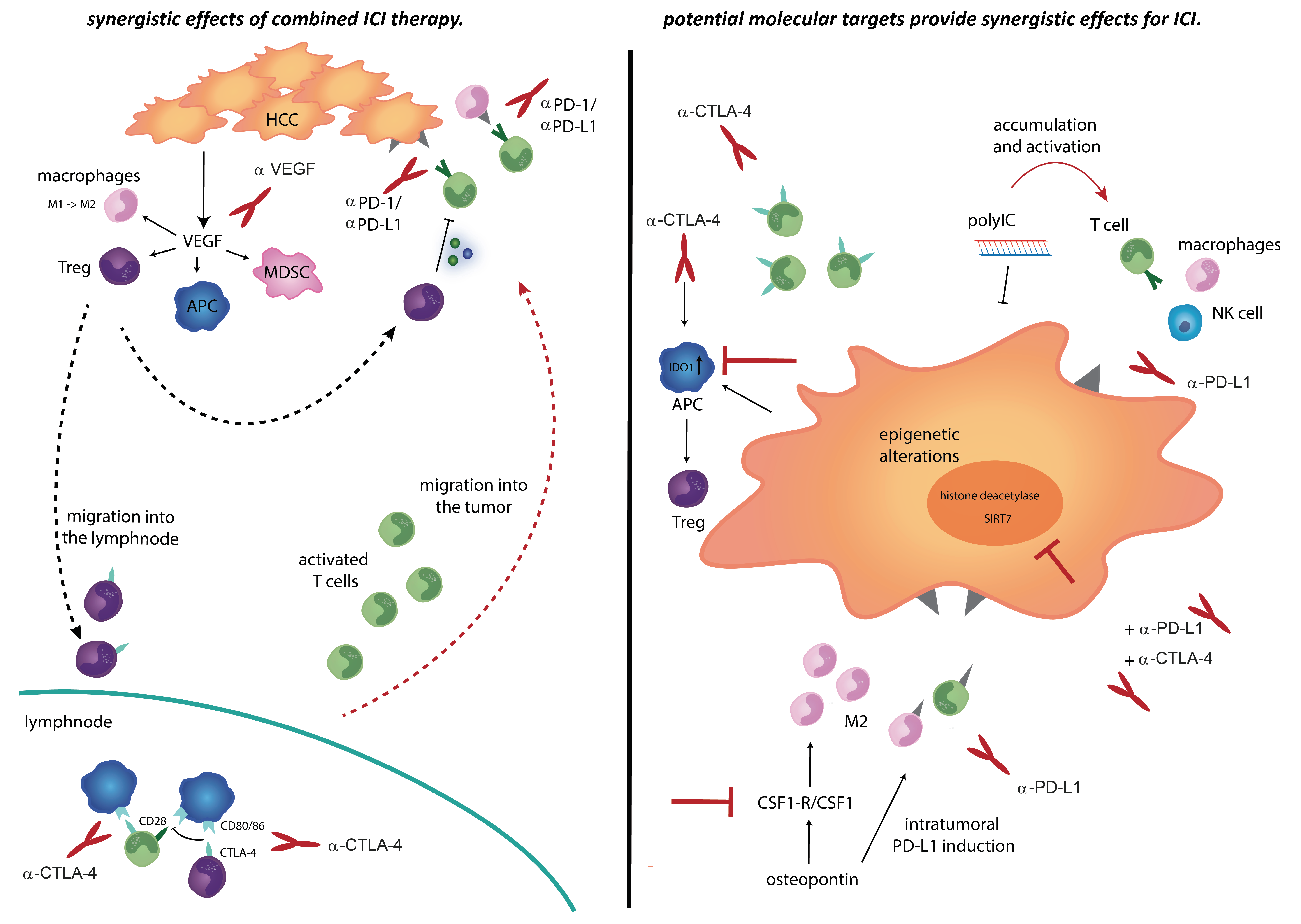
4.1. Combination Therapies of Checkpoint Inhibitors
4.2. Combination Therapies of Checkpoint Inhibitors and Anti-Angiogenesis
4.3. Combination of Immunotherapy and Locoregional Therapy
5. Translational Studies to Overcome Resistance to Immunotherapies
5.1. Checkpoint Inhibitors
5.2. Application of Neoantigens and Oncolytic Viruses in Immunotherapy
5.3. Targeting HCC Biomarkers–Vaccines, Antibodies, and Cytokines
5.4. Adoptive Cell Transfer and CAR T Cells
5.5. Targeting Cross-Communication between MDSCs and the TME
5.6. Targeting MDSC, TAMs, and Innate Immunity Interaction for HCC Prevention
6. Conclusions and Future Direction
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ab | Antibody |
| AFP | alpha-fetoprotein |
| APC | antigen presenting cells |
| CCRK | cell cycle-related kinase |
| CPS | combined positive score |
| CSF1 | colony stimulating factor 1 |
| CSF1R | colony stimulating factor receptor 1 |
| CTLA-4 | cytotoxic T lymphocyte antigen 4 |
| CXCR-4 | CXC receptor type 4 |
| DC | dendritic cell |
| GPC3 | glypican-3 |
| HCC | hepatocellular carcinoma |
| HSC | hepatic stellate cell |
| ICI | immune checkpoint inhibitor |
| IDO1 | indoleamine 2,3-dioxygenase 1 |
| LAG3 | lymphocyte activation gene 3 |
| M-MDSC | monocytic MDSC |
| MDSC | myeloid-derived suppressor cells |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steato hepatitis |
| NF-κB | nuclear factor-κB |
| NK cells | natural killer cells |
| NSCLC | non-small cell lung cancer |
| OR | overall response rate |
| OS | overall survival |
| PBMC | peripheral blood mononuclear cells |
| PD-1 | programmed death protein 1 |
| PD-L1 | programmed death ligand 1 |
| PFS | progression free survival |
| PSC | primary sclerosing cholangitis |
| TAM | tumor-associated macrophages |
| Th | T helper |
| TIL | tumor infiltrating lymphocytes |
| TIM3 | T cell membrane protein 3 |
| TKI | tyrosine kinase inhibitor |
| TME | tumor microenvironment |
| TPS | tumor proportion score |
| Treg | regulatory T cells |
| VEGF | vascular endothelial growth factor |
References
- Khan, S.A.; Taylor-Robinson, S.D.; Toledano, M.B.; Beck, A.; Elliott, P.; Thomas, H.C. Changing international trends in mortality rates for liver, biliary and pancreatic tumours. J. Hepatol. 2002, 37, 806–813. [Google Scholar] [CrossRef]
- Saha, S.K.; Zhu, A.X.; Fuchs, C.S.; Brooks, G.A. Forty-Year Trends in Cholangiocarcinoma Incidence in the U.S.: Intrahepatic Disease on the Rise. Oncologist 2016, 21, 594–599. [Google Scholar] [CrossRef] [Green Version]
- Ryerson, A.B.; Eheman, C.R.; Altekruse, S.F.; Ward, J.W.; Jemal, A.; Sherman, R.L.; Henley, S.J.; Holtzman, D.; Lake, A.; Noone, A.M.; et al. Annual Report to the Nation on the Status of Cancer, 1975-2012, featuring the increasing incidence of liver cancer. Cancer 2016, 122, 1312–1337. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Budhu, A.; Wang, X.W. The role of cytokines in hepatocellular carcinoma. J. Leukoc. Biol. 2006, 80, 1197–1213. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, Y.; Yuan, H.; Fang, Q.; Cai, N.; Suo, C.; Jin, L.; Zhang, T.; Chen, X. The trends in incidence of primary liver cancer caused by specific etiologies: Results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J. Hepatol. 2019, 70, 674–683. [Google Scholar] [CrossRef]
- Sun, B.; Karin, M. Obesity, inflammation, and liver cancer. J. Hepatol. 2012, 56, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Greten, T.F.; Wang, X.W.; Korangy, F. Current concepts of immune based treatments for patients with HCC: From basic science to novel treatment approaches. Gut 2015, 64, 842–848. [Google Scholar] [CrossRef]
- Ambade, A.; Satishchandran, A.; Saha, B.; Gyongyosi, B.; Lowe, P.; Kodys, K.; Catalano, D.; Szabo, G. Hepatocellular carcinoma is accelerated by NASH involving M2 macrophage polarization mediated by hif-1αinduced IL-10. Oncoimmunology 2016, 5, e1221557. [Google Scholar] [CrossRef] [Green Version]
- Galle, P.R.; Tovoli, F.; Foerster, F.; Worns, M.A.; Cucchetti, A.; Bolondi, L. The treatment of intermediate stage tumours beyond TACE: From surgery to systemic therapy. J. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Greten, T.F. Liver cancer: Regorafenib as second-line therapy in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Pinyol, R.; Sia, D.; Llovet, J.M. Immune Exclusion-Wnt/CTNNB1 Class Predicts Resistance to Immunotherapies in HCC. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2021–2023. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Evason, K.J.; Asahina, K.; Stainier, D.Y. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Investig. 2013, 123, 1902–1910. [Google Scholar] [CrossRef] [Green Version]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Makarova-Rusher, O.V.; Medina-Echeverz, J.; Duffy, A.G.; Greten, T.F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 2015, 62, 1420–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapanadze, T.; Gamrekelashvili, J.; Ma, C.; Chan, C.; Zhao, F.; Hewitt, S.; Zender, L.; Kapoor, V.; Felsher, D.W.; Manns, M.P.; et al. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J. Hepatol. 2013, 59, 1007–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizukoshi, E.; Yamashita, T.; Arai, K.; Terashima, T.; Kitahara, M.; Nakagawa, H.; Iida, N.; Fushimi, K.; Kaneko, S. Myeloid-derived suppressor cells correlate with patient outcomes in hepatic arterial infusion chemotherapy for hepatocellular carcinoma. Cancer Immunol. Immunother. 2016, 65, 715–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.H.; Yamagiwa, S.; Ichida, T.; Matsuda, Y.; Sugahara, S.; Watanabe, H.; Sato, Y.; Abo, T.; Horwitz, D.A.; Aoyagi, Y. Increase of CD4+ CD25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J. Hepatol. 2006, 45, 254–262. [Google Scholar] [CrossRef]
