Differential B-Cell Receptor Signaling Requirement for Adhesion of Mantle Cell Lymphoma Cells to Stromal Cells

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
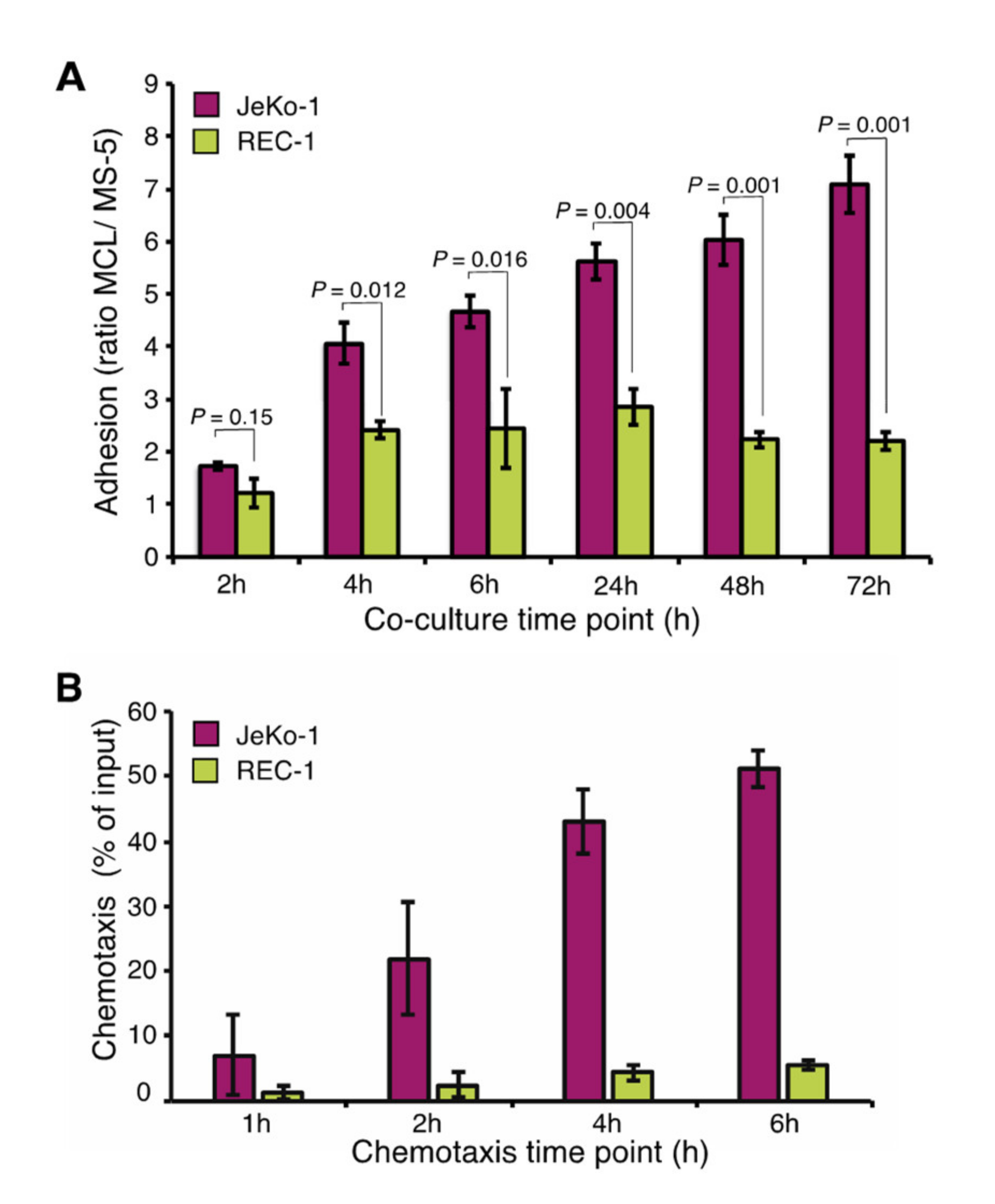
2.1. Different Adhesion of JeKo-1 and REC-1 MCL Cells to Stroma
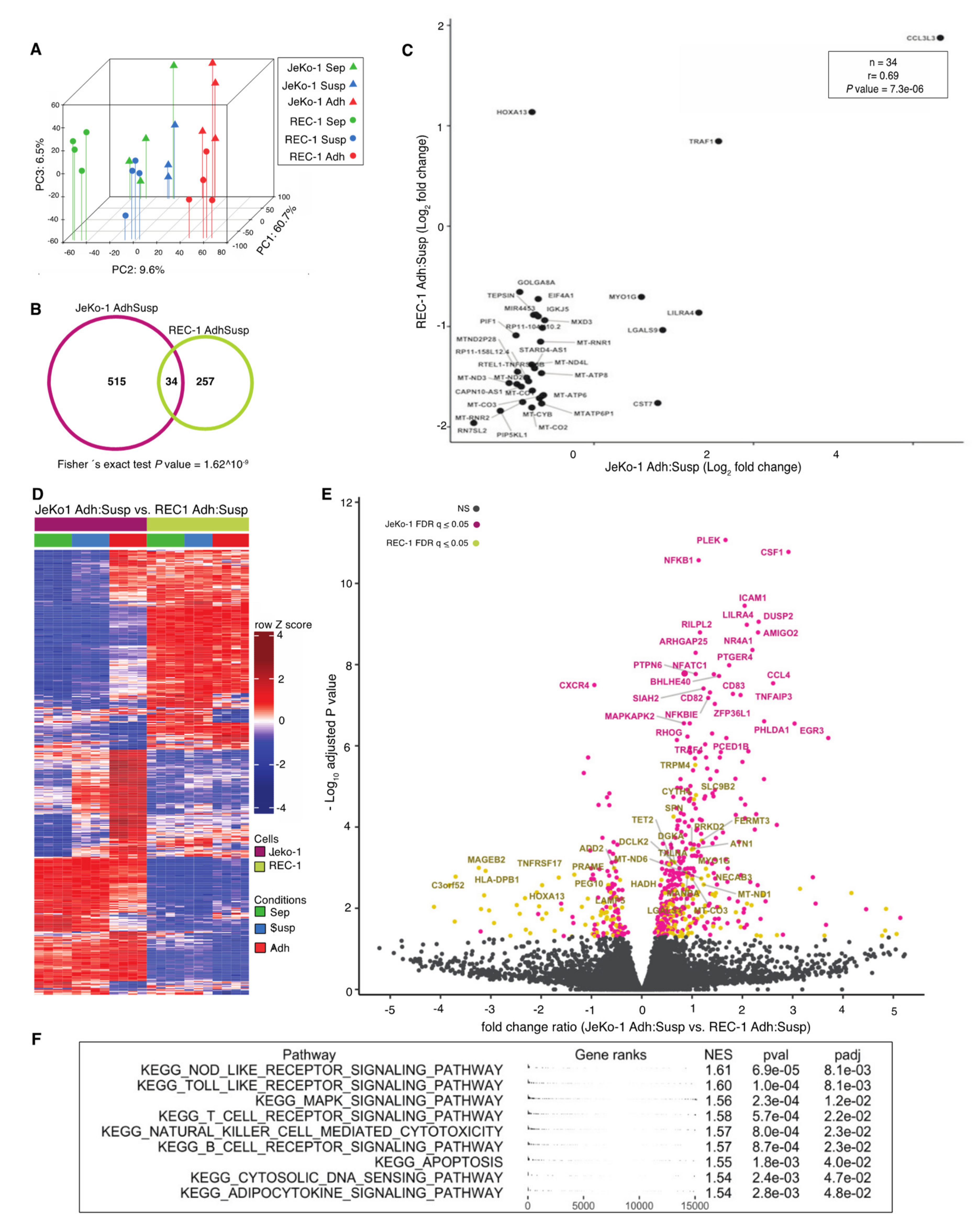
2.2. Adhesion to Stroma Affects Global Gene Expression Differently in JeKo-1 and REC-1 Cells
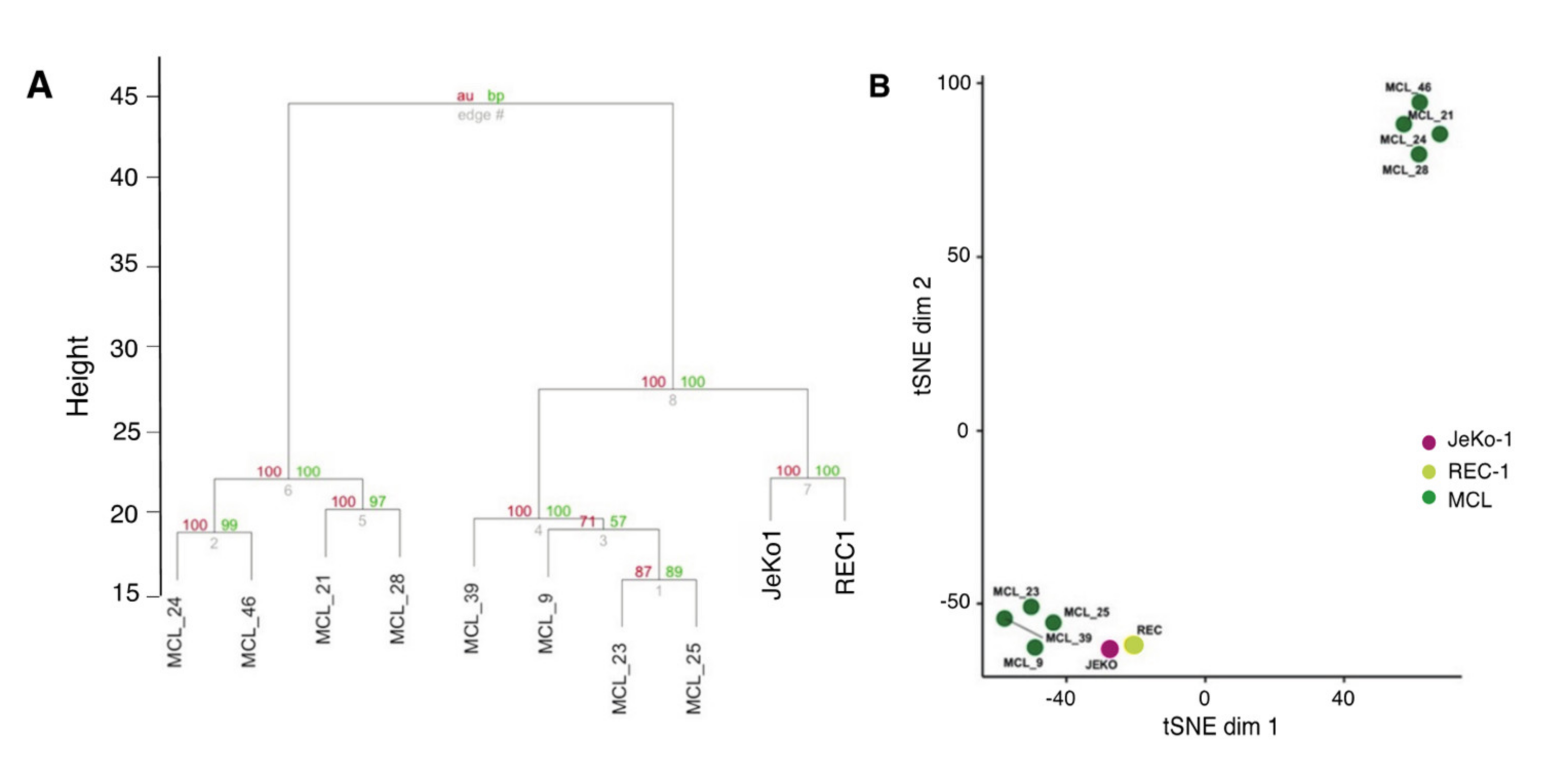
2.3. Adhesion-Regulated Gene Expression in Cell Lines Mimics Microenvironment-Regulated Gene Expression in MCL Patients
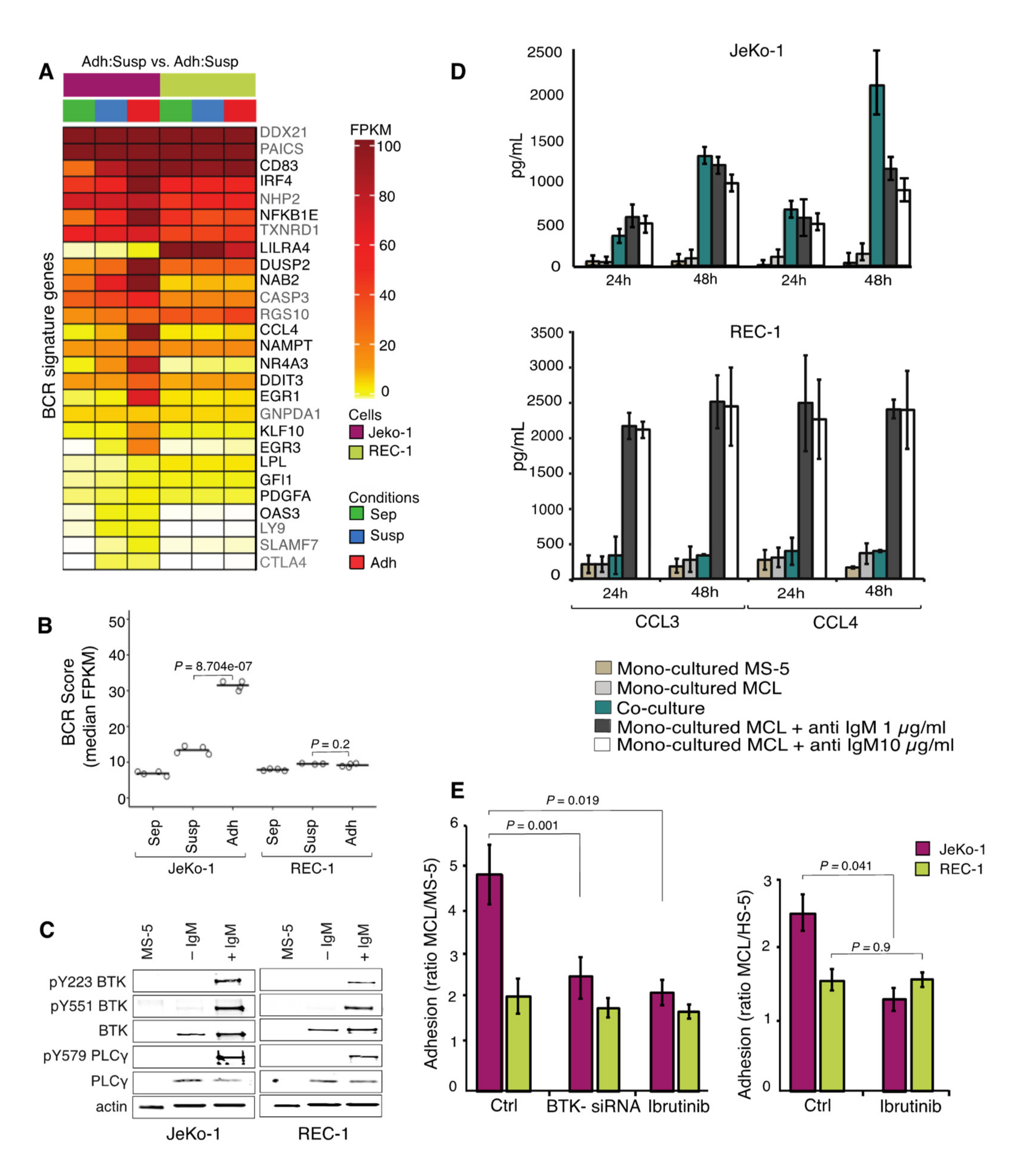
2.4. Differences in Gene Expression Changes for BCR Signature Genes in JeKo-1 and REC-1 Cells Upon Adhesion to Stromal Cells
2.5. BCR Signaling is Important for Adhesion of JeKo-1 but not REC-1 Cells to Stroma
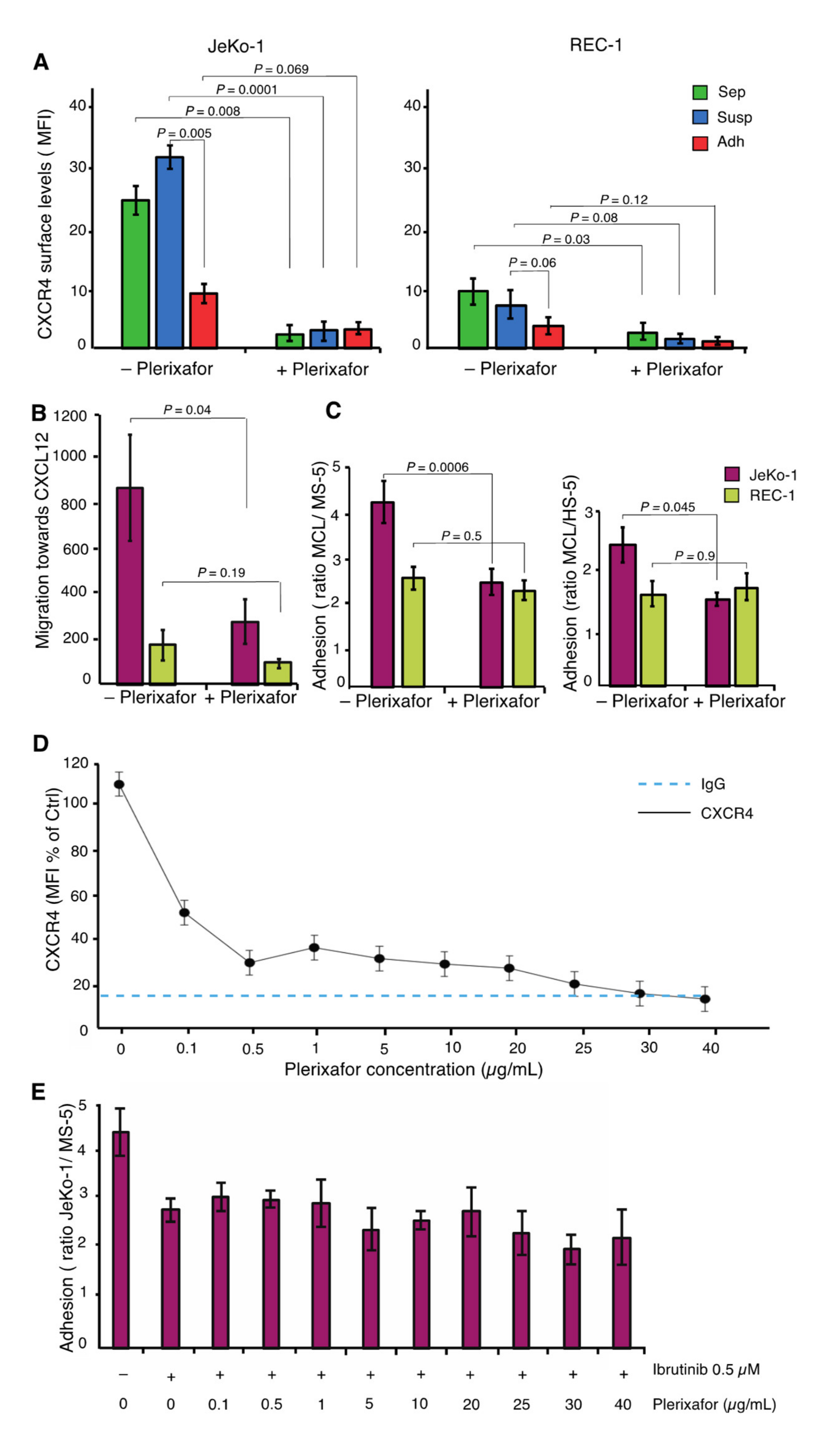
2.6. JeKo-1 and REC-1 Cells Express Different Cell Surface Levels of CXCR4
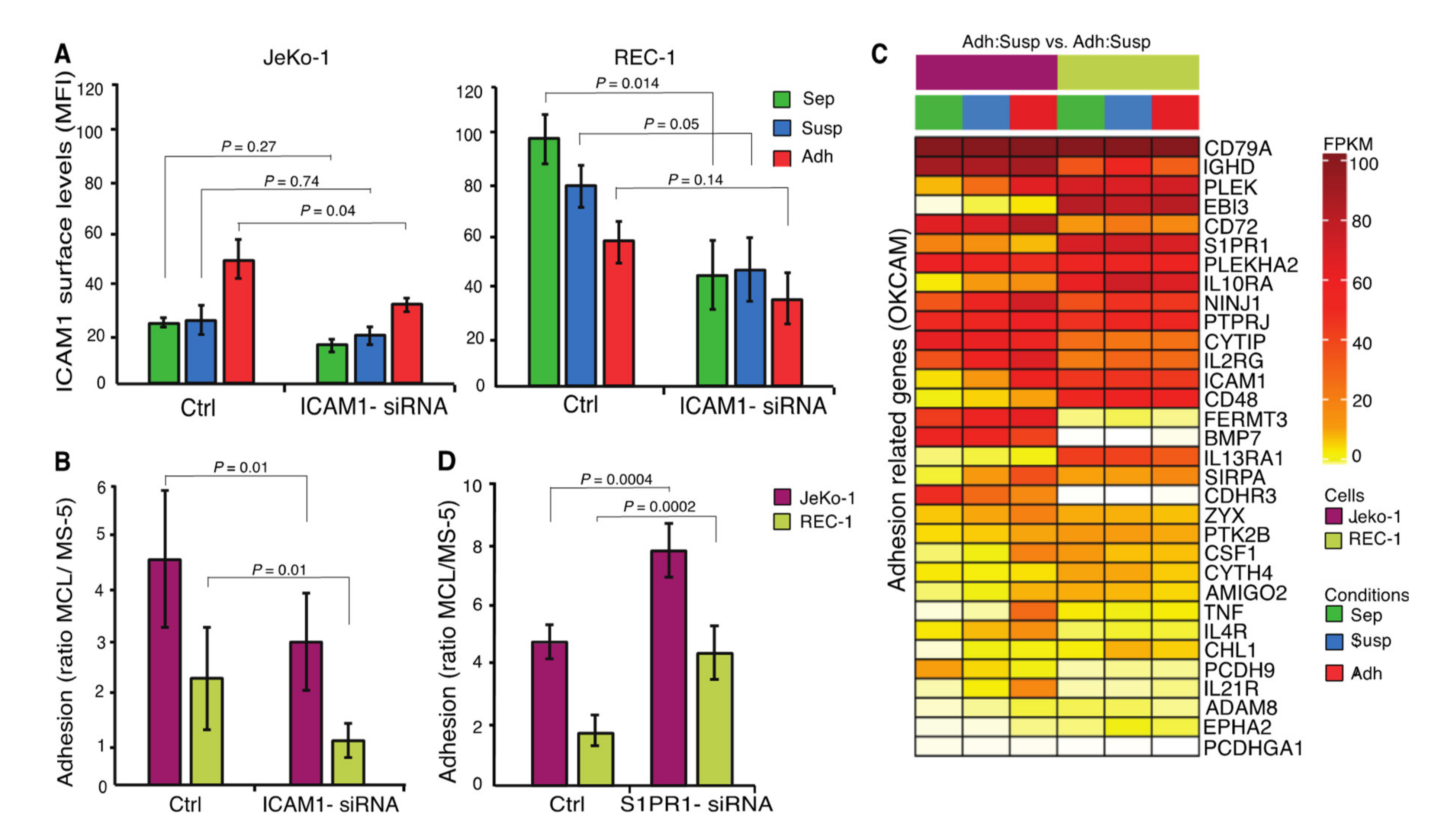
2.7. ICAM1 and S1PR1 are Important for the Adhesion of MCL Cells to Stroma
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Reagents, and Antibodies
4.2. Cell-Cell Binding Assay and Flow Cytometry
4.3. siRNA Transfection and Real-Time Quantitative PCR
4.4. RNA Extraction, Library Preparation, and Sequencing
4.5. Species-Based Read Separation and Mapping to Reference Genomes
4.6. Differential Gene Expression Analysis and GSEA
4.7. Data Availability
4.8. Microarray Data Analysis of Publicly Available Data Sets
4.9. Cytokine Levels in Conditioned Media
4.10. Cell Migration Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. Who Classification of Tumours of Haematopoetic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Puente, X.S.; Jares, P.; Campo, E. Chronic Lymphocytic Leukemia and Mantle Cell Lymphoma: Crossroads of Genetic and Microenvironment Interactions. Blood 2018, 131, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J.O. A Clinical Evaluation of the International Lymphoma Study Group Classification of Non-Hodgkin’s Lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 1997, 89, 3909–3918. [Google Scholar]
