Telomeres and Telomerase in the Development of Liver Cancer


1
Department of Internal Medicine I, University Hospital Ulm, 89081 Ulm, Germany
2
Department of Urology, University Hospital Ulm, 89081 Ulm, Germany
*
Author to whom correspondence should be addressed.
Cancers 2020, 12(8), 2048; https://doi.org/10.3390/cancers12082048
Submission received: 17 June 2020
/
Revised: 16 July 2020
/
Accepted: 22 July 2020
/
Published: 24 July 2020
(This article belongs to the Special Issue The Role of Telomeres and Telomerase in Cancer)
Abstract
:Liver cancer is one of the most common cancer types worldwide and the fourth leading cause of cancer-related death. Liver carcinoma is distinguished by a high heterogeneity in pathogenesis, histopathology and biological behavior. Dysregulated signaling pathways and various gene mutations are frequent in hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCCA), which represent the two most common types of liver tumors. Both tumor types are characterized by telomere shortening and reactivation of telomerase during carcinogenesis. Continuous cell proliferation, e.g., by oncogenic mutations, can cause extensive telomere shortening in the absence of sufficient telomerase activity, leading to dysfunctional telomeres and genome instability by breakage–fusion–bridge cycles, which induce senescence or apoptosis as a tumor suppressor mechanism. Telomerase reactivation is required to stabilize telomere functionality and for tumor cell survival, representing a genetic risk factor for the development of liver cirrhosis and liver carcinoma. Therefore, telomeres and telomerase could be useful targets in hepatocarcinogenesis. Here, we review similarities and differences between HCC and iCCA in telomere biology.
1. Introduction
Liver cancer is predicted to be the sixth most common tumor disease worldwide and the fourth leading cause of cancer-related death [1]. Liver cancer presents with a high heterogeneity in pathogenesis, histopathology and biological behavior. The heterogeneous disease in terms of etiologies reflects the poor prognosis of patients with liver cancer. The two most common types of liver cancer are hepatocellular carcinoma (HCC) (75–85% of cases) and intrahepatic cholangiocarcinoma (iCCA) (10–15% of cases) [1]. Most liver carcinomas are diagnosed at advanced stages despite the surveillance program of patients with liver cirrhosis to diagnose early liver tumors. To date, therapy options are limited to the multikinase inhibitors sorafenib and lenvatinib as first-line treatment options and regorafenib and cabozantinib as second-line treatment options for liver cancer patients [2,3,4]. A recent clinical trial revealed significantly longer overall and progression-free survival in patients with unresectable hepatocellular carcinoma, who received atezolizumab, a programmed death ligand 1 (PD-L1) inhibitor, combined with bevacizumab, a monoclonal antibody targeting the vascular endothelial growth factor (VEGF), in comparison to sorafenib only [5]. These findings point to new treatment options in patients with unresectable hepatocellular carcinoma and support the development of new therapy options.
The major risk factors in hepatocarcinogenesis are chronic viral infections with hepatitis B virus (HBV) and hepatitis C virus (HCV), heavy alcohol consumption, obesity, type 2 diabetes, smoking and long-term exposure to aflatoxin B [6]. Most liver tumors arise on the basis of chronic liver diseases and often result in liver fibrosis and cirrhosis formation, which itself represents a risk factor for tumor development. In western countries, HCC is mainly related to HCV, high alcohol consumption and non-alcoholic steatohepatitis (NASH) and connected with cirrhosis formation [7,8], whereas in Asia, most HCC patients are related to HBV infection; in addition, HCC can also develop in normal liver without fibrosis/cirrhosis or liver with limited fibrosis formation [9]. Interestingly, in Japan, chronic HCV infection is more common than HBV, and HCV infection accounts for the majority of HCC [10]. The burden of hepatocellular carcinoma is continuously growing due to increased rates of obesity, type 2 diabetes and nonalcoholic fatty liver disease (NAFLD), especially in low-risk HCC areas and thereby replaces viral- and alcohol-related chronic liver diseases [11]. Due to the diversity of risk factors, a high heterogeneity in liver tumors is reported. The molecular heterogeneity in terms of various gene mutations in liver cancer requires the identification of molecular targets for designing individualized therapies. Recent studies have described various sub-classification of HCC and iCCA tumor types [12,13,14]. Individual studies underline the importance of specific treatment options based on the tumor subtypes as a key to achieving a better overall survival of liver cancer patients [15,16,17]. The identification of dysregulated molecular pathways in premalignant lesions is required for an early disease detection in hepatocarcinogenesis [18]. A detailed description of molecular targets in the diversity of liver cancer subtypes would be beneficial for targeted therapies.
Telomere shortening and reactivation of telomerase, two common hallmarks of carcinogenesis, are described in a broad range of human cancers, including liver cancer [19,20,21]. Telomere shortening and reactivation of telomerase, through TERT promoter mutations, for example, represent genetic risk factors for the development of liver cirrhosis and liver cancer [12,22]. Therefore, disruptions in telomere biology could be a useful target for the treatment of liver cancer. In the following, we will present the current knowledge of telomere biology in HCC and iCCA.
2. Telomere Shortening in Liver Cirrhosis and Hepatocellular Carcinoma
Chronic liver disease is associated with chronic liver inflammation, which can lead to cell death and compensatory cell regeneration. The liver is characterized by a high regenerative reserve [23], which decreases in the context of chronic liver disease, consequently leading to telomere shortening and limiting the regenerative reserve of the liver. The frequent appearance of senescent hepatocytes in liver cirrhosis is the result of the loss of telomeric repeats and the extensive proliferation [21,24,25,26,27]. These senescent hepatocytes exhibit markers like p16INK4a and p21WAF1/Cip1 and are positive for senescence-associated β-galactosidase staining. A disruption of the p53-signaling pathway overcomes the senescence checkpoint and leads to further cell division of hepatocytes with already-shortened telomeres until the telomeres become critically short. At this point, the cells enter the crisis checkpoint, which is characterized by massive cell death [25,27,28].
In the liver, telomerase activity is downregulated during early embryonic development, and telomerase activity is absent in the adult liver. The healthy liver is a slowly proliferating organ, and most hepatocytes are in a quiescent stage (only one out of 20,000 cells (0.005%) is in the cell cycle [29]). Thus, telomerase activity seems not to be essential for hepatocyte function in a healthy liver. However, telomere shortening occurs in the absence of sufficient telomerase activity in hepatocytes under conditions of chronic liver diseases or upon injury [21,25]. Importantly, low levels of telomerase activity were observed under regenerative conditions, indicating the potential physiological activation of telomerase in adult hepatocytes [30,31]. So far, two distinct mechanisms were made responsible for telomere shortening upon increased proliferative signals, either due to oncogene activation (e.g., Ras mutations or c-Myc amplification) or expression of viral oncogenic proteins: (i) in most of the cases, absence of sufficient telomerase reactivation [32,33] and (ii) in some cases, germline mutations within the coding region of telomerase, which impair the enzymatic activity of telomerase in proliferating hepatocytes (see Section 5) [34,35].
Reactivation of telomerase activity has been shown in more than 80% of HCCs, which suggests that telomerase activation is a rate-limiting process for liver cancer formation [21,36,37]. The reactivation of telomerase correlates with the upregulation of both essential components TERT and TERC, respectively. It has been reported that the re-expression of TERT and activation of telomerase occurs at early premalignant stages in regenerative nodules and cirrhotic livers [31,38,39]. Importantly, telomerase activity was detected both in HCC and in iCCA to a similar extent (80–85%) [36,40]. Thus, it is important to emphasize at this point that the majority of both HCCs and iCCAs are similarly telomerase-positive, highlighting the necessity of telomerase activity for telomere functionality and tumor progression.
Irrespective of the mechanism, insufficient telomerase activity leads to accelerated telomere shortening in proliferating liver cells and as a result to genomic instability by breakage–fusion–bridge cycles (see review by Meena et al. [41]). In cells with intact DNA-damage response (DDR) checkpoints, telomere shortening leads to senescence/apoptosis and functions as a tumor suppressor mechanism. On the other hand, in cells lacking functional DDR, telomere shortening promotes genome instability and tumor formation.
Consequently, telomere shortening is an important risk factor for tumor initiation in liver carcinogenesis. The risk of tumor formation drastically increases at the cirrhosis stage, which is characterized by increased hepatocyte senescence, and upon further cell division at the crisis checkpoint by apoptosis of hepatocytes. Several studies have shown that telomere shortening is more pronounced in liver carcinoma compared to the surrounding liver tissue (see review by Satyanarayana et al. [42]). Furthermore, the progressive shortening of telomeres and the inactivation of cell cycle checkpoints in premalignant lesions led to the identification of a preneoplastic-sequence in human hepatocarcinogenesis, suggesting that small cell changes (SCC) are more advanced precursor lesions compared to large cell changes (LCC) [43]. In addition, telomere shortening was more pronounced in HCCs with a high degree of aneuploidy compared to diploid HCCs [44,45]. In fact, several studies provide evidence for a role of telomere shortening in the induction of chromosomal instability and increased risk for tumor formation [46,47,48,49]. The importance of telomere shortening and dysfunctional telomeres in HCC initiation was shown in transgenic mouse models (see Section 3).
3. Mouse Models of Telomere Dysfunction in Hepatocarcinogenesis
To understand the severe situation of dysfunctional telomeres and telomere shortening during chronic liver disease, transgenic mouse models were used to analyze the functions of telomeres. To this end, it is important to note that, firstly, there is a substantial difference in the regulation of telomerase between mouse and human liver. Telomerase activity is detectable in resting mouse liver but not in resting human liver [30,50,51,52,53]. The limiting component, restricting telomerase activity in human tissues, is the catalytic subunit of the telomerase TERT [54]. Concordantly, there is a marked difference in TERT mRNA levels in human and mouse livers [52,53,54]. In fact, in vivo experimental evidence supports the idea that the species-specific differential regulation is based on different promoter organization [30,53,55]. Secondly, the average telomere length in laboratory mice is about five times longer than that of human telomeres, to some extent due to constitutive telomerase activity in mouse cells [56,57].
The telomerase knockout mouse (Terc−/−) lacking the RNA component of the telomerase enzyme was used to analyze telomere shortening in liver regeneration, chronic liver disease, and hepatocarcinogenesis [24,58,59,60]. Mice are characterized by the existence of longer telomeres compared to humans [56]. Mice of different backgrounds differ a lot in telomere length [61]. For this reason, the Terc−/− mouse has to be crossed until the third to the sixth generation, depending on the used background strain to generate mice with critically short telomere lengths [58,60,62]. In an experimental model of liver regeneration involving the removal of two-thirds of the liver by partial hepatectomy of G3 Terc−/− and Terc+/+ mice, telomere shortening was observed to be a heterogeneous event at the cellular level, which led to the inhibition of a subpopulation of cells with critically short telomeres to enter the cell cycle and prevent those cells from participating in liver regeneration [58]. By comparing the mean telomere fluorescence intensities measured by FISH analysis, Satyanarayana and colleagues [58] observed no significant differences in TERC+/+ mice between BrdU-positive cells (952.53 ± 144.19) and BrdU-negative cells (957.44 ± 130.57) but saw a dramatic reduction of the mean telomere fluorescence intensity of BrdU-negative cells (364.94 ± 116.45) in comparison to BrdU-positive cells (509.65 ± 101.30) in G3 TERC−/− mice. In a mouse model of experimentally induced acute liver damage in which Terc−/− mice were subjected to genetic, surgical and chemical impairment of the liver, dysfunctional telomeres were associated with defective liver regeneration and accelerated formation of liver cirrhosis, which could be partly rescued by adenoviral delivery of the telomerase RNA [24]. In an approach comprising three different cancer-prone model systems—(1) treatment with CCl4 (carbon tetrachloride), (2) treatment with DEN (diethylnitrosamine) and (3) a genetic model (urokinase plasminogen activator transgenic mice) in Terc−/− mice—it could be shown that telomere dysfunction has a differential impact on tumor initiation and tumor progression. In all three model systems, dysfunctional telomeres were associated with higher amounts of tumor initiation and a decline in tumor progression [63]. In mouse models of chronic liver damage achieved by crossing HBsAg-expressing mice (the mice express the hepatitis B surface antigen under the liver-specific albumin promoter [64]) with Terc−/− mice, contrary effects of telomere shortening were shown between the beneficial effect on suppression of tumor growth and the negative effect on organismal survival [59]. In another mouse model of chronic liver disease, HBsAg mice were crossed with Terc−/− and Trp53 cKO mice. We generated mice with critically short telomeres by an intercross of Terc−/− and Terc+/− to generate siblings with loss of telomerase function in one group and telomerase expression in the other group. This study yielded the evidence for telomerase to be a critical component in the progression of Trp53-deficient hepatocellular carcinoma with short telomeres in the setting of chronic liver damage [60]. In addition, it was also shown that telomerase limits the accumulation of telomere dysfunction and the generation of aneuploidy by the activation of TRP53-independent checkpoints which suppress carcinogenesis [60]. Increased rates of chromosomal aberrations could be also shown in a DEN-induced liver cancer mouse model with dysfunctional telomeres. Telomerase knockout mice (Terc−/−) with chronic telomere dysfunction as well as a model of transient telomere dysfunction by inducing a dominant-negative variant of the TRF2 (telomeric repeat-binding factor 2) protein exhibited higher levels of chromosomal aberrations. In summary, the model of transient telomere dysfunction promotes chromosomal instability and liver carcinogenesis in telomerase-competent mice [65]. RAP1 (Ras-proximate-1 or Ras-related protein 1), like TRF2, is a component of the shelterin complex, which caps the telomere end for the protection of chromosome ends [66]. A recent publication suggested an important role of RAP1 in the protection of liver damage and liver carcinogenesis. DEN-induced Rap1−/− female mice were more prone to liver damage and hepatocellular carcinoma [67]. These models reflect the complexity and opposing roles of dysfunctional telomeres in hepatocarcinogenesis.