- Iwata, T.; Kondo, Y.; Kimura, O.; Morosawa, T.; Fujisaka, Y.; Umetsu, T.; Kogure, T.; Inoue, J.; Nakagome, Y.; Shimosegawa, T. PD-L1(+)MDSCs are increased in HCC patients and induced by soluble factor in the tumor microenvironment. Sci. Rep. 2016, 6, 39296. [Google Scholar] [CrossRef]
- Foerster, F.; Hess, M.; Gerhold-Ay, A.; Marquardt, J.U.; Becker, D.; Galle, P.R.; Schuppan, D.; Binder, H.; Bockamp, E. The immune contexture of hepatocellular carcinoma predicts clinical outcome. Sci. Rep. 2018, 8, 5351. [Google Scholar] [CrossRef]
- Brunner, S.M.; Rubner, C.; Kesselring, R.; Martin, M.; Griesshammer, E.; Ruemmele, P.; Stempfl, T.; Teufel, A.; Schlitt, H.J.; Fichtner-Feigl, S. Tumor-infiltrating, interleukin-33-producing effector-memory CD8(+) T cells in resected hepatocellular carcinoma prolong patient survival. Hepatology 2015, 61, 1957–1967. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto, J.; Melero, I.; Sangro, B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 681–700. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.X.; Ling, Y.; Wang, H.Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2018, 2, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprinzl, M.F.; Reisinger, F.; Puschnik, A.; Ringelhan, M.; Ackermann, K.; Hartmann, D.; Schiemann, M.; Weinmann, A.; Galle, P.R.; Schuchmann, M.; et al. Sorafenib perpetuates cellular anticancer effector functions by modulating the crosstalk between macrophages and natural killer cells. Hepatology 2013, 57, 2358–2368. [Google Scholar] [CrossRef]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, D.F.; Terabe, M.; Kapoor, V.; ElGindi, M.; et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016, 531, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Kuang, D.M.; Pan, W.D.; Wan, Y.L.; Lao, X.M.; Wang, D.; Li, X.F.; Zheng, L. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology 2013, 57, 1107–1116. [Google Scholar] [CrossRef]
- Sui, Q.; Zhang, J.; Sun, X.; Zhang, C.; Han, Q.; Tian, Z. NK cells are the crucial antitumor mediators when STAT3-mediated immunosuppression is blocked in hepatocellular carcinoma. J. Immunol. 2014, 193, 2016–2023. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ding, T.; Pan, W.; Zhu, L.Y.; Li, L.; Zheng, L. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int. J. Cancer 2009, 125, 1640–1648. [Google Scholar] [CrossRef]
- Dong, P.; Ma, L.; Liu, L.; Zhao, G.; Zhang, S.; Dong, L.; Xue, R.; Chen, S. CD86⁺/CD206⁺, Diametrically Polarized Tumor-Associated Macrophages, Predict Hepatocellular Carcinoma Patient Prognosis. Int. J. Mol. Sci. 2016, 17, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet. Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2019, 37, 4012. [Google Scholar] [CrossRef]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Inarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Furuse, J.; Galle, P.R.; Kelley, R.K.; Qin, S.; Armstrong, J.; Darilay, A.; Vlahovic, G.; Negro, A.; et al. A randomized, multicenter phase 3 study of durvalumab (D) and tremelimumab (T) as first-line treatment in patients with unresectable hepatocellular carcinoma (HCC): HIMALAYA study. J. Clin. Oncol. 2018, 36, TPS4144. [Google Scholar] [CrossRef]
- Kelley, R.K.; Sangro, B.; Harris, W.P.; Ikeda, M.; Okusaka, T.; Kang, Y.-K.; Qin, S.; Tai, W.M.D.; Lim, H.Y.; Yau, T.; et al. Efficacy, tolerability, and biologic activity of a novel regimen of tremelimumab (T) in combination with durvalumab (D) for patients (pts) with advanced hepatocellular carcinoma (aHCC). J. Clin. Oncol. 2020, 38, 4508. [Google Scholar] [CrossRef]
- Yau, T.; Zagonel, V.; Santoro, A.; Acosta-Rivera, M.; Choo, S.P.; Matilla, A.; He, A.R.; Gracián, A.C.; El-Khoueiry, A.B.; Sangro, B.; et al. Nivolumab (NIVO) + ipilimumab (IPI) + cabozantinib (CABO) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2020, 38, 478. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Brown, Z.J.; Greten, T.F.; Heinrich, B. Adjuvant Treatment of Hepatocellular Carcinoma: Prospect of Immunotherapy. Hepatology 2019, 70, 1437–1442. [Google Scholar] [CrossRef]
- Adcock, C.S.; Puneky, L.V.; Campbell, G.S. Favorable Response of Metastatic Hepatocellular Carcinoma to Treatment with Trans-arterial Radioembolization Followed by Sorafenib and Nivolumab. Cureus 2019, 11, e4083. [Google Scholar] [CrossRef] [Green Version]
- Crispe, I.N. The liver as a lymphoid organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, M.B.; Krishnan, S.; Hodge, J.W.; Chang, J.Y. Immunotherapy and stereotactic ablative radiotherapy (ISABR): A curative approach? Nat. Rev. Clin. Oncol. 2016, 13, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, E.; Yamashita, T.; Arai, K.; Sunagozaka, H.; Ueda, T.; Arihara, F.; Kagaya, T.; Yamashita, T.; Fushimi, K.; Kaneko, S. Enhancement of tumor-associated antigen-specific T cell responses by radiofrequency ablation of hepatocellular carcinoma. Hepatology 2013, 57, 1448–1457. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, Z.; Peng, M.; Fu, S.; Xue, Z.; Zhang, R. CAR-T cell therapy in gastrointestinal tumors and hepatic carcinoma: From bench to bedside. Oncoimmunology 2016, 5, e1251539. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.H.; Lim, Y.S.; Yeon, J.E.; Song, T.J.; Yu, S.J.; Gwak, G.Y.; Kim, K.M.; Kim, Y.J.; Lee, J.W.; et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 2015, 148, 1383–1391.e16. [Google Scholar] [CrossRef] [Green Version]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef]