- Zelenetz, A.D.; Gordon, L.I.; Wierda, W.G.; Abramson, J.S.; Advani, R.H.; Andreadis, N.C.B.; Bartlett, J.C.; Byrd, M.S.; Czuczman, L.E.; Fayad, R.I.; et al. Non-Hodgkin’s Lymphomas, Version 4.2014. J. Natl. Compr. Cancer Netw. 2014, 12, 1282–1303. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.; Jares, P.; Campo, E.; Lopez-Guillermo, A.; Piris, M.A.; Villamor, N.; Tassies, D.; Jaffe, E.S.; Montserrat, E.; Rozman, C.; et al. Prad-1/Cyclin D1 Gene Overexpression in Chronic Lymphoproliferative Disorders: A Highly Specific Marker of Mantle Cell Lymphoma. Blood 1994, 84, 2726–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffeld, M.; Jaffe, E.S. Bcl-1, T(11;14), and Mantle Cell-Derived Lymphomas. Blood 1991, 78, 259–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheah, C.Y.; Seymour, J.F.; Wang, M.L. Mantle Cell Lymphoma. J. Clin. Oncol. 2016, 34, 1256–1269. [Google Scholar] [CrossRef]
- Edlefsen, K.L.; Greisman, H.A.; Yi, H.S.; Mantei, K.M.; Fromm, J.R. Early Lymph Node Involvement by Mantle Cell Lymphoma Limited to the Germinal Center: Report of a Case with a Novel “Follicular In Situ” Growth Pattern. Am. J. Clin. Pathol. 2011, 136, 276–281. [Google Scholar] [CrossRef]
- Shain, K.H.; Dalton, W.S.; Tao, J. The Tumor Microenvironment Shapes Hallmarks of Mature B-Cell Malignancies. Oncogene 2015, 34, 4673–4682. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Ghia, P.; Rosenwald, A.; Caligaris-Cappio, F. The Microenvironment in Mature B-Cell Malignancies: A Target for New Treatment Strategies. Blood 2009, 114, 3367–3375. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A. Chemokines and Chemokine Receptors in Chronic Lymphocytic Leukemia (Cll): From Understanding the Basics Towards Therapeutic Targeting. Semin. Cancer Biol. 2010, 20, 424–430. [Google Scholar] [CrossRef]
- Zhao, X.; Lwin, T.; Silva, A.; Shah, B.; Tao, J.; Fang, B.; Zhang, L.; Fu, K.; Bi, C.; Li, J. Unification of De Novo and Acquired Ibrutinib Resistance in Mantle Cell Lymphoma. Nat. Commun. 2017, 8, 14920. [Google Scholar] [CrossRef] [PubMed]
- Rudelius, M.; Rosenfeldt, M.T.; Leich, E.; Rauert-Wunderlich, H.; Solimando, A.G.; Beilhack, A.; Ott, G.; Rosenwald, A. Inhibition of Focal Adhesion Kinase Overcomes Resistance of Mantle Cell Lymphoma to Ibrutinib in the Bone Marrow Microenvironment. Haematologica 2018, 103, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, J.A.; Kipps, T.J. Cxcr4: A Key Receptor in the Crosstalk between Tumor Cells and Their Microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Corcione, A.N.; Arduino, E.; Ferretti, L.; Raffaghello, S.; Roncella, D.; Rossi, F.; Fedeli, L.; Ottonello, L.; Trentin, F.; Dallegri, F. Ccl19 and Cxcl12 Trigger In Vitro Chemotaxis of Human Mantle Cell Lymphoma B Cells. Clin. Cancer Res. 2004, 10, 964–971. [Google Scholar] [CrossRef] [Green Version]