4. Telomere Shortening in Cholangiocarcinoma
Intrahepatic cholangiocarcinoma (iCCA) is the second most common malignant liver tumor which arises from the biliary tract and is characterized by a very poor prognosis with rising incidence and mortality in recent years [68,69]. The main risk factors described for HCC are also reported for iCCA. Additional risk factors are primary sclerosing cholangitis (PSC), hepatobiliary flukes, biliary duct cysts and hepatolithiasis [13]. A study by Verma and colleagues analyzed telomere shortening during aging in normal liver with no history of liver disease. Interestingly, they observed that the cholangiocytes exhibited the longest telomeres compared to all other analyzed intrahepatic lineages [70]. Similar to CD4+ and CD8+ lymphocytes, no significant telomere shortening was observed in cholangiocytes and hepatocytes of individuals without liver disease during aging. The authors only observed an age-related telomere shortening in Kupffer cells and stellate cells [70]. On the other hand, a consistent telomere shortening was reported during the development of biliary tract carcinoma, starting early in carcinogenesis in the inflamed biliary tract, metaplasia, dysplasia and carcinoma [71]. In contrast, the normal and the inflamed epithelium of the biliary tract showed a uniform telomere length [71]. Within cholangiocarcinoma, a frequent intratumoral heterogeneity of telomere length is reported [71]. As indicated above, telomere shortening in hepatocytes triggers cellular senescence in the context of intact DDR checkpoints. Similarly, an investigation of telomere shortening and senescence in the pathogenesis of primary biliary cirrhosis (PBC) showed telomere shortening. Moreover, DNA damage accumulation was detectable in biliary epithelial cells in the damaged small bile ducts and bile ductules in PBC in comparison to normal-looking bile ducts and bile ductules in PBC, chronic viral hepatitis and normal livers [72]. Of note, the accumulation of DNA damage foci correlated with increased expression of p16INK4a and p21WAF1/Cip1, which characterize biliary cellular senescence [72].
5. Loss of Function Mutations in Telomerase Components
Germline loss-of-function mutations in the telomerase components were found in a variety of human diseases, including dyskeratosis congenita, aplastic anemia, familial idiopathic fibrosis and acute myeloid leukemia [73,74,75,76,77,78,79,80]. These mutations provoked an impaired tissue regeneration due to telomere dysfunction and stem/progenitor cell exhaustion. Similar mutations were also reported in a subset of liver cancer samples [34,35]. The authors analyzed TERT and TERC mutations in buccal mucosa tissue and peripheral blood of patients with liver cirrhosis and compared them with healthy non-cirrhotic controls. An increased number of telomerase mutations were found in the group with liver cirrhosis. The study by Calado et al. [34] reported nine patients with a mutation in the TERT gene and one patient with a mutation in the TERC gene among 134 patients with liver cirrhosis. Similarly, Hartmann et al. [35] reported mutations in the TERT and TERC genes in 16 out of 521 patients. The Calado study reported a significantly higher allele frequency for the gene variants in the TERT and TERC genes in patients with cirrhosis (allele frequency 0.037) compared to controls (0.008; p = 0.0011). A similar result was shown by the Hartmann study, which stated an increased incidence of telomerase mutations detected in cirrhosis patients (allele frequency 0.017) compared to non-cirrhotic controls (0.003, p = 0.0007). The mutations in telomerase components led to decreased telomerase activity in comparison to wildtype telomerase enzyme activity. Consequently, patients with these mutations showed shorter telomeres in peripheral white blood cells [34,35]. Rare TERT mutations were also reported in patients with nonalcoholic fatty liver disease (NAFLD). Here, an enrichment of TERT mutations could be found in NAFLD-associated HCC [81]. Functional evaluation of these mutations exposed reduced protein synthesis from some of the mutations compared to the TERT wild-type protein. It is speculated that these TERT mutations could also impair the DNA-binding function of TERT. In summary, these results indicate that TERT mutations result in impaired telomerase activity, accelerated telomere shortening and impaired regeneration in chronic liver disease. These findings are supported by the above-mentioned studies indicating low/absent telomerase activity in resting liver and telomerase activation in the regenerating liver [30,31]. Taken together, these studies show that telomerase activity acts as a protective mechanism in chronic diseases to prevent telomere shortening during accelerated cell proliferation, whereas TERT mutations result in telomere shortening and may promote hepatocarcinogenesis by dysfunctional telomeres.
6. Telomerase Reactivation during Hepatocarcinogenesis
It has been proposed for a long time that up to 90% of human tumors can reactivate telomerase [20,82]. Telomerase reactivation is associated with the alteration of transcriptional regulators of the TERT promoter in cancer, TERT promoter mutations or rearrangements and DNA copy number amplifications [30,83,84,85,86,87,88]. Reactivation of the telomerase enzyme has been shown in more than 80% of HCCs, which suggests reactivation of telomerase as a rate-limiting process for liver cancer formation. The reactivation of telomerase correlates with the upregulation of both essential components, TERT and TERC. It has been reported that the re-expression of TERT and re-activation of telomerase occurs at early premalignant stages in regenerative nodules and cirrhotic livers [31,38,39]. Telomerase activation was also reported in iCCA (85%) [40]. Thus, it is important to note at this point that the majority of HCCs and iCCAs are telomerase-positive, highlighting the necessity of telomerase activity for telomere functionality and tumor progression in the two most common liver cancer entities.
6.1. TERT Promoter Mutations
TERT promoter mutations which result in increased TERT expression were first identified in melanoma and were subsequently reported in other cancers like bladder cancer, glioma, thyroid cancer and HCC [84,89,90,91]. In the following, we present data that report on TERT promoter mutations in HCC, iCCA and tumors with a mixed-differentiation HCC/iCCA (Table 1, Table 2 and Table 3). These data were achieved by PubMed literature search, using the keywords “TERT promoter mutations”, “liver carcinoma” and “biliary tract cancer”. Additionally, the list includes studies that analyzed TERT promoter mutations in different tumor entities. Due to limited sample sizes in the reported studies, we excluded studies/data on rare entities, such as fibrolamellar carcinoma (FLC) and cholangiolocellular carcinoma (coCC), as well as studies/data based on liquid biopsies. In HCCs, TERT promoter mutations were identified with an overall prevalence of 20–82% as the most frequent somatic genetic alterations (Table 1) [92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117].
However, it should be noted that in 80% of cases which lacked TERT promoter, mutations showed an enhanced TERT expression. Moreover, telomerase activity was detectable, indicating the existence of alternative mechanisms of telomerase reactivation during liver carcinogenesis [118]. For example, it was shown that the insertion of HBV in the TERT promoter region can activate TERT gene expression and telomerase promoting hepatocarcinogenesis in HBV-related HCC [119,120]. Unlike HBV, HCV is not able to integrate into the host genome, but HCV can also induce chromosomal instability by direct effects of its proteins [121]. Interestingly, mutations in the TERT promoter were identified to be significantly more common in HCV-related HCC tumors compared with tumors without HCV infection [93]. It is also notable that the TERT promoter mutation frequency is higher in HCV-related HCC (64%) compared to HBV-related HCC tumors (37%) [98].
The occurrence of TERT promoter mutations in liver carcinogenesis is identified in premalignant lesions, and the prevalence of mutations gradually increased with the degree of dysplasia, indicating that TERT promoter mutations are highly associated with the stepwise transformation from premalignant dysplastic nodules to malignant HCC. TERT promoter mutations were identified in 6% of low-grade dysplastic nodules, 19% of high-grade dysplastic nodules and 61% of early HCCs. Interestingly, TERT promoter mutations were not detected in cirrhotic liver [94,117]. TERT promoter mutations are so far the earliest recurrent genetic events in cirrhotic preneoplastic lesions and belong to the most frequent alterations in hepatocellular carcinoma [94,122]. These results support the idea that telomerase reactivation is required for the malignant transformation of liver cells from cirrhosis to cancer.
In HCC, TERT promoter mutations at several positions were described. The two most frequent mutations occur at positions −124 (G > A) and −146 (G > A) upstream of the ATG translation start site (Figure 1) [98,106]. Interestingly, the TERT promoter mutation at the −124 bp hotspot appears more often compared with the TERT promoter mutation at the −146 bp hotspot [93,94,96,97,98,100,101,106,108,111,112,114] was shown that the TERT promoter mutations generate a de novo consensus binding site for the E-twenty-six (ETS) transcription factor family [83,84], leading to an increase in TERT protein amounts, telomerase activity and telomere length [123]. Recently a new TERT promoter mutation was found in 7.5% of the analyzed HCCs at the position −297 (C > T) upstream of the ATG translation start site, which creates an AP2 consensus sequence [104].
Interestingly, the TERT promoter mutation frequency in HCC differs geographically. By comparing different studies, Pezzuto et al. [106] reported that TERT promoter mutations seem to be most common in Europe (56.6%). In Africa, TERT promoter mutations were identified in 53.3% and in Asia in 42.5% of HCCs. The overall TERT promoter mutation rate in America is around 40% [106].
In contrast to frequent mutations in the TERT promoter region in HCC, TERT promoter mutations are less frequently analyzed in cholangiocarcinoma [92,95,96,106,116,124,125,126]. These studies identified TERT promoter mutations in 31–47% of the HCCs. Nakamura et al. analyzed the largest dataset of 145 iCCAs (named as ICCs in the study). A TERT promoter mutation was detected only in one iCCA sample. Furthermore, the same study also analyzed a set of 86 extrahepatic cholangiocarcinomas, with none of them exhibiting a TERT promoter mutation [125].
While no TERT promoter mutation was detected in iCCA samples in the majority of studies [92,95,96,116], in some studies, performed on lower sample numbers, a low frequency of TERT promoter mutations were observed in iCCA [106,124,126] (Table 2).
The study by Fujimoto et al. was focused on different subtypes of liver cancer displaying biliary phenotype (LCB). They described TERT promoter mutations in 5.12% (4/78) of intrahepatic cholangiocarcinoma and in 53.3% (8/15) of combined hepatocellular cholangiocarcinoma [124].
The high percentage of TERT promoter mutations in HCC/iCCA may reflect the presence of the HCC part consisting of the TERT promoter mutation or indicate that both tumor types may arise from the same cell type of origin (Table 3).
In addition, a clear difference between hepatitis-positive LCBs and hepatitis-negative LCBs was observed (20% vs. 6%) [124]. Taken together, the fact that TERT promoter mutations are detectable only in a small subset of cholangiocarcinoma indicates that the mechanism of telomerase activation is evidently different from telomerase activation in many HCCs (Figure 2).
6.2. Amplification and Genomic Rearrangements of TERT
Structural and numerical aberrations in the organization of the genome can lead to TERT promoter activation and eventually to telomerase reactivation. Frequent amplifications of the TERT gene have been shown in human cancer cell lines and in human tumors [85,128]. The amplified TERT gene was found in around 22% of hepatocellular carcinoma and shows a higher incidence in poorly differentiated hepatocellular carcinoma [129]. Nevertheless, the TERT mRNA level did not correlate with the number of TERT gene copies, as was also reported in colorectal cancer [129,130]. Focal amplifications of 5p15.33 (TERT) were observed with a reduced overall survival, independently of other clinicopathological parameters in patients with hepatocellular carcinoma [131]. However, it should be noted that TERT amplifications do not always lead to increased TERT mRNA levels, which are by themselves not assignable to increased TERT translation levels.