- Hogdall, D.; Lewinska, M.; Andersen, J.B. Desmoplastic Tumor Microenvironment and Immunotherapy in Cholangiocarcinoma. Trends Cancer 2018, 4, 239–255. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Brown, Z.J.; Heinrich, B.; Greten, T.F. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 536–554. [Google Scholar] [CrossRef] [PubMed]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Duffy, A.G.; Mabry-Hrones, D.; Wood, B.; Levy, E.; Krishnasamy, V.; Khan, J.; Wei, J.S.; Agdashian, D.; Tyagi, M.; et al. Tremelimumab in Combination With Microwave Ablation in Patients With Refractory Biliary Tract Cancer. Hepatology 2019, 69, 2048–2060. [Google Scholar] [CrossRef]
- Gou, M.; Zhang, Y.; Si, H.; Dai, G. Efficacy and safety of nivolumab for metastatic biliary tract cancer. Onco Targets Ther. 2019, 12, 861–867. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.D.; Kim, D.W.; Alese, O.B.; Li, D.; Shah, N.; Schell, M.J.; Zhou, J.M.; Chung, V. A phase II study of nivolumab in patients with advanced refractory biliary tract cancers (BTC). J. Clin. Oncol. 2019, 37, 4097. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.-Y.; Breder, V.V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Results of KEYNOTE-240: Phase 3 study of pembrolizumab (Pembro) vs best supportive care (BSC) for second line therapy in advanced hepatocellular carcinoma (HCC). Am. Soc. Clin. Oncol. 2019, 37, 4004. [Google Scholar] [CrossRef]
- Li, G.; Liu, D.; Cooper, T.K.; Kimchi, E.T.; Qi, X.; Avella, D.M.; Li, N.; Yang, Q.X.; Kester, M.; Rountree, C.B.; et al. Successful chemoimmunotherapy against hepatocellular cancer in a novel murine model. J. Hepatol. 2017, 66, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, D.; Baird, J.R.; Young, K.H.; Cottam, B.; Crittenden, M.R.; Friedman, S.; Gough, M.J.; Newell, P. Programmed cell death-1 blockade enhances response to stereotactic radiation in an orthotopic murine model of hepatocellular carcinoma. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2017, 47, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.-H.; Harding, J.J.; Merle, P.; et al. LBA38_PRCheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Champiat, S.; Ferrara, R.; Massard, C.; Besse, B.; Marabelle, A.; Soria, J.C.; Ferte, C. Hyperprogressive disease: Recognizing a novel pattern to improve patient management. Nat. Rev. Clin. Oncol. 2018, 15, 748–762. [Google Scholar] [CrossRef]
- Kim, H.D.; Song, G.W.; Park, S.; Jung, M.K.; Kim, M.H.; Kang, H.J.; Yoo, C.; Yi, K.; Kim, K.H.; Eo, S.; et al. Association Between Expression Level of PD1 by Tumor-Infiltrating CD8(+) T Cells and Features of Hepatocellular Carcinoma. Gastroenterology 2018, 155, 1936–1950.e17. [Google Scholar] [CrossRef]
- Calderaro, J.; Rousseau, B.; Amaddeo, G.; Mercey, M.; Charpy, C.; Costentin, C.; Luciani, A.; Zafrani, E.S.; Laurent, A.; Azoulay, D.; et al. Programmed death ligand 1 expression in hepatocellular carcinoma: Relationship With clinical and pathological features. Hepatology 2016, 64, 2038–2046. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, X.Y.; Qiu, S.J.; Yamato, I.; Sho, M.; Nakajima, Y.; Zhou, J.; Li, B.Z.; Shi, Y.H.; Xiao, Y.S.; et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Thommen, D.S.; Koelzer, V.H.; Herzig, P.; Roller, A.; Trefny, M.; Dimeloe, S.; Kiialainen, A.; Hanhart, J.; Schill, C.; Hess, C.; et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 2018, 24, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.J.; Bao, J.J.; Wang, J.Z.; Wang, Y.; Jiang, M.; Xing, M.Y.; Zhang, W.G.; Qi, J.Y.; Roggendorf, M.; Lu, M.J.; et al. Immunostaining of PD-1/PD-Ls in liver tissues of patients with hepatitis and hepatocellular carcinoma. World J. Gastroenterol. 2011, 17, 3322–3329. [Google Scholar] [CrossRef]
- Shi, F.; Shi, M.; Zeng, Z.; Qi, R.Z.; Liu, Z.W.; Zhang, J.Y.; Yang, Y.P.; Tien, P.; Wang, F.S. PD-1 and PD-L1 upregulation promotes CD8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 2011, 128, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Sprengers, D.; Boor, P.P.C.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; de Jonge, J.; Gaspersz, M.; et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017, 153, 1107–1119.e10. [Google Scholar] [CrossRef] [PubMed]
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; von Felden, J.; Labgaa, I.; D’Avola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291. [Google Scholar] [CrossRef] [PubMed]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20. [Google Scholar] [CrossRef]
- Ma, L.; Hernandez, M.O.; Zhao, Y.; Mehta, M.; Tran, B.; Kelly, M.; Rae, Z.; Hernandez, J.M.; Davis, J.L.; Martin, S.P.; et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 2019, 36, 418–430.e6. [Google Scholar] [CrossRef] [Green Version]
- Kulangara, K.; Zhang, N.; Corigliano, E.; Guerrero, L.; Waldroup, S.; Jaiswal, D.; Ms, M.J.; Shah, S.; Hanks, D.; Wang, J.; et al. Clinical Utility of the Combined Positive Score for Programmed Death Ligand-1 Expression and the Approval of Pembrolizumab for Treatment of Gastric Cancer. Arch. Pathol. Lab. Med. 2019, 143, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Daud, A.I.; Wolchok, J.D.; Robert, C.; Hwu, W.J.; Weber, J.S.; Ribas, A.; Hodi, F.S.; Joshua, A.M.; Kefford, R.; Hersey, P.; et al. Programmed Death-Ligand 1 Expression and Response to the Anti-Programmed Death 1 Antibody Pembrolizumab in Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 4102–4109. [Google Scholar] [CrossRef] [PubMed]