- Kurtova, A.V.; Tamayo, A.T.; Ford, R.J.; Burger, J.A. Mantle Cell Lymphoma Cells Express High Levels of Cxcr4, Cxcr5, and Vla-4 (Cd49d): Importance for Interactions with the Stromal Microenvironment and Specific Targeting. Blood 2009, 113, 4604–4613. [Google Scholar] [CrossRef]
- Spoo, A.C.; Lubbert, M.; Wierda, W.G.; Burger, J.A. Cxcr4 Is a Prognostic Marker in Acute Myelogenous Leukemia. Blood 2007, 109, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Li, X.; You, S.; Bhuyan, S.S.; Dong, L. Effectiveness of Amd3100 in Treatment of Leukemia and Solid Tumors: From Original Discovery to Use in Current Clinical Practice. Exp. Hematol. Oncol. 2015, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Seda, V.; Mraz, M. B-Cell Receptor Signalling and Its Crosstalk with Other Pathways in Normal and Malignant Cells. Eur. J. Haematol. 2015, 94, 193–205. [Google Scholar] [CrossRef]
- Burger, J.A.; Chiorazzi, N.B. Cell Receptor Signaling in Chronic Lymphocytic Leukemia. Trends Immunol. 2013, 34, 592–601. [Google Scholar] [CrossRef] [Green Version]
- Fowler, N.; Davis, E. Targeting B-Cell Receptor Signaling: Changing the Paradigm. Hematol. Am. Soc. Hematol. Educ. Program B. 2013, 2013, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic Active B-Cell-Receptor Signalling in Diffuse Large B-Cell Lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herishanu, Y.; Perez-Galan, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The Lymph Node Microenvironment Promotes B-Cell Receptor Signaling, Nf-Kappab Activation, and Tumor Proliferation in Chronic Lymphocytic Leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.S.; Chang, B.Y.; Chang, S.; Tong, T.; Ham, S.; Sherry, B.; Burger, J.A.; Rai, K.R.; Chiorazzi, N. Btk Inhibition Results in Impaired Cxcr4 Chemokine Receptor Surface Expression, Signaling and Function in Chronic Lymphocytic Leukemia. Leukemia 2016, 30, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerkeman, M.; Hallek, M.; Dreyling, M.; Thieblemont, C.; Kimby, E.; Staudt, L. Targeting of B-Cell Receptor Signalling in B-Cell Malignancies. J. Intern. Med. 2017, 282, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, J.; Jimeno, A. Ibrutinib for the Treatment of Chronic Lymphocytic Leukemia and Mantle Cell Lymphoma. Drugs Today 2014, 50, 291–300. [Google Scholar] [CrossRef]