Structural rearrangements in the TERT gene have been shown in several cancers like neuroblastoma, renal cell carcinoma, sarcoma and prostate carcinoma, and also in liver carcinoma [82,86,132,133]. A specific event that occurs in hepatocellular carcinoma is integration of viral genomes. Integration of the Hepatitis B virus into the host genome has been described in hepatocellular carcinoma, frequently in the TERT promoter region [98,99,120,134,135,136]. The high frequency of HBV integration within the TERT promoter region might be a reason for a low rate of TERT promoter mutations in HBV-related HCC, which is known to induce telomerase transcription [94,119]. This could represent an alternative mechanism or two different mechanisms of telomerase reactivation.
AAV2 (adeno-associated virus type 2) is the second virus reported to integrate into HCC cell lines and hepatocellular carcinoma of human patients [137]. The insertion of AAV2 can take place in the TERT promoter region and thereby lead to overexpression of the TERT gene [137]. The AAV2 genome achieves a liver-specific enhancer-promoter activity in the 3′UTR and binding sites of hepatic transcription factors, which might be linked to TERT overexpression and telomerase reactivation [138].
6.3. Altered Transcriptional Regulation of TERT Gene Expression
Studies addressing telomerase activation in liver cells mainly focused on TERT gene regulation in hepatocytes and HCC. The RB/E2F gene regulatory circuit regulates TERT promoter activity during liver regeneration and cancer [30]. The role of the RB/E2F circuit in TERT regulation is supported by a recent report showing that the Human Krüppel related 3 (HKR3) is capable of repressing TERT gene expression in HCC cell lines, resulting in the subsequent activation of CDKN2A (encoding the p16 tumor suppressor factor) and cell cycle inhibition as a tumor suppressor mechanism [139]. In another study, the RNA-binding fox-1 homolog 3 (RBFOX3) protein cooperates with AP2β to activate TERT gene expression in HCC cell lines, indicating a role of telomerase activation in promotion of HCC [140]. Concurrently, impaired E2F1 binding to the TERT promoter has been shown to correlate with increased patient mortality [141].
On the other hand, the bromodomain PHD finger transcription factor (BPTF) seems to regulate TERT gene expression via nucleosome remodeling in HCC cell lines [142]. BPTF is required for c-MYC transcriptional activity in carcinogenesis, linking TERT gene activation to c-MYC, which was the first transcription factor shown to regulate TERT expression by direct promoter binding [143,144,145,146]. Of note, c-MYC amplification correlates with liver cancer progression, mainly HCC, and to a lesser extent, iCCA [147,148]. In a recent study using a genome-wide shRNA screening strategy in HepG2 cell line, Chr15orf55 (also known as NUTM1) and Chr7orf43, two regulatory factors with currently unknown functions, were found to activate TERT gene expression through SP1 or YAP1, respectively [149]. The exact nature of these factors and their role in HCC and iCCA remain yet to be clarified.
6.4. Epigenetic Mechanisms in the Regulation of TERT Gene Expression
There are a number of studies indicating that TERT promoter methylation and histone acetylation may be involved in the regulation of TERT transcription and hence telomerase activity. The mechanism of MAD1-mediated repression involves modification of the chromatin structure by histone deacetylation [146]. In fact, treatment of telomerase-negative human cell lines with the histone deacetylase inhibitor TSA (trichostatin A) results in the reactivation of TERT gene expression and telomerase activity [150], whereas the overexpression of HDAC1 causes the repression of the TERT gene and telomerase activity [151]. To what extent histone acetylation/deacetylation is involved in TERT regulation during carcinogenesis remains pending.
The overexpression of DNMTs is thought to be responsible for suppressing the expression of tumor suppressor proteins by methylating the promoters of their genes, which leads to the early switching off of these factors. DNMT3B in particular is strongly overexpressed in breast tumors and correlates with the degree of tumorigenesis [152]. It can therefore also be assumed that the epigenetic regulation of TERT gene expression takes place indirectly, namely via the promoter methylation of genes for transcription factors which are responsible for TERT gene expression. A recent report showed the cooperation between TERT and the transcription factor SP2 to stimulate DNMT3B transcription, while TERT depletion inhibited DNMT3B expression [153]. Higher levels of TERT and DNMT3B expression predicted shorter survival in HCC patients based on the TCGA database [153].
On the other hand, the methylation status of the TERT promoter was studied in some detail. Initial reports regarding the methylation state and the activity of the TERT promoter were contradictorily unequivocal: while Devereux et al. [154] and Dessain et al. [155] did not observe an impact of promoter methylation of TERT expression, Guilleret et al. [156] found a correlation between the methylation state and the activity of the TERT promoter. Moreover, studies on TERT promoter methylation in HCC revealed contradictory results. Zhang et al. [157] observed that aberrant methylation of TERT promoter in HCC patients of the Han Chinese population showed a nearly 56-fold increase of TERT expression from the hypermethylated promoter. In contrast to that study, an analysis of 106 patient tissues (64 with HCC and 42 without liver disorders) and hepatocarcinoma cell lines revealed that the TERT promoter was methylated in normal liver but was hypomethylated in most of the hepatocellular carcinomas [158].
Recent studies point out the importance of a comprehensive mapping of the methylation landscape within the TERT promoter using more advanced technologies, arguing that the methylation analysis of a small number of CpG sites may not represent the methylation landscape of the whole TERT promoter [159]. Using a next-generation sequencing technique, the authors performed a comprehensive methylation analysis and revealed a TERT hypermethylated oncological region (THOR) defining a 433 bp genomic region within the TERT promoter as a cancer-associated epigenetic mechanism of TERT upregulation. This region encompasses 52 CpG sites and is located upstream of the TERT promoter mutations (TPMs) [159]. Five CpG sites within THOR accurately presented the average THOR methylation and were used for a bigger screen of 1352 tumor samples and 80 normal samples. Of the tumor samples 91.4% exceeded the median THOR methylation level detected in normal samples. Low THOR methylation levels were detected in thyroid cancers, which was linked to a lower malignant potential and a better prognosis [159]. Tumors from skin and bone showed low and heterogeneous methylation levels, which indicates the use of other mechanisms of telomere maintenance like TERT promoter mutations or the alternative lengthening of telomeres (ALT). In line with these results, Esopi et al. [160] described hypermethylation in the region upstream of the recurrent C228T and C250T promoter mutations in immortalized and cancer cell lines (including hepatocarcinoma cell lines). In contrast, non-malignant primary cells were rather hypomethylated [160]. Interestingly, the authors could show on an allele level that the hypermethylation of TERT promoter sequences in cancer cells is associated with TERT repression, while the remaining unmethylated allele marked with an open chromatin is largely responsible for the TERT expression in cancer cells [160]. In cancers with TERT promoter mutations, the expressed allele is mutated, while the WT allele is silenced [160,161]. In summary, current data support the assumption that TERT promoter activity can be regulated by epigenetic mechanisms in a tissue- and cancer-type-specific manner.
7. Telomere and Telomerase-Based Cancer Therapy
The fact that telomerase is repressed in somatic tissues but active in cancer cells qualifies it as an ideal target for cancer therapy. On the other hand, impairment of telomere length and/or structure could be a target for tumor therapy as telomere length in cancer cells is usually shorter than in the corresponding normal tissue cells. Moreover, due to their high turnover, functional telomeres have to be rebuilt more often in tumor cells than in normal cells. Thus, deterioration of the intact telomere structure by compounds may represent an attractive target to cure cancer.
Several telomerase inhibitors have been used in preclinical studies and clinical trials, involving telomerase inhibitor molecules, such as the antisense oligonucleotide Imetelstat (GRN163L) or the small-molecule inhibitor BIBR1532, as well as the G-quadruplex stabilizers (BRACO, RHPS4, Telomestatin). There are also interesting alternative strategies such as telomerase vaccination (peptide-based: GV1001; mRNA-based: GRNVAC1) to induce anti-tumor responses or tumor cell lysis through TERT promoter-driven oncolytic adenovirus (Telomelysin (OBP-301)) or TERT promoter-driven pro-apoptotic protein (e.g., TRAIL) to take advantage of high telomerase activity in cancer cells for therapy options. For further reading, we refer to a recent excellent article, which discusses this topic in greater detail [162]. Furthermore, we refer to a recent review article, which summarizes clinical trials on telomerase-based cancer therapeutics [163].
8. Conclusions
The biology of telomere maintenance plays a major role in the process of cirrhosis formation as well as in initiation and progression in liver carcinoma. A shortening of telomeres occurs due to progressive chronic liver diseases and highly correlates with an increased tumor risk. Telomere shortening occurs in HCC as well as in iCCA. In addition, telomerase reactivation is detectable at a high frequency in both tumor entities. The mechanisms of telomerase activation seem, at least in part, to be different between the two liver cancer entities. TERT promoter mutations were more frequently observed in HCCs compared to iCCAs. The low frequency of TERT promoter mutations reported in iCCA might be due to a not-detected HCCs part (mixed differentiation) of the analyzed tumors. This links to the phenomenon that some tumor entities lacking specific mutations in the TERT promoter region and where other mechanisms leading to telomerase activation exist. As described above, the TERT promoter region contains binding sites for many transcription factors, i.e., c-MYC, E2Fs, and others, which contribute to the tissue-specific regulation of TERT gene expression [22]. It would be interesting to explore whether TERT expression is regulated differentially in HCC and iCCA. The identification of the regulatory mechanisms contributing to telomerase reactivation in HCC and iCCA could shed light on the differential tumor initiation and progression pathways and may provide alternative and specific therapy options.
The observation that TERT promoter mutations occur early during liver carcinogenesis highlights the importance of telomerase activity for tumor cell survival. Two possible scenarios are conceivable how telomerase contributes to tumorigenesis in liver cancer. On the one side, telomerase reactivation before entering the crisis checkpoint may stabilize critically short telomeres, providing growth advantage for cells with oncogenic mutations (Figure 3). In melanoma, a two-step mechanism was described, which showed that mutations in the TERT promoter region contribute to tumorigenesis [164]. This study showed that TERT promoter mutations are not sufficient to counteract telomere shortening, but they contribute to tumorigenesis by promoting the immortalization and chromosomal instability in two phases: (1) extend the cellular life span by healing the shortest telomeres without prevention of bulk telomere shortening, and (2) the existence of critically short telomeres conducts to genome instability and thereby, telomerase is further up-regulated to proceed cell proliferation.
On the other side, early reactivation of telomerase may be related to its non-canonical functions. There is strong experimental evidence indicating that oncogenic mutations or mutations which result in genome instability induce cellular senescence by inducing telomere replication stress [41,165]. This mechanism functions to limit continuous proliferation of cells carrying detrimental mutations, preventing tumorigenesis.
However, telomerase activity can alleviate telomere replication stress by a yet-unidentified mechanism and promote tumorigenesis [22,88,166,167].
Whether telomere maintenance or even elongation of telomeres during chronic liver diseases or an inactivation of the telomerase could be beneficial for the treatment of liver carcinoma remains to be elucidated in future studies.
Author Contributions
Drafting of the manuscript: L.i.d.S., U.T., C.G., A.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a project grant from the German Research Foundation (DFG/GRK 2254/C3—HEIST). Cagatay Günes is supported by the German Research Foundation (GU 569/6-1).