- Udall, M.; Rizzo, M.; Kenny, J.; Doherty, J.; Dahm, S.; Robbins, P.; Faulkner, E. PD-L1 diagnostic tests: A systematic literature review of scoring algorithms and test-validation metrics. Diagn. Pathol. 2018, 13, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinato, D.J.; Guerra, N.; Fessas, P.; Murphy, R.; Mineo, T.; Mauri, F.A.; Mukherjee, S.K.; Thursz, M.; Wong, C.N.; Sharma, R.; et al. Immune-based therapies for hepatocellular carcinoma. Oncogene 2020, 39, 3620–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Ang, C.; Klempner, S.J.; Ali, S.M.; Madison, R.; Ross, J.S.; Severson, E.A.; Fabrizio, D.; Goodman, A.; Kurzrock, R.; Suh, J.; et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget 2019, 10, 4018–4025. [Google Scholar] [CrossRef] [Green Version]
- Goumard, C.; Desbois-Mouthon, C.; Wendum, D.; Calmel, C.; Merabtene, F.; Scatton, O.; Praz, F. Low Levels of Microsatellite Instability at Simple Repeated Sequences Commonly Occur in Human Hepatocellular Carcinoma. Cancer Genom. Proteom. 2017, 14, 329–339. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Agdashian, D.; ElGindi, M.; Xie, C.; Sandhu, M.; Pratt, D.; Kleiner, D.E.; Figg, W.D.; Rytlewski, J.A.; Sanders, C.; Yusko, E.C.; et al. The effect of anti-CTLA4 treatment on peripheral and intra-tumoral T cells in patients with hepatocellular carcinoma. Cancer Immunol. Immunother. 2019, 68, 599–608. [Google Scholar] [CrossRef]
- Finkelmeier, F.; Canli, O.; Tal, A.; Pleli, T.; Trojan, J.; Schmidt, M.; Kronenberger, B.; Zeuzem, S.; Piiper, A.; Greten, F.R.; et al. High levels of the soluble programmed death-ligand (sPD-L1) identify hepatocellular carcinoma patients with a poor prognosis. Eur. J. Cancer 2016, 59, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Feun, L.G.; Li, Y.Y.; Wu, C.; Wangpaichitr, M.; Jones, P.D.; Richman, S.P.; Madrazo, B.; Kwon, D.; Garcia-Buitrago, M.; Martin, P.; et al. Phase 2 study of pembrolizumab and circulating biomarkers to predict anticancer response in advanced, unresectable hepatocellular carcinoma. Cancer 2019, 125, 3603–3614. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Mahoney, K.M.; Giobbie-Hurder, A.; Zhao, F.; Lee, S.; Liao, X.; Rodig, S.; Li, J.; Wu, X.; Butterfield, L.H.; et al. Soluble PD-L1 as a Biomarker in Malignant Melanoma Treated with Checkpoint Blockade. Cancer Immunol. Res. 2017, 5, 480–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE(2)-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J. Immunother Cancer 2019, 7, 193. [Google Scholar] [CrossRef] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360. [Google Scholar] [CrossRef] [Green Version]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [Green Version]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sánchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. β-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Qin, S.; Finn, R.S.; Kudo, M.; Meyer, T.; Vogel, A.; Ducreux, M.; Macarulla, T.M.; Tomasello, G.; Boisserie, F.; Hou, J.; et al. RATIONALE 301 study: Tislelizumab versus sorafenib as first-line treatment for unresectable hepatocellular carcinoma. Future Oncol. 2019, 15, 1811–1822. [Google Scholar] [CrossRef] [Green Version]
- Jimenez Exposito, M.J.; Akce, M.; Alvarez, J.L.M.; Assenat, E.; Balart, L.A.; Baron, A.D.; Decaens, T.; Heurgue-Berlot, A.; Martin, A.O.; Paik, S.W.; et al. CA209-9DX: Phase III, randomized, double-blind study of adjuvant nivolumab vs placebo for patients with hepatocellular carcinoma (HCC) at high risk of recurrence after curative resection or ablation. Ann. Oncol. 2018, 29, ix65. [Google Scholar] [CrossRef]
- Kaseb, A.O.; Pestana, R.C.; Vence, L.M.; Blando, J.M.; Singh, S.; Ikoma, N.; Vauthey, J.-N.; Allison, J.P.; Sharma, P. Randomized, open-label, perioperative phase II study evaluating nivolumab alone versus nivolumab plus ipilimumab in patients with resectable HCC. J. Clin. Oncol. 2019, 37, 185. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Golan, T.; Lin, C.-C.; Dahan, L.; Fu, S.; Moreno, V.; Geva, R.; Reck, M.; Wasserstrom, H.A.; Mi, G.; et al. Ramucirumab (Ram) and durvalumab (Durva) treatment of metastatic non-small cell lung cancer (NSCLC), gastric/gastroesophageal junction (G/GEJ) adenocarcinoma, and hepatocellular carcinoma (HCC) following progression on systemic treatment(s). J. Clin. Oncol. 2019, 37, 2528. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Ikeda, M.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. A phase Ib study of lenvatinib (LEN) plus pembrolizumab (PEMBRO) in unresectable hepatocellular carcinoma (uHCC). J. Clin. Oncol. 2020, 38, 4519. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kudo, M.; Cheng, A.-L.; Finn, R.S.; Galle, P.R.; Kaneko, S.; Meyer, T.; Qin, S.; Dutcus, C.E.; Chen, E.; et al. Lenvatinib (len) plus pembrolizumab (pembro) for the first-line treatment of patients (pts) with advanced hepatocellular carcinoma (HCC): Phase 3 LEAP-002 study. J. Clin. Oncol. 2019, 37, TPS4152. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Jia, R.; Yue, C.; Chang, L.; Liu, R.; Zhang, G.; Zhao, C.; Zhang, Y.; Chen, C.; et al. Anti-PD-1 Antibody SHR-1210 Combined with Apatinib for Advanced Hepatocellular Carcinoma, Gastric, or Esophagogastric Junction Cancer: An Open-label, Dose Escalation and Expansion Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Pishvaian, M.J.; Lee, M.S.; Ryoo, B.Y.; Stein, S.; Lee, K.H.; Verret, W.; Spahn, J.; Shao, H.; Liu, B.; Iizuka, K.; et al. Updated safety and clinical activity results from a phase Ib study of atezolizumab + bevacizumab in hepatocellular carcinoma (HCC). Ann. Oncol. 2018, 29, viii718–viii719. [Google Scholar] [CrossRef]
- Kudo, M.; Motomura, K.; Wada, Y.; Inaba, Y.; Sakamoto, Y.; Kurosaki, M.; Umeyama, Y.; Kamei, Y.; Yoshimitsu, J.; Fujii, Y.; et al. First-line avelumab + axitinib in patients with advanced hepatocellular carcinoma: Results from a phase 1b trial (VEGF Liver 100). J. Clin. Oncol. 2019, 37, 4072. [Google Scholar] [CrossRef]