- Wiestner, A. Targeting B-Cell Receptor Signaling for Anticancer Therapy: The Bruton’s Tyrosine Kinase Inhibitor Ibrutinib Induces Impressive Responses in B-Cell Malignancies. J. Clin. Oncol. 2013, 31, 128–130. [Google Scholar] [CrossRef] [Green Version]
- De Claro, R.A.; McGinn, K.M.; Verdun, N.; Lee, S.L.; Chiu, H.J.; Saber, H.; Brower, M.E.; Chang, C.J.; Pfuma, E.; Habtemariam, B.; et al. Fda Approval: Ibrutinib for Patients with Previously Treated Mantle Cell Lymphoma and Previously Treated Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2015, 21, 3586–3590. [Google Scholar] [CrossRef] [Green Version]
- Mittal, A.K.; Chaturvedi, N.K.; Rai, K.J.; Gilling-Cutucache, C.E.; Nordgren, T.M.; Moragues, M.; Lu, R.; Opavsky, R.; Bociek, G.R.; Weisenburger, D.; et al. Chronic Lymphocytic Leukemia Cells in a Lymph Node Microenvironment Depict Molecular Signature Associated with an Aggressive Disease. Mol. Med. 2014, 20, 290–301. [Google Scholar] [CrossRef]
- Palma, M.; Krstic, A.; Pena Perez, L.; Berglof, A.; Meinke, S.; Wang, Q.; Blomberg, E.M. Kamali-Moghaddam, M.; Shen, Q.; Jaremko, G.; et al. Ibrutinib Induces Rapid Down-Regulation of Inflammatory Markers and Altered Transcription of Chronic Lymphocytic Leukaemia-Related Genes in Blood and Lymph Nodes. Br. J. Haematol. 2018, 183, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.Y.; Francesco, M.; De Rooij, M.F.; Magadala, P.; Steggerda, S.M.; Huang, M.M.; Kuil, A.; Herman, S.E.M.; Chang, S.; Pals, S.T.; et al. Egress of Cd19(+)Cd5(+) Cells into Peripheral Blood Following Treatment with the Bruton Tyrosine Kinase Inhibitor Ibrutinib in Mantle Cell Lymphoma Patients. Blood 2013, 122, 2412–2424. [Google Scholar] [CrossRef]
- Peng, S.; Sorensen, S.; Pan, F.; Dorman, E.; Sun, S.; Van Sanden, S.; Sengupta, N.; Gaudig, M. Simulation Model of Ibrutinib in Treatment of Relapsed or Refractory Mantle Cell Lymphoma (Mcl). Value Health 2014, 17, A620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, D.J.; Goodell, L.; Glod, J.; Gelinas, C.; Rabson, A.B.; Strair, R.K. Mesenchymal Stromal Cells Protect Mantle Cell Lymphoma Cells from Spontaneous and Drug-Induced Apoptosis through Secretion of B-Cell Activating Factor and Activation of the Canonical and Non-Canonical Nuclear Factor Kappab Pathways. Haematologica 2012, 97, 