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.M.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- London, W.T.; Petrick, J.L.; McGlynn, K.A. Liver Cancer. In Cancer Epidemiology and Prevention, 4th ed.; Thun, M.J., Linet, M.S., Cerhan, J.R., Haiman, C.A., Schottenfeld, D., Eds.; Oxford University Press: Cambridge, MA, USA, 2018; pp. 635–660. [Google Scholar]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16019. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular Carcinoma. J. Clin. Gastroenterol. 2002, 35, S72–S78. [Google Scholar] [CrossRef]
- Nault, J.-C. Pathogenesis of hepatocellular carcinoma according to aetiology. Best Pr. Res. Clin. Gastroenterol. 2014, 28, 937–947. [Google Scholar] [CrossRef]
- Umemura, T.; Ichijo, T.; Yoshizawa, K.; Tanaka, E.; Kiyosawa, K. Epidemiology of hepatocellular carcinoma in Japan. J. Gastroenterol. 2009, 44, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeini, A.; Sia, D.; Bardeesy, N.; Mazzaferro, V.; Llovet, J.M. Molecular Pathogenesis and Targeted Therapies for Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2015, 22, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haga, H.; Patel, T. Molecular diagnosis of intrahepatic cholangiocarcinoma. J. Hepato-Biliary-Pancreat. Sci. 2014, 22, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.C.; Kang, T.W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L.; et al. A MYC–aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Yamaguchi, R.; Hasegawa, T.; Shimada, S.; Arihiro, K.; Hayashi, S.; Maejima, K.; Nakano, K.; Fujimoto, A.; Ono, A.; et al. Classification of primary liver cancer with immunosuppression mechanisms and correlation with genomic alterations. EBioMedicine 2020, 53, 102659. [Google Scholar] [CrossRef]
- Ahn, K.S.; O’Brien, D.; Kang, Y.N.; Mounajjed, T.; Kim, Y.H.; Kim, T.S.; Kocher, J.A.; Allotey, L.K.; Borad, M.J.; Roberts, L.R.; et al. Prognostic subclass of intrahepatic cholangiocarcinoma by integrative molecular-clinical analysis and potential targeted approach. Hepatol. Int. 2019, 13, 490–500. [Google Scholar] [CrossRef]
- Yang, Y.; Lin, X.; Lu, X.; Luo, G.; Zeng, T.; Tang, J.; Jiang, F.; Li, L.; Cui, X.; Huang, W.; et al. Interferon-microRNA signalling drives liver precancerous lesion formation and hepatocarcinogenesis. Gut 2016, 65, 1186–1201. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Shay, J.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Miura, N.; Horikawa, I.; Nishimoto, A.; Ohmura, H.; Ito, H.; Hirohashi, S.; Shay, J.W.; Oshimura, M. Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet. Cytogenet. 1997, 93, 56–62. [Google Scholar] [CrossRef]
- Kumar, M.; Lechel, A.; Günes, C. Telomerase: The Devil Inside. Genes 2016, 7, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K.L.; Chang, S.; Millard, M.; Schreiber-Agus, N.; Depinho, R.A. Inhibition of Experimental Liver Cirrhosis in Mice by Telomerase Gene Delivery. Science 2000, 287, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, S.U.; Satyanarayana, A.; Tsahuridu, M.; Tillmann, H.L.; Zender, L.; Klempnauer, J.; Flemming, P.; Franco, S.; Blasco, M.A.; Manns, M.P.; et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J. 2002, 16, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Seki, S.; Kawakita, N.; Kuroki, T.; Monna, T. Telomere Shortening in Chronic Liver Diseases. Biochem. Biophys. Res. Commun. 1995, 211, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Urabe, Y.; Nouso, K.; Higashi, T.; Nakatsukasa, H.; Hino, N.; Ashida, K.; Kinugasa, N.; Yoshida, K.; Uematsu, S.; Tsuji, T. Telomere length in human liver diseases. Liver Int. 2008, 16, 293–297. [Google Scholar] [CrossRef]
- Paradis, V.; Youssef, N.; Dargere, D.; Bâ, N.; Bonvoust, F.; Deschatrette, J.; Bedossa, P. Replicative senescence in normal liver, chronic hepatitis C, and hepatocellular carcinomas. Hum. Pathol. 2001, 32, 327–332. [Google Scholar] [CrossRef]
- Mangnall, D.; Bird, N.C.; Majeed, A.W. The molecular physiology of liver regeneration following partial hepatectomy. Liver Int. 2003, 23, 124–138. [Google Scholar] [CrossRef] [Green Version]
- Sirma, H.; Kumar, M.; Meena, J.K.; Witt, B.; Weise, J.M.; Lechel, A.; Ande, S.; Sakk, V.; Guguen-Guillouzo, C.; Zender, L.; et al. The Promoter of Human Telomerase Reverse Transcriptase Is Activated during Liver Regeneration and Hepatocyte Proliferation. Gastroenterology 2011, 141, 326–337.e3. [Google Scholar] [CrossRef]
- Kotoula, V.; Hytiroglou, P.; Pyrpasopoulou, A.; Saxena, R.; Thung, S.N.; Papadimitriou, C.S. Expression of human telomerase reverse transcriptase in regenerative and precancerous lesions of cirrhotic livers. Liver Int. 2002, 22, 57–69. [Google Scholar] [CrossRef]
- Allsopp, R.C.; Chang, E.; Kashefi-Aazam, M.; Rogaev, E.I.; Piatyszek, M.A.; Shay, J.W.; Harley, C.B. Telomere Shortening Is Associated with Cell Division in Vitro and in Vivo. Exp. Cell Res. 1995, 220, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.; Gollahon, L.; Willingham, T.; Barbosa, M.; Shay, J. p53 levels in human mammary epithelial cells expressing wild-type and mutant human papillomavirus type 16 (HPV-16) E6 proteins. Int. J. Oncol. 1996, 8, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Calado, R.T.; Brudno, J.; Mehta, P.; Kovacs, J.J.; Wu, C.; Zago, M.A.; Chanock, S.J.; Boyer, T.D.; Young, N.S. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology 2011, 53, 1600–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, D.; Srivastava, U.; Thaler, M.; Kleinhans, K.N.; N’Kontchou, G.; Scheffold, A.; Bauer, K.; Kratzer, R.F.; Kloos, N.; Katz, S.-F.; et al. Telomerase gene mutations are associated with cirrhosis formation. Hepatology 2011, 53, 1608–1617. [Google Scholar] [CrossRef] [Green Version]
- Tahara, H.; Nakanishi, T.; Kitamoto, M.; Nakashio, R.; Shay, J.W.; Tahara, E.; Kajiyama, G.; Ide, T. Telomerase activity in human liver tissues: Comparison between chronic liver disease and hepatocellular carcinomas. Cancer Res. 1995, 55, 2734–2736. [Google Scholar]
- Ide, T.; Tahara, H.; Nakashio, R.; Kitamoto, M.; Nakanishi, T.; Kajiyama, G. Telomerase in hepatocellular carcinogenesis. Hum. Cell 1996, 9, 283–286. [Google Scholar]
- Hytiroglou, P.; Kotoula, V.; Thung, S.N.; Tsokos, M.; Fiel, M.I.; Papadimitriou, C.S. Telomerase activity in precancerous hepatic nodules. Cancer 1998, 82, 1831–1838. [Google Scholar] [CrossRef]
- Youssef, N.; Paradis, V.; Ferlicot, S.; Bedossa, P. In situdetection of telomerase enzymatic activity in human hepatocellular carcinogenesis. J. Pathol. 2001, 194, 459–465. [Google Scholar] [CrossRef]
- Ozaki, S.; Harada, K.; Sanzen, T.; Watanabe, K.; Tsui, W.M.S.; Nakanuma, Y. In situ nucleic acid detection of human telomerase in intrahepatic cholangiocarcinoma and its preneoplastic lesion. Hepatology 1999, 30, 914–919. [Google Scholar] [CrossRef]
- Meena, J.; Rudolph, K.L.; Günes, C. Telomere Dysfunction, Chromosomal Instability and Cancer. Recent Results Cancer Res. 2015, 200, 61–79. [Google Scholar]
- Satyanarayana, A.; Manns, M.P.; Rudolph, K.L. Telomeres and telomerase: A dual role in hepatocarcinogenesis. Hepatology 2004, 40, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Plentz, R.R.; Park, Y.N.; Lechel, A.; Kim, H.; Nellessen, F.; Langkopf, B.H.E.; Wilkens, L.; Destro, A.; Fiamengo, B.; Manns, M.P.; et al. Telomere shortening and inactivation of cell cycle checkpoints characterize human hepatocarcinogenesis. Hepatology 2007, 45, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Plentz, R.R.; Caselitz, M.; Bleck, J.S.; Gebel, M.; Flemming, P.; Kubicka, S.; Manns, M.P.; Rudolph, K.L. Hepatocellular telomere shortening correlates with chromosomal instability and the development of human hepatoma. Hepatology 2004, 40, 80–86. [Google Scholar] [CrossRef]
- Plentz, R.R.; Schlegelberger, B.; Flemming, P.; Gebel, M.; Kreipe, H.; Manns, M.P.; Rudolph, K.L.; Wilkens, L. Telomere shortening correlates with increasing aneuploidy of chromosome 8 in human hepatocellular carcinoma. Hepatology 2005, 42, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Denchi, E.L.; De Lange, T. Persistent Telomere Damage Induces Bypass of Mitosis and Tetraploidy. Cell 2010, 141, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Davoli, T.; De Lange, T. Telomere-Driven Tetraploidization Occurs in Human Cells Undergoing Crisis and Promotes Transformation of Mouse Cells. Cancer Cell 2012, 21, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Pampalona, J.; Soler, D.; Genesca, A.; Tusell, L. Whole chromosome loss is promoted by telomere dysfunction in primary cells. Genes Chromosom. Cancer 2010, 49, 368–378. [Google Scholar] [CrossRef]
- Pampalona, J.; Frías, C.; Genesca, A.; Tusell, L. Progressive Telomere Dysfunction Causes Cytokinesis Failure and Leads to the Accumulation of Polyploid Cells. PLoS Genet. 2012, 8, e1002679. [Google Scholar] [CrossRef] [Green Version]
- Prowse, K.R.; Greider, C.W. Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc. Natl. Acad. Sci. USA 1995, 92, 4818–4822. [Google Scholar] [CrossRef] [Green Version]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Ritz, J.M.; Kühle, O.; Riethdorf, S.; Sipos, B.; Deppert, W.; Englert, C.; Gunes, C. A Novel Transgenic Mouse Model Reveals Humanlike Regulation of an 8-kbp Human TERT Gene Promoter Fragment in Normal and Tumor Tissues. Cancer Res. 2005, 65, 1187–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikawa, I.; Chiang, Y.J.; Patterson, T.; Feigenbaum, L.; Leem, S.-H.; Michishita, E.; Larionov, V.; Hodes, R.J.; Barrett, J.C. Differential cis-regulation of human versus mouse TERT gene expression in vivo: Identification of a human-specific repressive element. Proc. Natl. Acad. Sci. USA 2005, 102, 18437–18442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolquist, K.A.; Ellisen, L.W.; Counter, C.M.; Meyerson, M.M.; Tan, L.K.; Weinberg, R.A.; Haber, D.A.; Gerald, W.L. Expression of TERT in early premalignant lesions and a subset of cells in normal tissues. Nat. Genet. 1998, 19, 182–186. [Google Scholar] [CrossRef]
- Greenberg, R.A.; O’Hagan, R.C.; Deng, H.; Xiao, Q.; Hann, S.R.; Adams, R.R.; Lichtsteiner, S.; Chin, L.; Morin, G.B.; DePinho, R.A. Telomerase reverse transcriptase gene is a direct target of c-Myc but is not functionally equivalent in cellular transformation. Oncogene 1999, 18, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Kipling, D.; Cooke, H.J. Hypervariable ultra-long telomeres in mice. Nature 1990, 347, 400–402. [Google Scholar] [CrossRef]
- Kipling, D. Telomere structure and telomerase expression during mouse development and tumorigenesis. Eur. J. Cancer 1997, 33, 792–800. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Wiemann, S.; Buer, J.; Lauber, J.; Dittmar, K.; Wüstefeld, T.; Blasco, M.A.; Manns, M.; Rudolph, K.L. Telomere shortening impairs organ regeneration by inhibiting cell cycle re-entry of a subpopulation of cells. EMBO J. 2003, 22, 4003–4013. [Google Scholar] [CrossRef]
- Wiemann, S.U.; Satyanarayana, A.; Buer, J.; Kamino, K.; Manns, M.P.; Rudolph, K.L. Contrasting effects of telomere shortening on organ homeostasis, tumor suppression, and survival during chronic liver damage. Oncogene 2004, 24, 1501–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechel, A.; Holstege, H.; Begus, Y.; Schienke, A.; Kamino, K.; Lehmann, U.; Kubicka, S.; Schirmacher, P.; Jonkers, J.; Rudolph, K.L. Telomerase Deletion Limits Progression of p53-Mutant Hepatocellular Carcinoma With Short Telomeres in Chronic Liver Disease. Gastroenterology 2007, 132, 1465–1475. [Google Scholar] [CrossRef]
