Tetraploidy-Associated Genetic Heterogeneity Confers Chemo-Radiotherapy Resistance to Colorectal Cancer Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
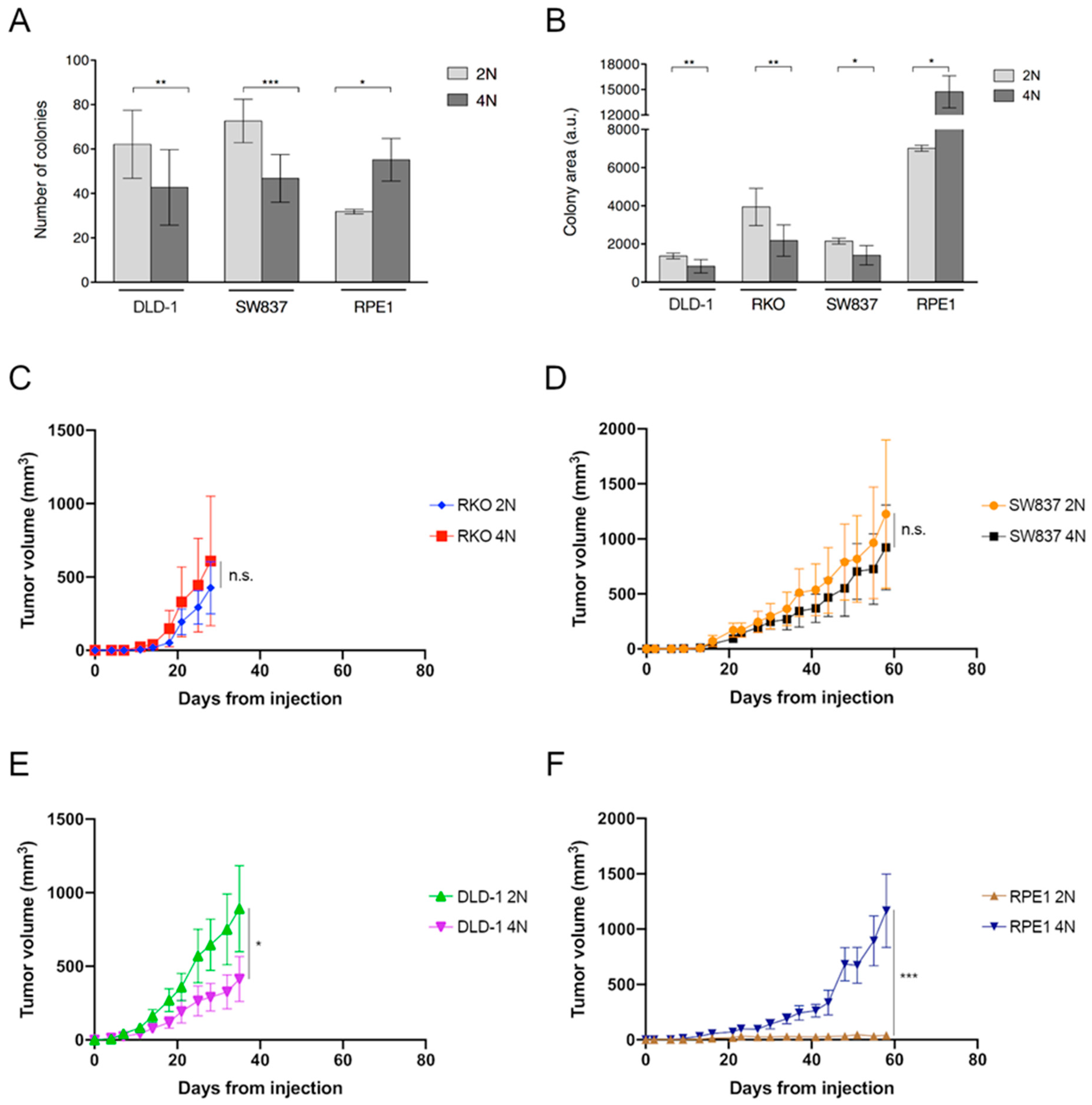
2.1. Growth Retardation of Near-Tetraploid Cells in Untreated Conditions and Diverse Tumorigenic Potential
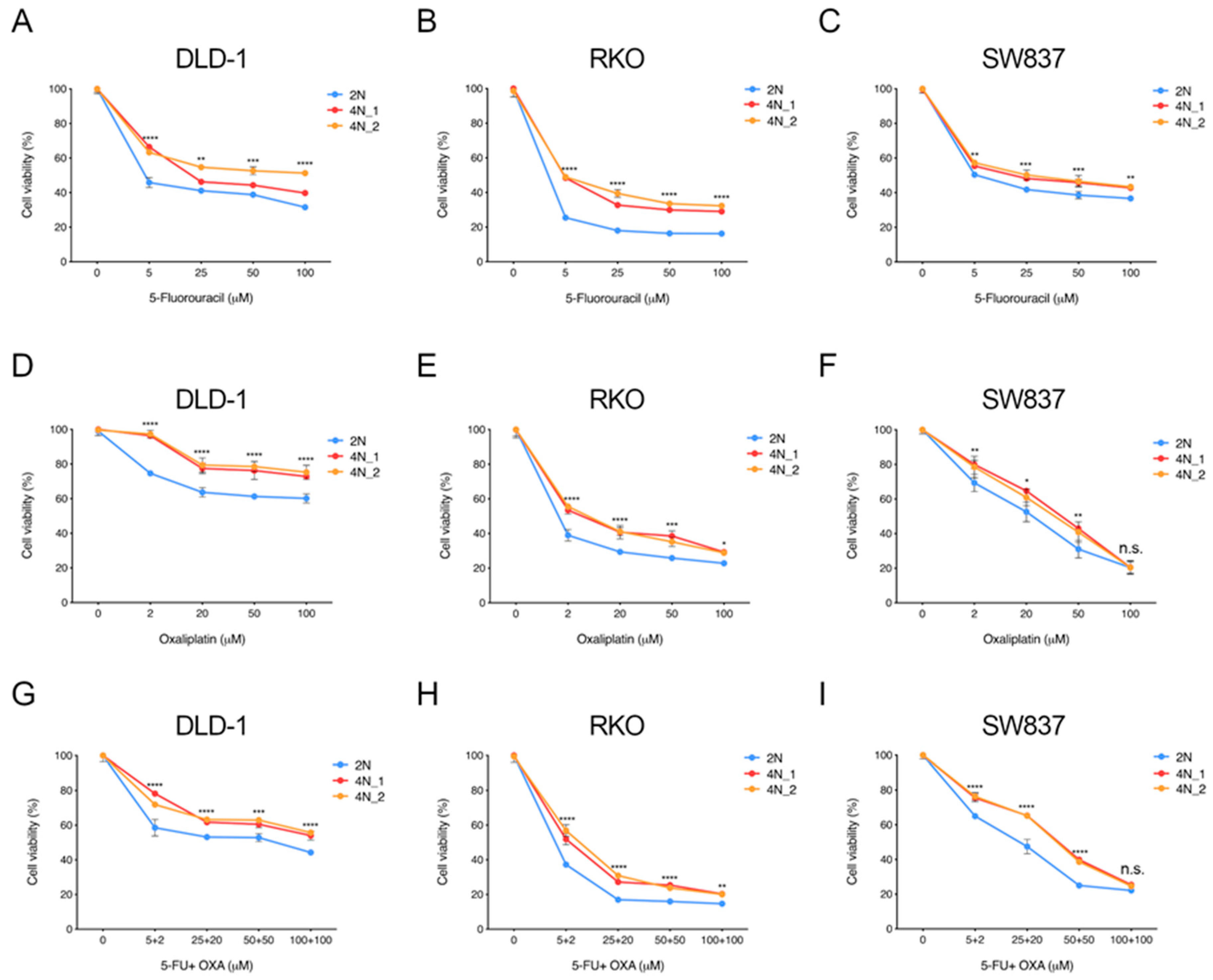
2.2. Near-Tetraploid Cells Exhibit Tolerance to Treatment with First-Line and Other Chemotherapeutic Agents
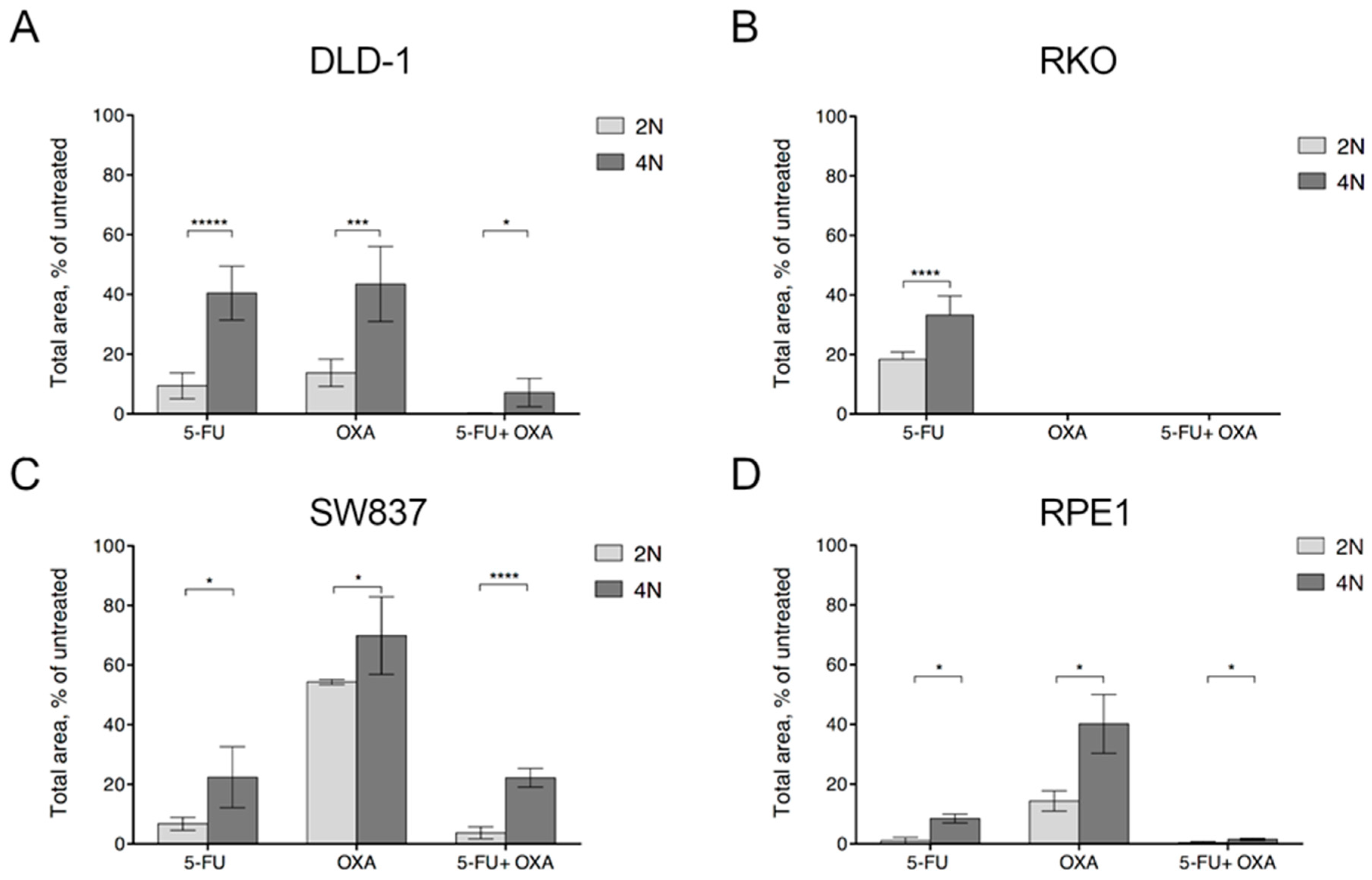
2.3. Apoptosis is Ubiquitously Activated by First-Line Chemotherapy Treatments
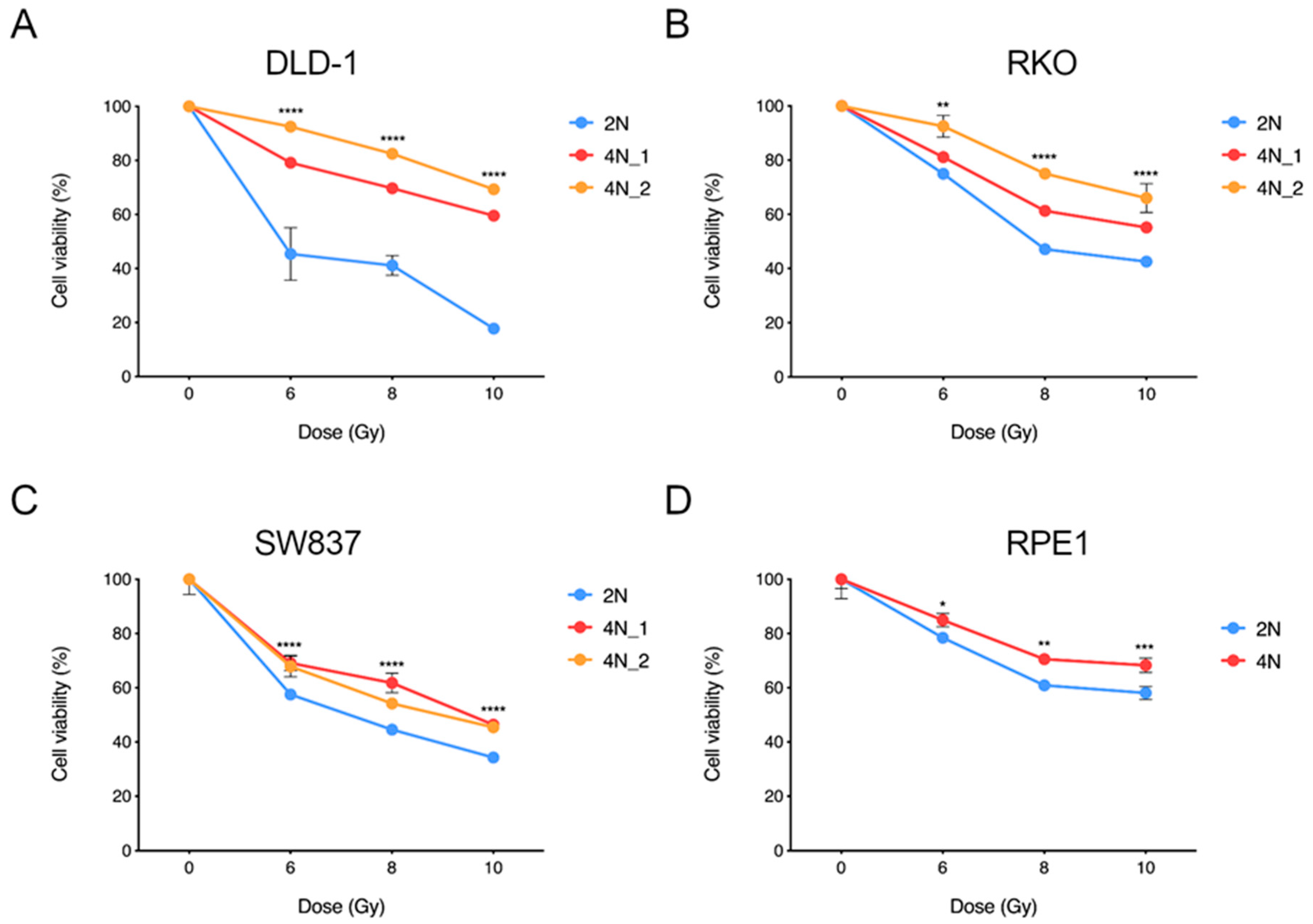