1255–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvidsson, G.; Henriksson, J.; Sander, B.; Wright, A.P. Mixed-Species Rnaseq Analysis of Human Lymphoma Cells Adhering to Mouse Stromal Cells Identifies a Core Gene Set That Is Also Differentially Expressed in the Lymph Node Microenvironment of Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia Patients. Haematologica 2018, 103, 666–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, N.S.; Liu, D.; Herman, S.E.M.; Underbayev, C.; Tian, X.; Behrend, D.; Weniger, M.A.; Skarzynski, M.; Gyamfi, J.; Fontan, L.; et al. Pathogenic Role of B-Cell Receptor Signaling and Canonical Nf-Kappab Activation in Mantle Cell Lymphoma. Blood 2016, 128, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Lwin, T.; Hazlehurst, L.A.; Dessureault, S.; Lai, R.; Bai, W.; Sotomayor, E.; Moscinski, L.C.; Dalton, W.S.; Tao, J. Cell Adhesion Induces P27kip1-Associated Cell-Cycle Arrest through Down-Regulation of the Scfskp2 Ubiquitin Ligase Pathway in Mantle-Cell and Other Non-Hodgkin B-Cell Lymphomas. Blood 2007, 110, 1631–1638. [Google Scholar] [CrossRef]
- Kanduri, M.; Sander, B.; Ntoufa, S.; Papakonstantinou, N.; Sutton, L.A.; Stamatopoulos, K.; Kanduri, C.; Rosenquist, R. A Key Role for Ezh2 in Epigenetic Silencing of Hox Genes in Mantle Cell Lymphoma. Epigenetics 2013, 8, 1280–1288. [Google Scholar] [CrossRef] [Green Version]
- Papin, A.; Tessoulin, B.; Bellanger, C.; Moreau, A.; Le Bris, Y.; Maisonneuve, H.; Moreau, P.; Touzeau, C.; Amiot, M.; Pellat-Deceunynck, C.; et al. Csf1r and Btk Inhibitions as Novel Strategies to Disrupt the Dialog between Mantle Cell Lymphoma and Macrophages. Leukemia 2019, 33, 2442–2453. [Google Scholar] [CrossRef]
- Takahashi, K.; Sivina, M.; Hoellenriegel, J.; Oki, Y.; Hagemeister, F.B.; Fayad, L.; Romaguera, J.E.; Fowler, N.; Fanale, M.A.; Kwak, L.W.; et al. Ccl3 and Ccl4 Are Biomarkers for B Cell Receptor Pathway Activation and Prognostic Serum Markers in Diffuse Large B Cell Lymphoma. Br. J. Haematol. 2015, 171, 726–735. [Google Scholar] [CrossRef]
- Arvidsson, G.; Wright, A.P.H. A Protein Intrinsic Disorder Approach for Characterising Differentially Expressed Genes in Transcriptome Data: Analysis of Cell-Adhesion Regulated Gene Expression in Lymphoma Cells. Int. J. Mol. Sci. 2018, 19, 3101. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, J.; Grahn, M.; Wright, A.P. Proteome-Wide Evidence for Enhanced Positive Darwinian Selection within Intrinsically Disordered Regions in Proteins. Genome Biol. 2011, 12, R65. [Google Scholar] [CrossRef] [Green Version]