- Hemann, M.T. Wild-derived inbred mouse strains have short telomeres. Nucleic Acids Res. 2000, 28, 4474–4478. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, K.L.; Chang, S.; Lee, H.-W.; Blasco, M.A.; Gottlieb, G.J.; Greider, C.W.; Depinho, R.A. Longevity, Stress Response, and Cancer in Aging Telomerase-Deficient Mice. Cell 1999, 96, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Farazi, P.A.; Glickman, J.; Jiang, S.; Yu, A.; Rudolph, K.L.; DePinho, R.A. Differential impact of telomere dysfunction on initiation and progression of hepatocellular carcinoma. Cancer Res. 2003, 63, 5021–5027. [Google Scholar] [PubMed]
- Chisari, F.V.; Klopchin, K.; Moriyama, T.; Pasquinelli, C.; Dunsford, H.A.; Sell, S.; Pinkert, C.; Brinster, R.L.; Palmiter, R.D. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 1989, 59, 1145–1156. [Google Scholar] [CrossRef]
- Begus-Nahrmann, Y.; Hartmann, D.; Kraus, J.; Eshraghi, P.; Scheffold, A.; Grieb, M.; Rasche, V.; Schirmacher, P.; Lee, H.-W.; Kestler, H.A.; et al. Transient telomere dysfunction induces chromosomal instability and promotes carcinogenesis. J. Clin. Investig. 2012, 122, 2283–2288. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara-Romeo, I.; Martínez, P.; Blasco, M.A. Mice lacking RAP1 show early onset and higher rates of DEN-induced hepatocellular carcinomas in female mice. PLoS ONE 2018, 13, e0204909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.-W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [Green Version]
- Njei, B. Changing pattern of epidemiology in intrahepatic cholangiocarcinoma. Hepatology 2014, 60, 1107–1108. [Google Scholar] [CrossRef]
- Verma, S.; Tachtatzis, P.; Penrhyn-Lowe, S.; Scarpini, C.G.; Jurk, D.; Zglinicki, T.; Coleman, N.; Alexander, G.J.M. Sustained telomere length in hepatocytes and cholangiocytes with increasing age in normal liver. Hepatology 2012, 56, 1510–1520. [Google Scholar] [CrossRef] [Green Version]
- Hansel, D.E.; Meeker, A.K.; Hicks, J.; De Marzo, A.M.; Lillemoe, K.D.; Schulick, R.; Hruban, R.H.; Maitra, A.; Argani, P. Telomere length variation in biliary tract metaplasia, dysplasia, and carcinoma. Mod. Pathol. 2006, 19, 772–779. [Google Scholar] [CrossRef]
- Sasaki, M.; Ikeda, H.; Yamaguchi, J.; Nakada, S.; Nakanuma, Y. Telomere shortening in the damaged small bile ducts in primary biliary cirrhosis reflects ongoing cellular senescence. Hepatològy 2008, 48, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Heiss, N.S.; Knight, S.W.; Vulliamy, T.; Klauck, S.M.; Wiemann, S.; Mason, P.J.; Poustka, A.; Dokal, I. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 1998, 19, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Vulliamy, T.; Marrone, A.; Goldman, F.; Dearlove, A.; Bessler, M.; Mason, P.J.; Dokal, I. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 2001, 413, 432–435. [Google Scholar] [CrossRef]
- Fogarty, P.F.; Yamaguchi, H.; Wiestner, A.; Baerlocher, G.M.; Sloand, E.; Zeng, W.; Read, E.J.; Lansdorp, P.M.; Young, N.S. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet 2003, 362, 1628–1630. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Calado, R.T.; Ly, H.; Kajigaya, S.; Baerlocher, G.M.; Chanock, S.J.; Lansdorp, P.M.; Young, N.S. Mutations inTERT, the Gene for Telomerase Reverse Transcriptase, in Aplastic Anemia. N. Engl. J. Med. 2005, 352, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- Armanios, M.Y.; Chen, J.J.-L.; Cogan, J.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A.; et al. Telomerase Mutations in Families with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [Green Version]
- Calado, R.T.; Regal, J.A.; Hills, M.; Yewdell, W.T.; Dalmazzo, L.F.; Zago, M.A.; Lansdorp, P.M.; Hogge, N.; Chanock, S.J.; Estey, E.H.; et al. Constitutional hypomorphic telomerase mutations in patients with acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Kirwan, M.; Vulliamy, T.; Marrone, A.; Walne, A.J.; Beswick, R.; Hillmen, P.; Kelly, R.; Stewart, A.; Bowen, D.; Schönland, S.; et al. Defining the pathogenic role of telomerase mutations in myelodysplastic syndrome and acute myeloid leukemia. Hum. Mutat. 2009, 30, 1567–1573. [Google Scholar] [CrossRef]
- Donati, B.; Pietrelli, A.; Pingitore, P.; Dongiovanni, P.; Caddeo, A.; Walker, L.; Baselli, G.; Pelusi, S.; Rosso, C.; Vanni, E.; et al. Telomerase reverse transcriptase germline mutations and hepatocellular carcinoma in patients with nonalcoholic fatty liver disease. Cancer Med. 2017, 6, 1930–1940. [Google Scholar] [CrossRef]
- Barthel, F.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Zheng, C.; Lindvall, C.; Hou, M.; Ekedahl, J.; Lewensohn, R.; Yan, Z.; Yang, X.; Henriksson, M.; Blennow, E.; et al. Frequent amplification of the telomerase reverse transcriptase gene in human tumors. Cancer Res. 2000, 60, 6230–6235. [Google Scholar]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Krämer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef]
- Kumar, M.; Witt, B.; Knippschild, U.; Koch, S.; Meena, J.K.; Heinlein, C.; Weise, J.M.; Krepulat, F.; Kuchenbauer, F.; Iben, S.; et al. CEBP factors regulate telomerase reverse transcriptase promoter activity in whey acidic protein-T mice during mammary carcinogenesis. Int. J. Cancer 2013, 132, 2032–2043. [Google Scholar] [CrossRef]
- Günes, C.; Wezel, F.; Southgate, J.; Bolenz, C. Implications of TERT promoter mutations and telomerase activity in urothelial carcinogenesis. Nat. Rev. Urol. 2018, 15, 386–393. [Google Scholar] [CrossRef] [Green Version]
- Landa, I.; Ganly, I.; Chan, T.A.; Mitsutake, N.; Matsuse, M.; Ibrahimpasic, T.; Ghossein, R.A.; Fagin, J.A. Frequent somatic TERT promoter mutations in thyroid cancer: Higher prevalence in advanced forms of the disease. J. Clin. Endocrinol. Metab. 2013, 98, E1562–E1566. [Google Scholar] [CrossRef] [Green Version]
- Rachakonda, P.S.; Hosen, I.; De Verdier, P.J.; Fallah, M.; Heidenreich, B.; Ryk, C.; Wiklund, N.P.; Steineck, G.; Schadendorf, D.; Hemminki, K.; et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc. Natl. Acad. Sci. USA 2013, 110, 17426–17431. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.; Hosen, I.; Gousias, K.; Rachakonda, S.; Heidenreich, B.; Gessi, M.; Schramm, J.; Hemminki, K.; Waha, A.; Kumar, R. TERT promoter mutations: A novel independent prognostic factor in primary glioblastomas. Neuro-Oncol. 2014, 17, 45–52. [Google Scholar] [CrossRef]
- Huang, D.-S.; Wang, Z.; He, X.-J.; Diplas, B.H.; Yang, R.; Killela, P.J.; Meng, Q.; Ye, Z.-Y.; Wang, W.; Jiang, X.-T.; et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur. J. Cancer 2015, 51, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.E.; Chang, S.-H.; Kim, W.Y.; Lim, S.D.; Kim, W.S.; Hwang, T.S.; Han, H.S. Frequent somatic TERT promoter mutations and CTNNB1 mutations in hepatocellular carcinoma. Oncotarget 2016, 7, 69267–69275. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [Green Version]
- Quaas, A.; Oldopp, T.; Tharun, L.; Klingenfeld, C.; Krech, T.; Sauter, G.; Grob, T.J. Frequency of TERT promoter mutations in primary tumors of the liver. Virchows Arch. 2014, 465, 673–677. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [Green Version]
- Chianchiano, P.; Pezhouh, M.K.; Kim, A.; Luchini, C.; Cameron, A.; Weiss, M.J.; He, J.; Voltaggio, L.; Oshima, K.; Anders, R.A.; et al. Distinction of intrahepatic metastasis from multicentric carcinogenesis in multifocal hepatocellular carcinoma using molecular alterations. Hum. Pathol. 2017, 72, 127–134. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Kawai-Kitahata, F.; Asahina, Y.; Tanaka, S.; Kakinuma, S.; Murakawa, M.; Nitta, S.; Watanabe, T.; Otani, S.; Taniguchi, M.; Goto, F.; et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J. Gastroenterol. 2015, 51, 473–486. [Google Scholar] [CrossRef]
- Lee, H.W.; Park, T.I.; Jang, S.Y.; Park, S.Y.; Park, W.-J.; Jung, S.-J.; Lee, J.-H. Clinicopathological characteristics of TERT promoter mutation and telomere length in hepatocellular carcinoma. Medicine 2017, 96, e5766. [Google Scholar] [CrossRef]
- Cevik, D.; Yildiz, G.; Ozturk, M. Common telomerase reverse transcriptase promoter mutations in hepatocellular carcinomas from different geographical locations. World J. Gastroenterol. 2015, 21, 311–317. [Google Scholar] [CrossRef]
- Kwa, W.T.; Effendi, K.; Yamazaki, K.; Kubota, N.; Hatano, M.; Ueno, A.; Masugi, Y.; Sakamoto, M. Telomerase reverse transcriptase (TERT) promoter mutation correlated with intratumoral heterogeneity in hepatocellular carcinoma. Pathol. Int. 2020. [Google Scholar] [CrossRef]
- Rudini, N.; Novello, C.; Destro, A.; Riboldi, E.; Donadon, M.; Viganò, L.; Morenghi, E.; Roncalli, M.; Di Tommaso, L. Phenotypic and molecular changes in nodule-in-nodule hepatocellular carcinoma with pathogenetic implications. Histopathology 2018, 73, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Saitta, C.; Giosa, D.; Di Tocco, F.C.; Musolino, C.; Caminiti, G.; Chines, V.; Franzè, M.S.; Alibrandi, A.; Navarra, G.; et al. Frequency of somatic mutations in TERT promoter, TP53 and CTNNB1 genes in patients with hepatocellular carcinoma from Southern Italy. Oncol. Lett. 2020, 19, 2368–2374. [Google Scholar] [PubMed]
- Joseph, N.M.; Umetsu, S.E.; Shafizadeh, N.; Ferrell, L.; Kakar, S. Genomic profiling of well-differentiated hepatocellular neoplasms with diffuse glutamine synthetase staining reveals similar genetics across the adenoma to carcinoma spectrum. Mod. Pathol. 2019, 32, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, F.; Izzo, F.; Buonaguro, L.; Annunziata, C.; Tatangelo, F.; Botti, G.; Buonaguro, F.M.; Tornesello, M.L. Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget 2016, 7, 54253–54262. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Ueda, Y.; Hatano, E.; Kakiuchi, N.; Takeda, H.; Goto, T.; Shimizu, T.; Yoshida, K.; Ikura, Y.; Shiraishi, Y.; et al. TERT promoter mutations and chromosome 8p loss are characteristic of nonalcoholic fatty liver disease-related hepatocellular carcinoma. Int. J. Cancer 2016, 139, 2512–2518. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Cheng, G.; Yu, J.; Zheng, S.; Sun, C.; Sun, Q.; Li, K.; Lin, Z.; Liu, T.; Li, P.; et al. The TERT promoter mutation incidence is modified by germline TERT rs2736098 and rs2736100 polymorphisms in hepatocellular carcinoma. Oncotarget 2017, 8, 23120–23129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilati, C.; Letouzé, E.; Nault, J.-C.; Imbeaud, S.; Boulai, A.; Calderaro, J.; Poussin, K.; Franconi, A.; Couchy, G.; Morcrette, G.; et al. Genomic Profiling of Hepatocellular Adenomas Reveals Recurrent FRK-Activating Mutations and the Mechanisms of Malignant Transformation. Cancer Cell 2014, 25, 428–441. [Google Scholar] [CrossRef] [Green Version]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Yang, X.; Guo, X.; Chen, Y.; Chen, G.; Ma, Y.; Huang, K.; Zhang, Y.; Zhao, Q.; Winkler, C.A.; An, P.; et al. Telomerase reverse transcriptase promoter mutations in hepatitis B virus-associated hepatocellular carcinoma. Oncotarget 2016, 7, 27838–27847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Zhou, W.; Li, C.; Yang, Y.; Shang, Y.-K.; Chen, C.; Zhang, J.; Yao, R.; Wang, P.; Wen, W.; et al. Promoter mutations and cellular distribution of telomerase in non-clear cell and clear cell hepatocellular carcinoma. Oncotarget 2017, 8, 26288–26297. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouzé, E.; Blanc, J.-F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Jeng, Y.M.; Chang, C.N.; Lee, H.J.; Hsu, H.C.; Lai, P.L.; Yuan, R.-H. TERT promoter mutation in resectable hepatocellular carcinomas: A strong association with hepatitis C infection and absence of hepatitis B infection. Int. J. Surg. 2014, 12, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Akita, M.; Ajiki, T.; Fukumoto, T.; Itoh, T.; Zen, Y. Keratin 19-expressing hepatocellular carcinoma and small-duct type intrahepatic cholangiocarcinoma show a similar postoperative clinical course but have distinct genetic features. Histopathology 2019, 75, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.-C.; Calderaro, J.; Di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef]