- Kelley, R.K.; Cheng, A.-L.; Braiteh, F.S.; Park, J.-W.; Benzaghou, F.; Milwee, S.; Borgman, A.; El-Khoueiry, A.B.; Kayali, Z.K.; Zhu, A.X.; et al. Phase 3 (COSMIC-312) study of cabozantinib (C) in combination with atezolizumab (A) versus sorafenib (S) in patients (pts) with advanced hepatocellular carcinoma (aHCC) who have not received previous systemic anticancer therapy. J. Clin. Oncol. 2019, 37, TPS4157. [Google Scholar] [CrossRef]
- Knox, J.; Cheng, A.; Cleary, S.; Galle, P.; Kokudo, N.; Lencioni, R.; Park, J.; Zhou, J.; Mann, H.; Morgan, S.; et al. A phase 3 study of durvalumab with or without bevacizumab as adjuvant therapy in patients with hepatocellular carcinoma (HCC) who are at high risk of recurrence after curative hepatic resection. Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Sangro, B.; Kudo, M.; Qin, S.; Ren, Z.; Chan, S.; Erinjeri, J.; Arai, Y.; He, P.; Morgan, S.; Cohen, G.; et al. P-347 A phase 3, randomized, double-blind, placebo-controlled study of transarterial chemoembolization combined with durvalumab or durvalumab plus bevacizumab therapy in patients with locoregional hepatocellular carcinoma: EMERALD-1. Ann. Oncol. 2020, 31, S202–S203. [Google Scholar] [CrossRef]
- Floudas, C.S.; Xie, C.; Brar, G.; Morelli, M.P.; Fioravanti, S.; Walker, M.; Mabry-Hrones, D.; Wood, B.J.; Levy, E.B.; Krishnasamy, V.P.; et al. Combined immune checkpoint inhibition (ICI) with tremelimumab and durvalumab in patients with advanced hepatocellular carcinoma (HCC) or biliary tract carcinomas (BTC). J. Clin. Oncol. 2019, 37, 336. [Google Scholar] [CrossRef]
- Popovic, A.; Sugar, E.; Ferguson, A.; Wilt, B.; Durham, J.N.; Kamel, I.R.; Kim, A.; Philosophe, B.; Anders, R.A.; Jaffee, E.M.; et al. Abstract CT207: Feasibility of neoadjuvant cabozantinib plus nivolumab followed by definitive resection for patients with locally advanced hepatocellular carcinoma: A Phase Ib trial (NCT03299946). Cancer Res. 2019, 79, CT207. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthelemy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Kaseb, A.; Vence, L.; Blando, J.; Yadav, S.; Ikoma, N.; Pestana, R.; Vauthey, J.; Cao, H.; Chun, Y.; Sakamura, D.; et al. Randomized, open-label, perioperative phase II study evaluating nivolumab alone versus nivolumab plus ipilimumab in patients with resectable HCC. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30 (Suppl. 4), iv112. [Google Scholar] [CrossRef]
- Kelley, R.K.; Abou-Alfa, G.K.; Bendell, J.C.; Kim, T.-Y.; Borad, M.J.; Yong, W.-P.; Morse, M.; Kang, Y.-K.; Rebelatto, M.; Makowsky, M.; et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. J. Clin. Oncol. 2017, 35, 4073. [Google Scholar] [CrossRef]
- Deng, Y.R.; Liu, W.B.; Lian, Z.X.; Li, X.; Hou, X. Sorafenib inhibits macrophage-mediated epithelial-mesenchymal transition in hepatocellular carcinoma. Oncotarget 2016, 7, 38292–38305. [Google Scholar] [CrossRef]
- Kato, Y.; Tabata, K.; Kimura, T.; Yachie-Kinoshita, A.; Ozawa, Y.; Yamada, K.; Ito, J.; Tachino, S.; Hori, Y.; Matsuki, M.; et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS ONE 2019, 14, e0212513. [Google Scholar] [CrossRef]
- Zerbini, A.; Pilli, M.; Laccabue, D.; Pelosi, G.; Molinari, A.; Negri, E.; Cerioni, S.; Fagnoni, F.; Soliani, P.; Ferrari, C.; et al. Radiofrequency thermal ablation for hepatocellular carcinoma stimulates autologous NK-cell response. Gastroenterology 2010, 138, 1931–1942. [Google Scholar] [CrossRef]
- Nobuoka, D.; Motomura, Y.; Shirakawa, H.; Yoshikawa, T.; Kuronuma, T.; Takahashi, M.; Nakachi, K.; Ishii, H.; Furuse, J.; Gotohda, N.; et al. Radiofrequency ablation for hepatocellular carcinoma induces glypican-3 peptide-specific cytotoxic T lymphocytes. Int. J. Oncol. 2012, 40, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Hansler, J.; Wissniowski, T.T.; Schuppan, D.; Witte, A.; Bernatik, T.; Hahn, E.G.; Strobel, D. Activation and dramatically increased cytolytic activity of tumor specific T lymphocytes after radio-frequency ablation in patients with hepatocellular carcinoma and colorectal liver metastases. World J. Gastroenterol. 2006, 12, 3716–3721. [Google Scholar] [CrossRef] [PubMed]
- den Brok, M.H.; Sutmuller, R.P.; van der Voort, R.; Bennink, E.J.; Figdor, C.G.; Ruers, T.J.; Adema, G.J. In situ tumor ablation creates an antigen source for the generation of antitumor immunity. Cancer Res. 2004, 64, 4024–4029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Brok, M.H.; Sutmuller, R.P.; Nierkens, S.; Bennink, E.J.; Frielink, C.; Toonen, L.W.; Boerman, O.C.; Figdor, C.G.; Ruers, T.J.; Adema, G.J. Efficient loading of dendritic cells following cryo and radiofrequency ablation in combination with immune modulation induces anti-tumour immunity. Br. J. Cancer 2006, 95, 896–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, T.F.; Mauda-Havakuk, M.; Heinrich, B.; Korangy, F.; Wood, B.J. Combined locoregional-immunotherapy for liver cancer. J. Hepatol. 2019, 70, 999–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apetoh, L.; Mignot, G.; Panaretakis, T.; Kroemer, G.; Zitvogel, L. Immunogenicity of anthracyclines: Moving towards more personalized medicine. Trends Mol. Med. 2008, 14, 141–151. [Google Scholar] [CrossRef]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Kruger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef]
- Arihara, F.; Mizukoshi, E.; Kitahara, M.; Takata, Y.; Arai, K.; Yamashita, T.; Nakamoto, Y.; Kaneko, S. Increase in CD14+HLA-DR -/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol. Immunother. 2013, 62, 1421–1430. [Google Scholar] [CrossRef] [Green Version]
- Chew, V.; Lee, Y.H.; Pan, L.; Nasir, N.J.M.; Lim, C.J.; Chua, C.; Lai, L.; Hazirah, S.N.; Lim, T.K.H.; Goh, B.K.P.; et al. Immune activation underlies a sustained clinical response to Yttrium-90 radioembolisation in hepatocellular carcinoma. Gut 2019, 68, 335–346. [Google Scholar] [CrossRef]