- Macossay-Castillo, M.; Marvelli, G.; Guharoy, M.; Jain, A.; Kihara, D.; Tompa, P.; Wodak, S.J. The Balancing Act of Intrinsically Disordered Proteins: Enabling Functional Diversity While Minimizing Promiscuity. J. Mol. Biol. 2019, 431, 1650–1670. [Google Scholar] [CrossRef] [PubMed]
- Merolle, M.I.; Ahmed, M.; Nomie, K.; Wang, M.L. The B Cell Receptor Signaling Pathway in Mantle Cell Lymphoma. Oncotarget 2018, 9, 25332–25341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, S.E.M.; Montraveta, A.; Niemann, C.U.; Mora-Jensen, H.; Gulrajani, M.; Krantz, F.; Mantel, R.; Smith, L.L.; McClanahan, F.; Harrington, B.K.; et al. The Bruton Tyrosine Kinase (Btk) Inhibitor Acalabrutinib Demonstrates Potent on-Target Effects and Efficacy in Two Mouse Models of Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2017, 23, 2831–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, A.W.; Wawryk, S.O.; Burns, G.F.; Fecondo, J.V. Intercellular Adhesion Molecule 1 (Icam-1) Has a Central Role in Cell-Cell Contact-Mediated Immune Mechanisms. Proc. Natl. Acad. Sci. USA 1988, 85, 3095–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.Y.; Liu, Q.R.; Zhang, P.W.; Li, X.M.; Wei, L.; Uhl, G.R. Okcam: An Ontology-Based, Human-Centered Knowledgebase for Cell Adhesion Molecules. Nucleic Acids Res. 2009, 37, D251–D260. [Google Scholar] [CrossRef] [PubMed]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.W.; Huang, Y.; Cyster, J.G.; et al. Promotion of Lymphocyte Egress into Blood and Lymph by Distinct Sources of Sphingosine-1-Phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef]
- Nishimura, H.; Akiyama, T.; Monobe, Y.; Matsubara, K.; Igarashi, Y.; Abe, M.; Sugihara, T.; Sadahira, Y. Expression of Sphingosine-1-Phosphate Receptor 1 in Mantle Cell Lymphoma. Modern Pathol. 2010, 23, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Nie, Y.; Waite, J.; Brewer, F.; Sunshine, M.J.; Littman, D.R.; Zou, Y.R. The Role of Cxcr4 in Maintaining Peripheral B Cell Compartments and Humoral Immunity. J. Exp. Med. 2004, 200, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Mohle, R.; Failenschmid, C.; Bautz, F.; Kanz, L. Overexpression of the Chemokine Receptor Cxcr4 in B Cell Chronic Lymphocytic Leukemia Is Associated with Increased Functional Response to Stromal Cell-Derived Factor-1 (Sdf-1). Leukemia 1999, 13, 1954–1959. [Google Scholar] [CrossRef] [Green Version]