- Pinyol, R.; Tovar, V.; Llovet, J.M. TERT promoter mutations: Gatekeeper and driver of hepatocellular carcinoma. J. Hepatol. 2014, 61, 685–687. [Google Scholar] [CrossRef] [Green Version]
- Paterlini-Bréchot, P.; Saigo, K.; Murakami, Y.; Chami, M.; Gozuacik, D.; Mugnier, C.; Lagorce, D.; Bréchot, C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 2003, 22, 3911–3916. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef]
- Wu, S.C.; Chang, S.C.; Wu, H.Y.; Liao, P.J.; Chang, M.F. Hepatitis C virus NS5A protein down-regulates the expression of spindle gene Aspm through PKR-p38 signaling pathway. J. Biol. Chem. 2008, 283, 29396–29404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, J.-C.; Zucman-Rossi, J. TERT promoter mutations in primary liver tumors. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 9–14. [Google Scholar] [CrossRef]
- Borah, S.; Xi, L.; Zaug, A.J.; Powell, N.M.; Dancik, G.M.; Cohen, S.B.; Costello, J.C.; Theodorescu, D.; Cech, T.R. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 2015, 347, 1006–1010. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, A.; Furuta, M.; Shiraishi, Y.; Gotoh, K.; Kawakami, Y.; Arihiro, K.; Nakamura, T.; Ueno, M.; Ariizumi, S.-I.; Nguyen, H.H.; et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 2015, 6, 6120. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; ElZawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Joseph, N.M.; Tsokos, C.G.; Umetsu, S.E.; Shain, A.H.; Kelley, R.K.; Onodera, C.; Bowman, S.; Talevich, E.; Ferrell, L.D.; Kakar, S.; et al. Genomic profiling of combined hepatocellular-cholangiocarcinoma reveals similar genetics to hepatocellular carcinoma. J. Pathol. 2019, 248, 164–178. [Google Scholar] [CrossRef]
- Sasaki, M.; Sato, Y.; Nakanuma, Y. Mutational landscape of combined hepatocellular carcinoma and cholangiocarcinoma, and its clinicopathological significance. Histopathology 2016, 70, 423–434. [Google Scholar] [CrossRef]
- Fan, X.; Wang, Y.; Kratz, J.; Brat, D.J.; Robitaille, Y.; Moghrabi, A.; Perlman, E.J.; Dang, C.V.; Burger, P.C.; Eberhart, C.G. hTERT Gene Amplification and Increased mRNA Expression in Central Nervous System Embryonal Tumors. Am. J. Pathol. 2003, 162, 1763–1769. [Google Scholar] [CrossRef] [Green Version]
- Takuma, Y.; Nouso, K.; Kobayashi, Y.; Nakamura, S.; Tanaka, H.; Matsumoto, E.; Fujikawa, T.; Suzuki, M.; Hanafusa, T.; Shiratori, Y. Telomerase reverse transcriptase gene amplification in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2004, 19, 1300–1304. [Google Scholar] [CrossRef]
- Palmqvist, R.; Zhang, A.; Xu, D.; Golovleva, I.; Norrback, K.-F.; Gruber, A.; Öberg, Å.; Stenling, R.; Roos, G. hTERT gene copy number is not associated with hTERT RNA expression or telomerase activity in colorectal cancer. Int. J. Cancer 2005, 116, 395–400. [Google Scholar] [CrossRef]
- Li, X.; Xu, W.; Kang, W.; Wong, S.H.; Wang, M.; Zhou, Y.; Fang, X.; Zhang, X.; Yang, H.; Wong, C.H.; et al. Genomic analysis of liver cancer unveils novel driver genes and distinct prognostic features. Theranostics 2018, 8, 1740–1751. [Google Scholar] [CrossRef] [Green Version]
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; Van Sluis, P.; Volckmann, R.; Van Noesel, M.M.; George, R.E.; Tytgat, G.A.M.; Molenaar, J.J.; et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef]
- Davis, C.F.; Ricketts, C.J.; Wang, M.; Yang, L.; Cherniack, A.D.; Shen, H.; Buhay, C.; Kang, H.; Kim, S.C.; Fahey, C.C.; et al. The Somatic Genomic Landscape of Chromophobe Renal Cell Carcinoma. Cancer Cell 2014, 26, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Xue, R.; Li, R.; Guo, H.; Guo, L.; Su, Z.; Ni, X.; Qi, L.; Zhang, T.; Li, Q.; Zhang, Z.; et al. Variable Intra-Tumor Genomic Heterogeneity of Multiple Lesions in Patients With Hepatocellular Carcinoma. Gastroenterology 2016, 150, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.-H.; Liu, X.; Yan, H.-X.; Li, W.-Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.-P.; Zhuang, X.-H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef]
- Khoury, J.D.; Tannir, N.M.; Williams, M.D.; Chen, Y.; Yao, H.; Zhang, J.; Thompson, E.J.; Meric-Bernstam, F.; Medeiros, L.J.; Weinstein, J.N.; et al. Landscape of DNA Virus Associations across Human Malignant Cancers: Analysis of 3,775 Cases Using RNA-Seq. J. Virol. 2013, 87, 8916–8926. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouzé, E.; Pilati, C.; Verret, B.; Blanc, J.F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef]
- Logan, G.J.; Dane, A.P.; Hallwirth, C.V.; Smyth, C.M.; Wilkie, E.E.; Amaya, A.K.; Zhu, E.; Khandekar, N.; Ginn, S.L.; Liao, S.H.Y.; et al. Identification of liver-specific enhancer–promoter activity in the 3′ untranslated region of the wild-type AAV2 genome. Nat. Genet. 2017, 49, 1267–1273. [Google Scholar] [CrossRef]
- Choi, S.H.; Cho, K.J.; Yun, S.H.; Jin, B.; Lee, H.Y.; Ro, S.W.; Kim, D.Y.; Ahn, S.H.; Han, K.-H.; Park, J.Y. HKR3 regulates cell cycle through the inhibition of hTERT in hepatocellular carcinoma cell lines. J. Cancer 2020, 11, 2442–2452. [Google Scholar] [CrossRef]
- Liu, T.; Li, W.; Lu, W.; Chen, M.; Luo, M.; Zhang, C.; Li, Y.; Qin, G.; Shi, D.; Xiao, B.; et al. RBFOX3 Promotes Tumor Growth and Progression via hTERT Signaling and Predicts a Poor Prognosis in Hepatocellular Carcinoma. Theranostics 2017, 7, 3138–3154. [Google Scholar] [CrossRef]
- Ko, E.; Seo, H.-W.; Jung, E.S.; Kim, B.-H.; Jung, G. The TERT promoter SNP rs2853669 decreases E2F1 transcription factor binding and increases mortality and recurrence risks in liver cancer. Oncotarget 2015, 7, 684–699. [Google Scholar] [CrossRef]
- Zhao, X.; Zheng, F.; Li, Y.; Hao, J.; Tang, Z.; Tian, C.; Yang, Q.; Zhu, T.; Diao, C.; Zhang, C.; et al. BPTF promotes hepatocellular carcinoma growth by modulating hTERT signaling and cancer stem cell traits. Redox Boil. 2019, 20, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Günes, C.; Lichtsteiner, S.; Vasserot, A.P.; Englert, C. Expression of the hTERT gene is regulated at the level of transcriptional initiation and repressed by Mad1. Cancer Res. 2000, 60, 2116–2121. [Google Scholar] [PubMed]
- Wang, J.; Xie, L.Y.; Allan, S.; Beach, D.; Hannon, G.J. Myc activates telomerase. Genes Dev. 1998, 12, 1769–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.-J.; Grandori, C.; Amacker, M.; Simon-Vermot, N.; Polack, A.; Lingner, J.; Dalla-Favera, R. Direct activation of TERT transcription by c-MYC. Nat. Genet. 1999, 21, 220–224. [Google Scholar] [CrossRef]
- Xu, D.; Popov, N.; Hou, M.; Wang, Q.; Björkholm, M.; Gruber, A.; Menkel, A.R.; Henriksson, M. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3826–3831. [Google Scholar] [CrossRef] [Green Version]
- Abou-Elella, A.; Gramlich, T.; Fritsch, C.; Gansler, T. c-myc amplification in hepatocellular carcinoma predicts unfavorable prognosis. Mod. Pathol. 1996, 9, 95–98. [Google Scholar]
- Takahashi, Y.; Kawate, S.; Watanabe, M.; Fukushima, J.-I.; Mori, S.; Fukusato, T. Amplification of c-myc and cyclin D1 genes in primary and metastatic carcinomas of the liver. Pathol. Int. 2007, 57, 437–442. [Google Scholar] [CrossRef]
- Amisaki, M.; Tsuchiya, H.; Sakabe, T.; Fujiwara, Y.; Shiota, G. Identification of genes involved in the regulation of TERT in hepatocellular carcinoma. Cancer Sci. 2019, 110, 550–560. [Google Scholar] [CrossRef]
- Takakura, M.; Kyo, S.; Sowa, Y.; Wang, Z.; Yatabe, N.; Maida, Y.; Tanaka, M.; Inoue, M. Telomerase activation by histone deacetylase inhibitor in normal cells. Nucleic Acids Res. 2001, 29, 3006–3011. [Google Scholar] [CrossRef] [Green Version]
- Hou, M.; Wang, X.; Popov, N.; Zhang, A.; Zhao, X.; Zhou, R.; Zetterberg, A.; Björkholm, M.; Henriksson, M.; Gruber, A.; et al. The Histone Deacetylase Inhibitor Trichostatin A Derepresses the Telomerase Reverse Transcriptase (hTERT) Gene in Human Cells. Exp. Cell Res. 2002, 274, 25–34. [Google Scholar] [CrossRef]
- Girault, I.; Tozlu, S.; Lidereau, R.; Bièche, I. Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin. Cancer Res. 2003, 9, 4415–4422. [Google Scholar]
- Yu, J.; Yuan, X.; Sjöholm, L.; Liu, T.; Kong, F.; Ekström, T.J.; Björkholm, M.; Xu, D. Telomerase reverse transcriptase regulates DNMT3B expression/aberrant DNA methylation phenotype and AKT activation in hepatocellular carcinoma. Cancer Lett. 2018, 434, 33–41. [Google Scholar] [CrossRef]
- Devereux, T.R.; Horikawa, I.; Anna, C.H.; Annab, L.A.; Afshari, C.A.; Barrett, J.C. DNA methylation analysis of the promoter region of the human telomerase reverse transcriptase (hTERT) gene. Cancer Res. 1999, 59, 6087–6090. [Google Scholar]
- Dessain, S.K.; Yu, H.; Reddel, R.R.; Beijersbergen, R.L.; Weinberg, R.A. Methylation of the human telomerase gene CpG island. Cancer Res. 2000, 60, 537–541. [Google Scholar]
- Guilleret, I.; Yan, P.; Grange, F.; Braunschweig, R.; Bosman, F.T.; Benhattar, J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int. J. Cancer 2002, 101, 335–341. [Google Scholar] [CrossRef]
- Zhang, H.; Weng, X.; Ye, J.; He, L.; Zhou, D.; Liu, Y. Promoter hypermethylation of TERT is associated with hepatocellular carcinoma in the Han Chinese population. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Satra, M.; Drakaki, A.; Poultsides, G.A.; Tsezou, A. Epigenetic regulation of hTERT promoter in hepatocellular carcinomas. Int. J. Oncol. 2009, 34, 391–399. [Google Scholar]
- Lee, D.D.; Leão, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolónio, J.D.; et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Investig. 2019, 129, 1801. [Google Scholar] [CrossRef]
- Esopi, D.; Graham, M.K.; Brosnan-Cashman, J.A.; Meyers, J.; Vaghasia, A.; Gupta, A.; Kumar, B.; Haffner, M.C.; Heaphy, C.M.; De Marzo, A.M.; et al. Pervasive promoter hypermethylation of silenced TERT alleles in human cancers. Cell. Oncol. 2020. [Google Scholar] [CrossRef]
- Stern, J.L.; Paucek, R.D.; Huang, F.W.; Ghandi, M.; Nwumeh, R.; Costello, J.C.; Cech, T.R. Allele-Specific DNA Methylation and Its Interplay with Repressive Histone Marks at Promoter-Mutant TERT Genes. Cell Rep. 2017, 21, 3700–3707. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.-C.; Ningarhari, M.; Rebouissou, S.; Zucman-Rossi, J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Relitti, N.; Saraswati, A.P.; Federico, S.; Khan, T.; Brindisi, M.; Zisterer, D.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Telomerase-based Cancer Therapeutics: A Review on their Clinical Trials. Curr. Top. Med. Chem. 2020, 20, 433–457. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Lorbeer, F.K.; Shain, A.H.; McSwiggen, D.T.; Schruf, E.; Oh, A.; Ryu, J.; Darzacq, X.; Bastian, B.C.; Hockemeyer, D. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science 2017, 357, 1416–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suram, A.; Kaplunov, J.; Patel, P.L.; Ruan, H.; Cerutti, A.; Boccardi, V.; Fumagalli, M.; Di Micco, R.; Mirani, N.; Gurung, R.L.; et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 2012, 31, 2839–2851. [Google Scholar] [CrossRef] [PubMed]
- Günes, C.; Rudolph, K.L. Telomere dysfunction putss the brakes on oncogene-induced cancers. EMBO J. 2012, 31, 2833–2904. [Google Scholar] [CrossRef]
- Günes, C.; Rudolph, K.L. The Role of Telomeres in Stem Cells and Cancer. Cell 2013, 152, 390–393. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Identification of TERT promoter mutations in HCCs. Two hotspot mutations in the TERT promoter region generate de novo E-twenty-six (ETS) binding sites for transcription factors of the ETS family by exchange of nucleotides.