- Bresnahan, E.; Lindblad, K.E.; Ruiz de Galarreta, M.; Lujambio, A. Mouse models of oncoimmunology in hepatocellular carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D. PD-1 Blockers. Cell 2015, 162, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Dika, I.; Khalil, D.N.; Abou-Alfa, G.K. Immune checkpoint inhibitors for hepatocellular carcinoma. Cancer 2019, 125, 3312–3319. [Google Scholar] [CrossRef]
- Brown, Z.J.; Yu, S.J.; Heinrich, B.; Ma, C.; Fu, Q.; Sandhu, M.; Agdashian, D.; Zhang, Q.; Korangy, F.; Greten, T.F. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol. Immunother. 2018, 67, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kato, Y.; Ozawa, Y.; Kodama, K.; Ito, J.; Ichikawa, K.; Yamada, K.; Hori, Y.; Tabata, K.; Takase, K.; et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1-6 hepatocellular carcinoma model. Cancer Sci. 2018, 109, 3993–4002. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Liao, R.; Wang, G.; Yang, B.H.; Luo, X.; Varki, N.M.; Qiu, S.J.; Ren, B.; Fu, W.; Feng, G.S. Preventive Inhibition of Liver Tumorigenesis by Systemic Activation of Innate Immune Functions. Cell Rep. 2017, 21, 1870–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, L.; Xin, B.; Wu, P.; Lin, C.H.; Peng, C.; Wang, G.; Lee, J.; Lu, L.F.; Feng, G.S. An Efficient Combination Immunotherapy for Primary Liver Cancer by Harmonized Activation of Innate and Adaptive Immunity in Mice. Hepatology 2019, 69, 2518–2532. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, J.; Xu, D.; Gao, X.M.; Zhang, Z.; Hsu, J.L.; Li, C.W.; Lim, S.O.; Sheng, Y.Y.; Zhang, Y.; et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut 2019, 68, 1653–1666. [Google Scholar] [CrossRef]
- Llopiz, D.; Ruiz, M.; Villanueva, L.; Iglesias, T.; Silva, L.; Egea, J.; Lasarte, J.J.; Pivette, P.; Trochon-Joseph, V.; Vasseur, B.; et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 2019, 68, 379–393. [Google Scholar] [CrossRef]
- Xiang, J.; Zhang, N.; Sun, H.; Su, L.; Zhang, C.; Xu, H.; Feng, J.; Wang, M.; Chen, J.; Liu, L.; et al. Disruption of SIRT7 Increases the Efficacy of Checkpoint Inhibitor via MEF2D Regulation of Programmed Cell Death 1 Ligand 1 in Hepatocellular Carcinoma Cells. Gastroenterology 2020, 158, 664–678.e24. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Yoo, S.Y.; Badrinath, N.; Woo, H.Y.; Heo, J. Oncolytic Virus-Based Immunotherapies for Hepatocellular Carcinoma. Mediat. Inflamm. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Nakatake, R.; Kaibori, M.; Nakamura, Y.; Tanaka, Y.; Matushima, H.; Okumura, T.; Murakami, T.; Ino, Y.; Todo, T.; Kon, M. Third-generation oncolytic herpes simplex virus inhibits the growth of liver tumors in mice. Cancer Sci. 2018, 109, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef]
- Luo, Y.; Lin, C.; Ren, W.; Ju, F.; Xu, Z.; Liu, H.; Yu, Z.; Chen, J.; Zhang, J.; Liu, P.; et al. Intravenous Injections of a Rationally Selected Oncolytic Herpes Virus as a Potent Virotherapy for Hepatocellular Carcinoma. Mol. Ther. Oncolytics 2019, 15, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Maeng, H.; Terabe, M.; Berzofsky, J.A. Cancer vaccines: Translation from mice to human clinical trials. Curr. Opin. Immunol. 2018, 51, 111–122. [Google Scholar] [CrossRef]
- Si, C.; Xu, M.; Lu, M.; Yu, Y.; Yang, M.; Yan, M.; Zhou, L.; Yang, X. In vivo antitumor activity evaluation of cancer vaccines prepared by various antigen forms in a murine hepatocellular carcinoma model. Oncol. Lett. 2017, 14, 7391–7397. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Chen, J.; Lan, R.; Wang, Z.; Chen, R.; Lin, J.; Zhang, L.; Fu, L. delta-Catenin peptide vaccines repress hepatocellular carcinoma growth via CD8(+) T cell activation. Oncoimmunology 2018, 7, e1450713. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Pi, L.; Le Trinh, T.; Zuo, C.; Xia, M.; Jiao, Y.; Hou, Z.; Jo, S.; Puszyk, W.; Pham, K.; et al. A Novel Vaccine Targeting Glypican-3 as a Treatment for Hepatocellular Carcinoma. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2299–2308. [Google Scholar] [CrossRef] [Green Version]
- Hanke, P.; Serwe, M.; Dombrowski, F.; Sauerbruch, T.; Caselmann, W.H. DNA vaccination with AFP-encoding plasmid DNA prevents growth of subcutaneous AFP-expressing tumors and does not interfere with liver regeneration in mice. Cancer Gene Ther. 2002, 9, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Tian, G.; Yi, J.L.; Xiong, P. Specific cellular immunity and antitumor responses in C57BL/6 mice induced by DNA vaccine encoding murine AFP. Hepatobiliary Pancreat. Dis. Int. 2004, 3, 440–443. [Google Scholar] [PubMed]
- Tan, X.H.; Zhu, Q.; Liu, C.; Liu, X.L.; Shao, X.T.; Wei, B. Immunization with dendritic cells infected with human AFP adenovirus vector effectively elicits immunity against mouse hepatocellular carcinomas. Zhonghua Zhong Liu Za Zhi 2006, 28, 13–16. [Google Scholar] [PubMed]
- Lan, Y.H.; Li, Y.G.; Liang, Z.W.; Chen, M.; Peng, M.L.; Tang, L.; Hu, H.D.; Ren, H. A DNA vaccine against chimeric AFP enhanced by HSP70 suppresses growth of hepatocellular carcinoma. Cancer Immunol. Immunother. 2007, 56, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Peng, Y.; Guo, Z.S.; Guevara-Patino, J.; Pang, J.; Butterfield, L.H.; Mivechi, N.F.; Munn, D.H.; Bartlett, D.L.; He, Y. Epitope-optimized alpha-fetoprotein genetic vaccines prevent carcinogen-induced murine autochthonous hepatocellular carcinoma. Hepatology 2014, 59, 1448–1458. [Google Scholar] [CrossRef] [Green Version]
- Ahn, H.M.; Ryu, J.; Song, J.M.; Lee, Y.; Kim, H.J.; Ko, D.; Choi, I.; Kim, S.J.; Lee, J.W.; Kim, S. Anti-cancer Activity of Novel TM4SF5-Targeting Antibodies through TM4SF5 Neutralization and Immune Cell-Mediated Cytotoxicity. Theranostics 2017, 7, 594–613. [Google Scholar] [CrossRef] [Green Version]