- Andritsos, L.A.; Byrd, J.C.; Cheverton, P.; Wu, J.; Sivina, M.; Kipps, T.J.; Burger, J.A. A Multicenter Phase 1 Study of Plerixafor and Rituximab in Patients with Chronic Lymphocytic Leukemia. Leuk. Lymphoma 2019, 60, 3461–3469. [Google Scholar] [CrossRef]
- Uy, G.L.; Rettig, M.P.; Motabi, I.H.; McFarland, K.; Trinkaus, K.M.; Hladnik, L.M.; Kulkarni, S.; Abboud, C.N.; Cashen, A.F.; Stockerl-Goldstein, K.E.; et al. A Phase 1/2 Study of Chemosensitization with the Cxcr4 Antagonist Plerixafor in Relapsed or Refractory Acute Myeloid Leukemia. Blood 2012, 119, 3917–3924. [Google Scholar] [CrossRef] [PubMed]
- Ryser, M.F.; Ugarte, F.; Lehmann, R.; Bornhauser, M.; Brenner, S. S1p(1) Overexpression Stimulates S1p-Dependent Chemotaxis of Human Cd34+ Hematopoietic Progenitor Cells but Strongly Inhibits Sdf-1/Cxcr4-Dependent Migration and In Vivo Homing. Mol. Immunol. 2008, 46, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Wasik, A.M.; Wu, C.; Mansouri, L.; Rosenquist, R.; Pan-Hammarstrom, Q.; Sander, B. Clinical and Functional Impact of Recurrent S1pr1 Mutations in Mantle Cell Lymphoma. Blood Adv. 2018, 2, 621–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golan, K.; Kollet, O.; Lapidot, T. Dynamic Cross Talk between S1p and Cxcl12 Regulates Hematopoietic Stem Cells Migration, Development and Bone Remodeling. Pharmaceuticals 2013, 6, 1145–1169. [Google Scholar] [CrossRef]
- Golan, K.; Vagima, Y.; Ludin, A.; Itkin, T.; Cohen-Gur, S.; Kalinkovich, A.; Kollet, O.; Kim, C.; Schajnovitz, A.; Ovadya, Y.; et al. S1p Promotes Murine Progenitor Cell Egress and Mobilization Via S1p1-Mediated Ros Signaling and Sdf-1 Release. Blood 2012, 119, 2478–2488. [Google Scholar] [CrossRef] [Green Version]
- Campbell, I.D.; Humphries, M.J. Integrin Structure, Activation, and Interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef] [Green Version]
- Jacob, M.C.; Agrawal, S.; Chaperot, L.; Giroux, C.; Gressin, R.; Le Marc’Hadour, F.; Favre, M.; Sotto, J.J.; Bensa, J.C.; Plumas, J. Quantification of Cellular Adhesion Molecules on Malignant B Cells from Non-Hodgkin’s Lymphoma. Leukemia 1999, 13, 1428–1433. [Google Scholar] [CrossRef] [Green Version]