Figure 1.
Identification of TERT promoter mutations in HCCs. Two hotspot mutations in the TERT promoter region generate de novo E-twenty-six (ETS) binding sites for transcription factors of the ETS family by exchange of nucleotides.

Figure 2.
Distribution of TERT promoter mutations in liver carcinoma.

Figure 3.
TERT promoter mutations are frequently involved in carcinogenesis and represent one way of telomerase activation.
Figure 3.
TERT promoter mutations are frequently involved in carcinogenesis and represent one way of telomerase activation.


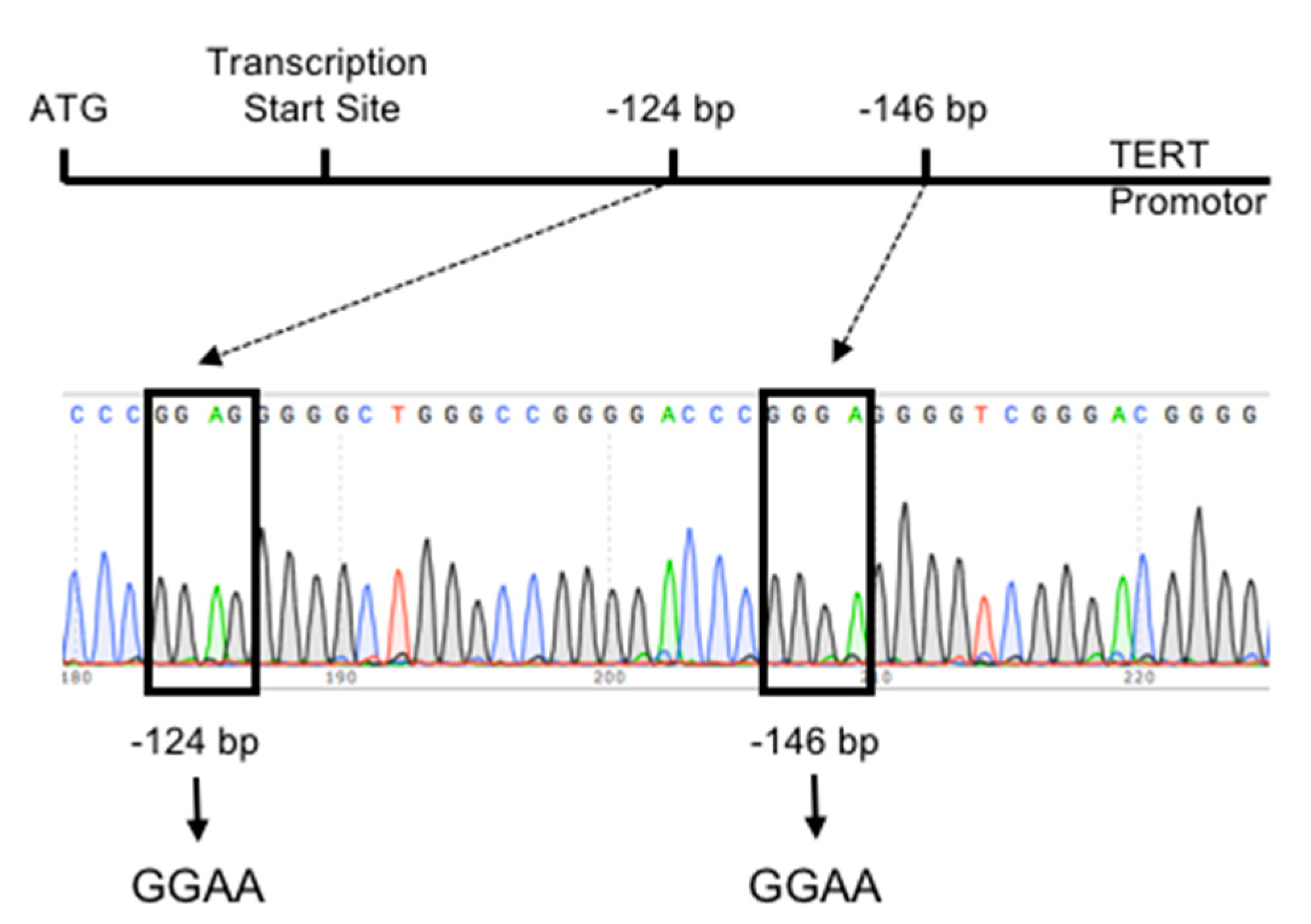{kind=link}
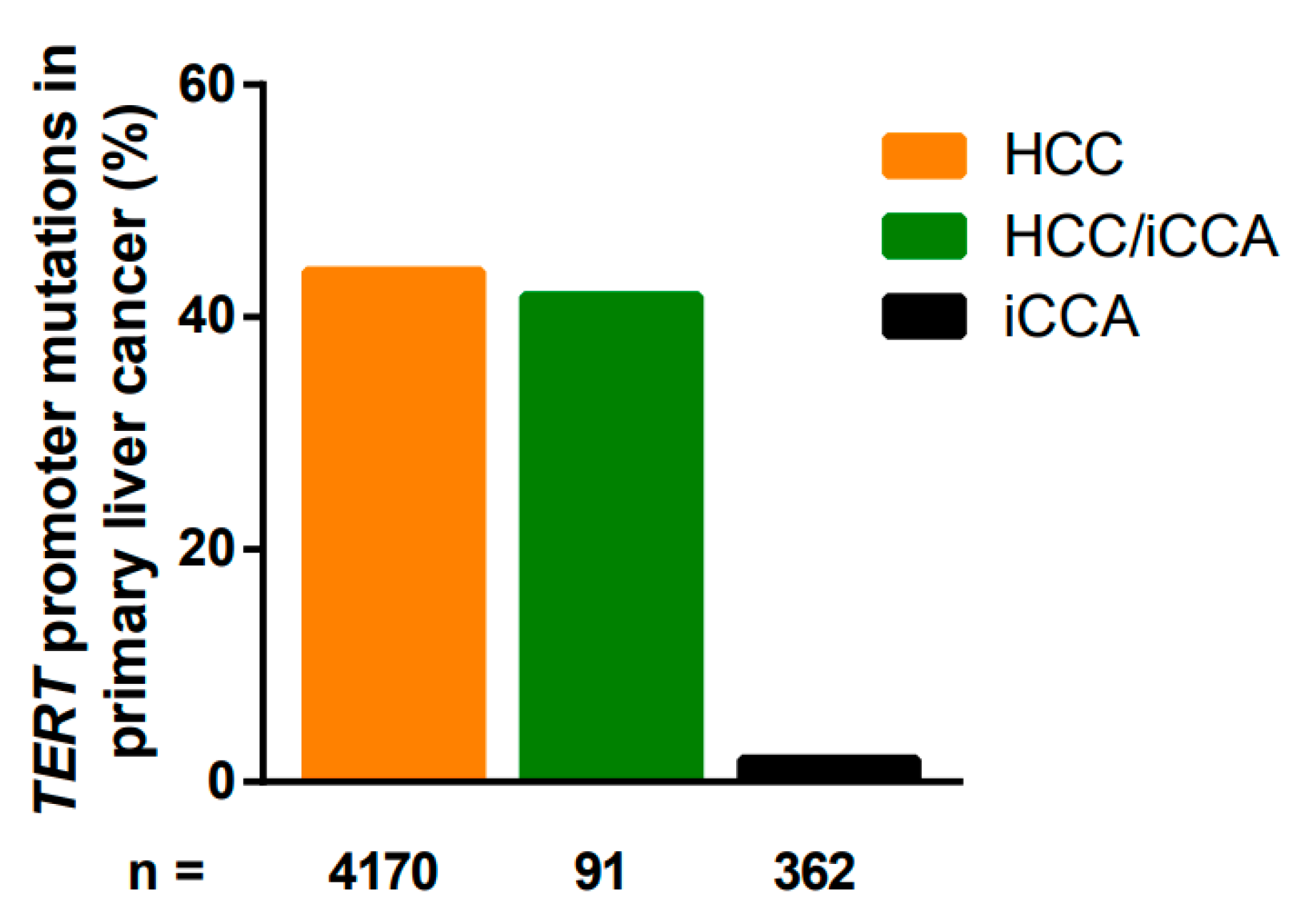{kind=link}
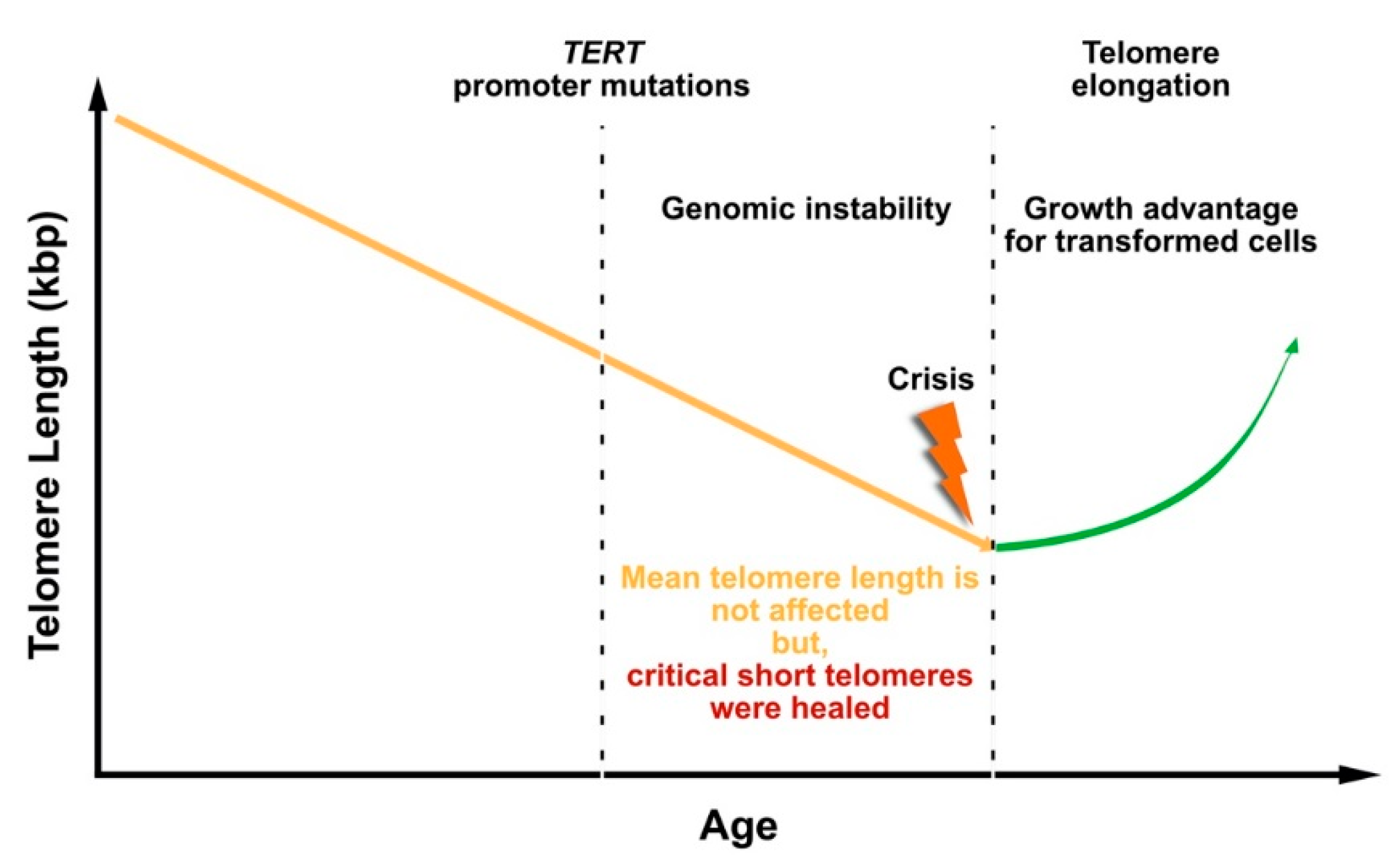{kind=link}
Table 1.