- Piotrowska, D.; Baczynska, K. Incidence of positive Waaler-Rose test in women exposed to urogenital infections. Reumatologia 1978, 16, 21–24. [Google Scholar]
- Feng, M.; Gao, W.; Wang, R.; Chen, W.; Man, Y.G.; Figg, W.D.; Wang, X.W.; Dimitrov, D.S.; Ho, M. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, E1083–E1091. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Lei, L.; Lin, D.; Liu, Y.; Song, Y.; Gong, H.; Zhu, Y.; Mei, Y.; Hu, B.; Wu, Y.; et al. IL-33 Released in the Liver Inhibits Tumor Growth via Promotion of CD4(+) and CD8(+) T Cell Responses in Hepatocellular Carcinoma. J. Immunol. 2018, 201, 3770–3779. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, L.; Fulton, R.J.; Rettman, P.; Sayan, A.E.; Coad, J.; Al-Shamkhani, A.; Khakoo, S.I. Activity of IL-12/15/18 primed natural killer cells against hepatocellular carcinoma. Hepatol. Int. 2019, 13, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Subleski, J.J.; Hall, V.L.; Back, T.C.; Ortaldo, J.R.; Wiltrout, R.H. Enhanced antitumor response by divergent modulation of natural killer and natural killer T cells in the liver. Cancer Res. 2006, 66, 11005–11012. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhang, Z.; Liu, Z.; Wei, D.; Wu, X.; Bian, H.; Chen, Z. Adoptive cell transfer therapy for hepatocellular carcinoma. Front. Med. 2019, 13, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dargel, C.; Bassani-Sternberg, M.; Hasreiter, J.; Zani, F.; Bockmann, J.H.; Thiele, F.; Bohne, F.; Wisskirchen, K.; Wilde, S.; Sprinzl, M.F.; et al. T Cells Engineered to Express a T-Cell Receptor Specific for Glypican-3 to Recognize and Kill Hepatoma Cells In Vitro and in Mice. Gastroenterology 2015, 149, 1042–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Li, K.; Tu, H.; Pan, X.; Jiang, H.; Shi, B.; Kong, J.; Wang, H.; Yang, S.; Gu, J.; et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6418–6428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Luo, H.; Fan, M.; Wu, X.; Shi, B.; Di, S.; Liu, Y.; Pan, Z.; Jiang, H.; Li, Z. Development of GPC3-Specific Chimeric Antigen Receptor-Engineered Natural Killer Cells for the Treatment of Hepatocellular Carcinoma. Mol. Ther. 2018, 26, 366–378. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T Cells Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front. Immunol. 2016, 7, 690. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Luo, H.; Shi, B.; Di, S.; Sun, R.; Su, J.; Liu, Y.; Li, H.; Jiang, H.; Li, Z. Combined Antitumor Effects of Sorafenib and GPC3-CAR T Cells in Mouse Models of Hepatocellular Carcinoma. Mol. Ther. 2019, 27, 1483–1494. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhou, J.; Liu, X.; Feng, Y.; Yang, W.; Wu, F.; Cheung, O.K.; Sun, H.; Zeng, X.; Tang, W.; et al. Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut 2020, 69, 365–379. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, M.; Sun, H.; Feng, Y.; Xu, L.; Chan, A.W.H.; Tong, J.H.; Wong, J.; Chong, C.C.N.; Lai, P.B.S.; et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut 2018, 67, 931–944. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Reviews. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Degroote, H.; Van Dierendonck, A.; Geerts, A.; Van Vlierberghe, H.; Devisscher, L. Preclinical and Clinical Therapeutic Strategies Affecting Tumor-Associated Macrophages in Hepatocellular Carcinoma. J. Immunol. Res. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Turkowski, K.; Mora, J.; Brüne, B.; Seeger, W.; Weigert, A.; Savai, R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 2017, 8, 48436–48452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ao, J.Y.; Zhu, X.D.; Chai, Z.T.; Cai, H.; Zhang, Y.Y.; Zhang, K.Z.; Kong, L.Q.; Zhang, N.; Ye, B.G.; Ma, D.N.; et al. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 1544–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Ba, Q.; Li, X.; Li, H.; Zhang, S.; Yuan, Y.; Wang, F.; Duan, X.; Li, J.; Zhang, W.; et al. A Natural CCR2 Antagonist Relieves Tumor-associated Macrophage-mediated Immunosuppression to Produce a Therapeutic Effect for Liver Cancer. EBioMedicine 2017, 22, 58–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, Z.T.; Zhu, X.D.; Ao, J.Y.; Wang, W.Q.; Gao, D.M.; Kong, J.; Zhang, N.; Zhang, Y.Y.; Ye, B.G.; Ma, D.N.; et al. microRNA-26a suppresses recruitment of macrophages by down-regulating macrophage colony-stimulating factor expression through the PI3K/Akt pathway in hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.Y.; Wang, N.; Man, K.; Tsao, S.W.; Che, C.M.; Feng, Y. Autophagy-induced RelB/p52 activation mediates tumour-associated macrophage repolarisation and suppression of hepatocellular carcinoma by natural compound baicalin. Cell Death Dis. 2015, 6, e1942. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Chen, L.; Zhang, H. Natural Killer Cells in Liver Disease and Hepatocellular Carcinoma and the NK Cell-Based Immunotherapy. J. Immunol. Res. 2018, 2018, 1206737. [Google Scholar] [CrossRef] [Green Version]
- Mikulak, J.; Bruni, E.; Oriolo, F.; Di Vito, C.; Mavilio, D. Hepatic Natural Killer Cells: Organ-Specific Sentinels of Liver Immune Homeostasis and Physiopathology. Front. Immunol. 2019, 10, 946. [Google Scholar] [CrossRef] [Green Version]
- Juengpanich, S.; Shi, L.; Iranmanesh, Y.; Chen, J.; Cheng, Z.; Khoo, A.K.; Pan, L.; Wang, Y.; Cai, X. The role of natural killer cells in hepatocellular carcinoma development and treatment: A narrative review. Transl. Oncol. 2019, 12, 1092–1107. [Google Scholar] [CrossRef]