- Terol, M.J.; Lopez-Guillermo, A.; Bosch, F.; Villamor, N.; Cid, M.C.; Rozman, C.; Campo, E.; Montserrat, E. Expression of the Adhesion Molecule Icam-1 in Non-Hodgkin’s Lymphoma: Relationship with Tumor Dissemination and Prognostic Importance. J. Clin. Oncol. 1998, 16, 35–40. [Google Scholar] [CrossRef]
- Molica, S.; Dattilo, A.; Mannella, A.; Levato, D.; Levato, L. Expression on Leukemic Cells and Serum Circulating Levels of Intercellular Adhesion Molecule-1 (Icam-1) in B-Cell Chronic Lymphocytic Leukemia: Implications for Prognosis. Leuk. Res. 1995, 19, 573–580. [Google Scholar] [CrossRef]
- Yaris, N.; Buyukpamukcu, M.; Kansu, E.; Canpinar, H. Expression of Adhesion Molecules Lfa-1, Icam-1, Cd44, and L-Selectin in Childhood Non-Hodgkin Lymphomas. Med. Pediatr. Oncol. 2001, 36, 359–364. [Google Scholar] [CrossRef]
- Novero, A.; Ravella, P.M.; Chen, Y.; Dous, G.; Liu, D. Ibrutinib for B Cell Malignancies. Exp. Hematol. Oncol. 2014, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershkovitz-Rokah, O.; Pulver, D.; Lenz, G.; Shpilberg, O. Ibrutinib Resistance in Mantle Cell Lymphoma: Clinical, Molecular and Treatment Aspects. Br. J. Haematol. 2018, 181, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Conway, T.; Wazny, J.; Bromage, A.; Tymms, M.; Sooraj, D.; Williams, E.D.; Beresford-Smith, B. Xenome—A Tool for Classifying Reads from Xenograft Samples. Bioinformatics 2012, 28, i172–i178. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast Universal Rna-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: Ncbi Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadeghi, L.; Arvidsson, G.; Merrien, M.; Wasik, A.M.; Görgens, A.; Smith, C.I.E.; Sander, B.; P. Wright, A. Differential B-Cell Receptor Signaling Requirement for Adhesion of Mantle Cell Lymphoma Cells to Stromal Cells. Cancers 2020, 12, 1143. https://doi.org/10.3390/cancers12051143
Sadeghi L, Arvidsson G, Merrien M, Wasik AM, Görgens A, Smith CIE, Sander B, P. Wright A. Differential B-Cell Receptor Signaling Requirement for Adhesion of Mantle Cell Lymphoma Cells to Stromal Cells. Cancers. 2020; 12(5):1143. https://doi.org/10.3390/cancers12051143
Chicago/Turabian StyleSadeghi, Laia, Gustav Arvidsson, Magali Merrien, Agata M. Wasik, André Görgens, C.I. Edvard Smith, Birgitta Sander, and Anthony P. Wright. 2020. "Differential B-Cell Receptor Signaling Requirement for Adhesion of Mantle Cell Lymphoma Cells to Stromal Cells" Cancers 12, no. 5: 1143. https://doi.org/10.3390/cancers12051143