TERT promoter mutations in hepatocellular carcinoma.
| Tumor Type | Number of Samples | TERT Promoter Mutation | Etiology | Reference | |
|---|---|---|---|---|---|
| −124 bp | −146 bp | ||||
| HCC * | 61 | 44.2% (27/61) | HCV | Killela et al., 2013 [96] | |
| 62.5% (10/16) | 0 (0/16) | ||||
| HBV | |||||
| 26.6% (4/15) | 0 (0/15) | ||||
| ETOH | |||||
| 100% (2/2) | 0 | ||||
| cyptogenic liver disease | |||||
| 100% (1/1) | 0 | ||||
| unknown | |||||
| 50% (8/16) | 0 (0/16) | ||||
| HCC | 70 # | 71% (50/70) | HCV | Chianchiano et al., 2018 [97] | |
| 87.5% (35/40) | 2.5% (1/40) | ||||
| HBV | |||||
| 0 (0/7) | 0 (0/7) | ||||
| ETOH | |||||
| 16.6% (1/6) | 0 (0/6) | ||||
| HCV/HBV | |||||
| 100% (2/2) | 0 | ||||
| unknown | |||||
| 73.3% (11/15) | 0 (0/15) | ||||
| HCC | 457 | 54.2% (248/457) | HCV | Totoki et al., 2014 [98] | |
| 62.2% (117/188) | 1.6% (3/188) | ||||
| HBV | |||||
| 28.7% (31/108) | 3.7% (4/108) | ||||
| HCV/HBV | |||||
| 66.6% (8/12) | 0 (0/12) | ||||
| NBNC † | |||||
| 53.6% (80/149) | 3.3% (5/149) | ||||
| HCC | 104 | 65% ‡ (68/104) | HCV | Kawai-Kitahata et al., 2016 [99] | |
| 80% (40/50) | |||||
| HBV | |||||
| 32% (9/28) | |||||
| ETOH | |||||
| 83% (10/12) | |||||
| unknown | |||||
| 64% (9/14) | |||||
| HCC | 160 | 28.8% § (46/160) | HCV | Lee et al., 2017 [100] | |
| 60% (3/5) | |||||
| HBV | |||||
| 32.7% (19/58) | |||||
| ETOH | |||||
| 28.5% (6/21) | |||||
| others | |||||
| 23.6% (18/76) | |||||
| HCC | 105 | 39% (41/105) | HCV | Lee et al., 2016 [93] | |
| 83.3% (5/6) | |||||
| HBV | |||||
| 29.4% (23/78) | |||||
| ETOH | |||||
| 37.5% (3/8) | |||||
| unknown | |||||
| 76.9% (10/13) | |||||
| HCC | 44 | 34% (15/44) | HBV | Cevik et al., 2015 [101] | |
| 13% (3/23) | 13% (3/23) | ||||
| unknown | |||||
| 33.3% (7/21) | 9.5% (2/21) | ||||
| HCC | 97 | 54.6% (53/97) | HCV | Kwa et al., 2020 [102] | |
| 71% (22/31) | |||||
| HBV | |||||
| 36.4% (8/22) | |||||
| NBNC | |||||
| 52.3% (23/44) | |||||
| HCC | 10 | 50% (5/10) | HCV | Rudini et al., 2018 [103] | |
| 42.9% (3/7) | 14.3% (1/7) | ||||
| ETOH | |||||
| 0 (0/2) | 0 (0/2) | ||||
| unknown | |||||
| 100% (1/1) | 0 (0/1) | ||||
| HCC | 67 | 43.3% (29/67) | ESC-NA | Lombardo et al., 2020 [104] | |
| HCC | 14 | 21.4% (3/14) | ESC-NA | Jospeh et al., 2019 [105] | |
| HCC | 127 | 50.4% (64/127) | HCV | Pezzuto et al., 2016 [106] | |
| 53.6% (59/110) | |||||
| HBV | |||||
| 41.7% (5/12) | |||||
| NBNC | |||||
| 0 (0/5) | |||||
| HCC | 11 | 81.9% (9/11) | NAFLD | Kim et al., 2016 [107] | |
| 100% (9/9) | 0 (0/9) | ||||
| HCC | 190 | 30% (57/190) | HBV | Yuan et al., 2017 [108] | |
| 32.7% (50/153) | |||||
| unknown | |||||
| 18.9% (7/37) | |||||
| HCC | 375 | 20.3% (76/375) | classical HCC | Pilati et al., 2014 [109] | |
| 54% (68/125) | |||||
| HCC derived from adenomas | |||||
| 56% (5/9) | |||||
| borderline lesions HCA/HCC | |||||
| 17% (3/18) | |||||
| classical adenomas | |||||
| 0 (0/223) | |||||
| HCC | 196 | 44.4% (87/196) | HCV | The Cancer Genome Atlas Research Network 2017 [110] | |
| 61.3% (19/31) | 3.2% (1/31) | ||||
| HBV | |||||
| 22.5% (9/40) | 2.5% (1/40) | ||||
| HCV/HBV | |||||
| 50% (2/4) | 0 (0/4) | ||||
| NBNC | |||||
| 40.5% (49/121) | 5% (6/121) | ||||
| HCC | 88 | 29.6% (26/88) | low-grade dysplastic nodules | Nault et al., 2014 [117] | |
| 6.3% (2/32) | |||||
| high-grade dysplastic nodules | |||||
| 18.8% (3/16) | |||||
| early HCC | |||||
| 60.9% (14/23) | |||||
| progressed HCC | |||||
| 41.2% (7/17) | |||||
| HCC | 276 | 31% (85/276) | HBV | Yang et al., 2016 [111] | |
| 98.8% (84/85) | 1.2% (1/85) | ||||
| non-clear cell HCC | 259 | 33.2% (86/259) | 94.2% (81/86) | 5.8% (5/86) | Huang et al., 2017 [112] |
| clear cell HCC | 57 | 26.3% (15/57) | 100% (15/15) | 0 (0/15) | |
| HCC | 322 | 64.5% (208/322) | combined etiology | Calderaro et al., 2017 [113] | |
| 64.5% (208/322) | |||||
| HCC | 195 | 29.2% (57/195) | 94.7% (54/57) | 5.3% (3/57) | Chen et al., 2014 [114] |
| HCC | 235 | 60.4% (142/235) | combined etiology | Schulze et al., 2015 [115] | |
| 60.4% (142/235) | |||||
| HCC | 35 ** | 31.4% (11/35) | 81.8% (9/11) | 18.2% (2/11) | Huang et al., 2015 [92] |
| HCC | 78 ** | 47% (37/78) | 100% (37/37) | 0 (0/37) | Quaas et al., 2014 [95] |
| HCC | 305 ** | 58.6% (179/305) | 93.8% (168/179) | 6.1% (11/179) | Nault et al., 2013 [94] |
| HCC | 162 ** | 45% (73/162) | NA | NA | Barthel et al., 2017 [82] |
| HCC (K19−) | 44 *** | 59% (26/44) | 100% (26/26) | 0 | Akita et al., 2019 [116] |
| HCC (K19+) | 26 *** | 31% (8/26) | 100% (8/8) | 0 | |
| Total | 4170 | 43.9% (1831/4170) | |||
* Only for hepatocellular carcinoma (HCC) patients with known clinical information; NA = data not available; # tumors from 24 patients; † no hepatitis infection; ‡ 66/68 tumors show mutation at −124 bp, and 2/68 patients show mutation at −146 bp; § 32/46 tumors show mutation at −124 bp, and 14/46 tumors show mutation at −146 bp; ** etiology was not described; *** etiology was not correlated; HBV: Hepatitis B virus; HCV: Hepatitis C virus; ETOH: Alcohol; HCV/HBV: Hepatitis c and Hepatitis C virus; ASH: Alcoholic steatohepatitis; NASH: Non-alcoholic steatohepatitis; PBC: Primary biliary cholangitis; PSC: Primary sclerosing cholangitis; NBNC: No hepatitis virus infected; ESC-NA: Etiology-specific classification is not available; NAFLD: non-alcoholic fatty liver disease.
Table 2.
TERT promoter mutations in intrahepatic cholangiocarcinoma (iCCA).
| Tumor Type | Number of Samples | TERT Promoter Mutation | Etiology | Reference | |
|---|---|---|---|---|---|
| −124 bp | −146 bp | ||||
| iCCA | 145 | 0.70% (1/145) | HCV | Nakamura et al., 2015 [125] | |
| 0 (0/10) | 0 (0/10) | ||||
| HBV | |||||
| 0 (0/7) | 0 (0/7) | ||||
| NBNC † | |||||
| 0.8% (1/122) | 0 (0/122) | ||||
| unknown | |||||
| 0 (0/6) | 0 (0/6) | ||||
| iCCA | 78 | 5.12% (4/78) | HCV | Fujimoto et al., 2015 [124] | |
| 22.2% (2/9) | |||||
| HBV | |||||
| 9% (1/11) | |||||
| NBNC † | |||||
| 1.7% (1/58) | |||||
| iCCA | 10 | 10% (1/10) | HCV | Joseph et al., 2019 [126] | |
| 0 (0/5) | |||||
| HBV | |||||
| 0 (0/2) | |||||
| NASH | |||||
| 50% (1/2) | |||||
| PBC | |||||
| 0 (0/1) | |||||
| CC | 4 | 25% (1/4) | HCV | Pezzuto et al., 2016 [106] | |
| 25% (1/4) | |||||
| iCCA | 9 ** | 0 (0/9) | 0 (0/9) | 0 (0/9) | Huang et al., 2015 [92] |
| iCCA | 52 ** | 0 (0/52) | 0 (0/52) | 0 (0/52) | Quaas et al., 2014 [95] |
| iCCA | 28 ** | 0 (0/28) | 0 (0/28) | 0 (0/28) | Killela et al., 2013 [96] |
| S-iCCA | 36 *** | 0 (0/36) | 0 (0/36) | 0 (0/36) | Akita et al., 2019 [116] |
| Total | 362 | 1.9% (7/362) | |||
† No hepatitis infection; ** etiology was not described; *** etiology was not correlated.
Table 3.
TERT promoter mutations in combined HCC/iCCA.
| Tumor Type | Number of Samples | TERT Promoter Mutation | Etiology | Reference | |
|---|---|---|---|---|---|
| −124 bp | −146 bp | ||||
| cHCC/CC | 15 | 53.3% (8/15) | HCV | Fujimoto et al., 2015 [124] | |
| 83.3% (5/6) | |||||
| HBV | |||||
| 0 (0/3) | |||||
| NBNC * | |||||
| 50% (3/6) | |||||
| combined HCC-CC | 20 | 70% (14/20) | HCV | Joseph et al., 2019 [126] | |
| 81.8% (9/11) | |||||
| HBV | |||||
| 0 (0/1) | |||||
| HCV/HBV | |||||
| 100% (1/1) | |||||
| ASH | |||||
| 100% (1/1) | |||||
| NASH | |||||
| 100% (1/1) | |||||
| ASH/NASH | |||||
| 0% (0/1) | |||||
| PSC | |||||
| 100% (1/1) | |||||
| unknown | |||||
| 33.3% (1/3) | |||||
| cHC-CC | 53 | 30.2% (16/53) | HCV | Sasaki et al., 2017 [127] | |
| 31.8% (7/22) | |||||
| HBV | |||||
| 44.5% (4/9) | |||||
| ETOH | |||||
| 40% (2/5) | |||||
| NAFLD | |||||
| 0 (0/8) | |||||
| unknown | |||||
| 33.4% (3/9) | |||||
| HCC-CC | 3 | 0 (0/3) | HCV | Pezzuto et al., 2016 [106] | |
| 0 (0/2) | |||||
| HBV | |||||
| 0 (0/1) | |||||
| Total | 91 | 41.8% (38/91) | |||
* No hepatitis infection.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
in der Stroth, L.; Tharehalli, U.; Günes, C.; Lechel, A. Telomeres and Telomerase in the Development of Liver Cancer. Cancers 2020, 12, 2048. https://doi.org/10.3390/cancers12082048
AMA Style
in der Stroth L, Tharehalli U, Günes C, Lechel A. Telomeres and Telomerase in the Development of Liver Cancer. Cancers. 2020; 12(8):2048. https://doi.org/10.3390/cancers12082048
Chicago/Turabian Stylein der Stroth, Lena, Umesh Tharehalli, Cagatay Günes, and André Lechel. 2020. "Telomeres and Telomerase in the Development of Liver Cancer" Cancers 12, no. 8: 2048. https://doi.org/10.3390/cancers12082048
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.