- Hoechst, B.; Voigtlaender, T.; Ormandy, L.; Gamrekelashvili, J.; Zhao, F.; Wedemeyer, H.; Lehner, F.; Manns, M.P.; Greten, T.F.; Korangy, F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009, 50, 799–807. [Google Scholar] [CrossRef]
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.R.; Zhang, H.G. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target Molecule | Drug Name | Company |
|---|---|---|
| PD-1 | Nivolumab | Bristol Meyer Squibb |
| PD-1 | Pembrolizumab | Merck |
| PD-L1 | Atezolizumab (in combination with bevacizumab) | Roche |
| CTLA-4 | Ipilimumab | Bristol Meyer Squibb/Medarex |
| Author (Year) | Phase/Trial Name | Target | Therapy Regimen | ORR | PFS (Months) | pts | DCR | Additional Information | Status |
|---|---|---|---|---|---|---|---|---|---|
| Monotherapy | |||||||||
| Sangro (2013) [44] | II | CTLA-4 | Tremilimumab | 17.6% (3 PR) | 6.48 | 21 | 76.4% | completed | |
| El-Khoueiry (2017) [40] | II CHECKMATE-40 Second line | PD-1 | Nivolumab | 20% (3 CR, 39 PR) | 4.0 | 214 | 64% (37% over 6 months) | PD-1high ORR 26%, PD-1low ORR 19%, 9 months OS 74%, KM median not reached yet | completed |
| Zhu (2018) [41] | II KEYNOTE-224 Second line | PD-1 | Pembrolizumab | 17% (1 CR, 17 PR) | NR | 104 | 64% | Positive correlation of ORR and TPS score | completed |
| Finn (2020) [117] | III KEYNOTE-240 Second line | PD-1 | Pembrolizumab | 18.3% (6 CR, 45 PR) | 3.0 | 413 | 62.2%, (31% over 6 months) | OS 13.9 months | negative trial |
| Yau (2019) [73] | III CHECKMATE459 First line | PD-1 | Nivolumab vs. Sorafenib | 15% (14 CR, 43 PR) | 3.7 | 743 | - | PD-L1high ORR 28%, PD-L1low ORR 12% | negative trial |
| Qin (2019) [118] | III Rationale 301 First line | PD-L1/PD-L2 | Tislelizumab vs. Sorafenib | - | - | - | - | ongoing | |
| Exposito (2018) [119] | III CHECKMATE-9DX Adjuvant | PD-1 | Nivolumab | - | - | 530 | - | ongoing | |
| Combination of immunotherapies | |||||||||
| Yau (2019) [42] | I/II CHECKMATE-40 | PD-1 + CTLA-4 | Nivolumab + Ipilimumab | 32% (4 CR, 12 PR) | - | 148 | 54% | 12 months OS 61% PD-1high and PD-1low: no difference | ongoing |
| Kelley (2020) [46] | I/II | PD-L1, CTLA-4 | Durvalumab + Tremelimumab | 22% | - | 75 | Median OS 18.7 months | ongoing | |
| Abou-Alfa (2018) [45] | III HIMALAYA | PD-L1, CTLA-4 | Durvalumab + Tremelimumab vs. Durvalumab vs. Sorafenib | - | - | - | - | ongoing | |
| Kaseb (2019) [120] | II Neoadjuvant + adjuvant | PD-1, CTLA-4 | Nivolumab + Ipilimumab | 37.5% (3 CR) | - | 8 | - | ongoing | |
| Combination with MKI | |||||||||
| Bang (2019) [121] | Ib | PD-L1 + VEGF | Durvalumab + Ramucirumab | 11% (3 CR+PR) | 4.4 | 28 | 61% | PD-L1high ORR 18%, DCR 73% | ongoing |
| Zhu (2020) [122] | Ib KEYNOTE 524 First line | PD-1 + MKI | Pembrolizumab + Lenvatinib | 36% (1 CR, 35 PR) | 8.6 | 30 | 60% | ongoing | |
| Llovet (2019) [123] | III LEAP002 First line | PD-1 + MKI | Lenvatinib + Pembrolizumab vs. Lenvatinib | - | - | 750 | - | ongoing | |
| Xu (2019) [124] | I Second line | PD-1 + MKI | Camrelizumab + Apatinib | 50% (8 PR) | 5.8 | 39 (16 HCC) | 93.8% | OS NR | ongoing |
| II IMMUNIB First line | PD-1 + MKI | Nivolumab + Lenvatinib | - | - | est. 50 | - | ongoing | ||
| Pishvaian (2018) [125] | Ib | PD-L1 + VEGF | Atezolizumab + Bevacizumab | 34% (1 CR, 22 PR) | 14.9 | 68 | 78% (50% over 6 months) | ongoing | |
| Finn (2018) [48] | III IMbrave150 First line | PD-L1 + VEGF | Atezolizumab + Bevacizumab vs. Sorafenib | 33% (33 CR, 75 PR) | 6.8 | 325 | 72.3% | ongoing | |
| Yau (2020) [47] | II CHECKMATE 40 | PD-1 + CTLA-4 + MKI | Nivolumab + Cabozantinib vs. Nivolumab + Ipilimumab + Cabozantinib | 26% (9 PR) | 6.8 | 71 | 83% | 71% grade III-IV AEs, discontinuation in 20% | ongoing |
| Kudo (2019) [126] | Ib VEGF Liver 100 First line | PD-L1 + MKI | Avelumab + Axitinib | 13.6% | 5.5 | 22 | 68.2% | OS 12.7 months | ongoing |
| Kelley (2019) [127] | III COSMIC-312 First line | PD-L1 + MKI | Atezolizumab + Cabozantinib vs. Sorafenib | - | - | 640 | - | ongoing | |
| Knox (2019) [128] | III EMERALD 2 Adjuvant | PD-L1 + VEGF | Durvalumab + Bevacizumab | - | - | - | - | ongoing | |
| Combination with locoregional therapy | |||||||||
| Duffy (2017) [43] | I/II | CTLA-4 + locoregional | Tremilimumab + TACE/RFA | 26.3% | 7.4 | 32 | - | OS 12.3 months | completed |
| Sangro (2020) [129] | III EMERALD 1 adjuvant | PD-L1 + VEGF + locoregional | Durvalumab + Bevacizumab + TACE | - | - | 600 | - | ongoing | |
| Charalampos (2019) [130] | II adjuvant | PD-L1 + CTLA-4 + locoregional | Durvalumab + Tremilimumab + TACE/RFA/cryoablation | 20% (2 PR) | 7.8 | 22 (10 HCC) | 60% | OS 15.9 months | ongoing |
| II IMMULAB | PD-1 + locoregional | Pembrolizumab + RFA/MWA | - | - | - | - | ongoing | ||
| II PLTHCC | PD-1 + MKI+ locoregional | Immunotherapy + Lenvatinib + TCAE | - | - | - | - | ongoing | ||
| Popovic (2019) [131] | Ib CaboNivo Neoadjuvant in locally advanced HCC | PD-1 + MKI + resection | Nivolumab + Cabozantinib | - | - | 15 | - | ongoing | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heinrich, S.; Castven, D.; Galle, P.R.; Marquardt, J.U. Translational Considerations to Improve Response and Overcome Therapy Resistance in Immunotherapy for Hepatocellular Carcinoma. Cancers 2020, 12, 2495. https://doi.org/10.3390/cancers12092495
Heinrich S, Castven D, Galle PR, Marquardt JU. Translational Considerations to Improve Response and Overcome Therapy Resistance in Immunotherapy for Hepatocellular Carcinoma. Cancers. 2020; 12(9):2495. https://doi.org/10.3390/cancers12092495
Chicago/Turabian StyleHeinrich, Sophia, Darko Castven, Peter R. Galle, and Jens U. Marquardt. 2020. "Translational Considerations to Improve Response and Overcome Therapy Resistance in Immunotherapy for Hepatocellular Carcinoma" Cancers 12, no. 9: 2495. https://doi.org/10.3390/cancers12092495