2.4. Near-Tetraploid Cells Display Resistance to Ionizing Radiation Treatment
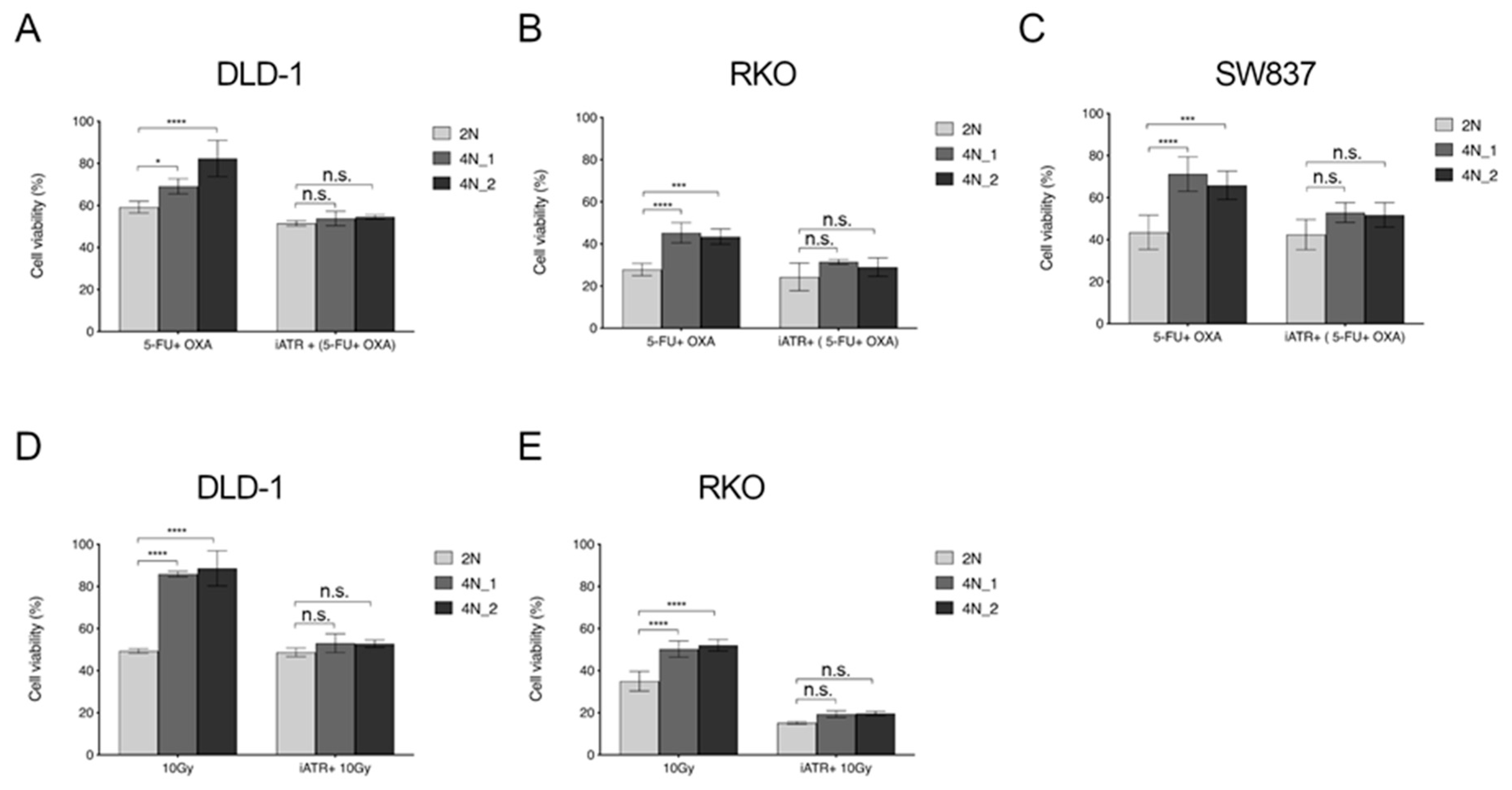
2.5. ATR Inhibitors Sensitize Near-Tetraploid Cells to Chemo-Radiation Treatment
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Irradiation
4.3. Cell Viability Assay
4.4. Colony Formation
4.5. Apoptosis Assay
4.6. Lysate Preparation and Western Blot Analysis
4.7. Tumorigenic in vivo Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zack, T.; Schumacher, S.; Carter, S. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotillo, R.; Hernando, E.; Díaz-Rodríguez, E.; Teruya-Feldstein, J.; Cordón-Cardo, C.; Lowe, S.W.; Benezra, R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007, 11, 9–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storchova, Z.; Kuffer, C. The consequences of tetraploidy and aneuploidy. J. Cell Sci. 2008, 121, 3859–3866. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Gronroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.L.; et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014, 4, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the evolution of non–small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Z.; Chan, Y.J.A.; Chua, Y.J.K.; Rutledge, S.D.; Pavelka, N.; Cimini, D.; Rancatia, G. Environmental stresses induce karyotypic instability in colorectal cancer cells. Mol. Biol. Cell 2019, 30, 42–55. [Google Scholar] [CrossRef]
- Sheltzer, J.M.; Ko, J.H.; Replogle, J.M.; Habibe Burgos, N.C.; Chung, E.S.; Meehl, C.M.; Sayles, N.M.; Passerini, V.; Storchova, Z.; Amon, A. Single-chromosome gains Commonly function as tumor suppressors. Cancer Cell 2017, 31, 240–255. [Google Scholar] [CrossRef] [Green Version]
- Ganem, N.J.; Cornils, H.; Chiu, S.-Y.; O’Rourke, K.P.; Arnaud, J.; Yimlamai, D.; Théry, M.; Camargo, F.D.; Pellman, D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 2014, 158, 833–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.-C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [Green Version]
- Wangsa, D.; Quintanilla, I.; Torabi, K.; Vila-Casadesús, M.; Ercilla, A.; Klus, G.; Yuce, Z.; Galofré, C.; Cuatrecasas, M.; Lozano, J.J.; et al. Near-tetraploid cancer cells show chromosome instability triggered by replication stress and exhibit enhanced invasiveness. FASEB J. 2018, 32, 3502–3517. [Google Scholar] [CrossRef] [Green Version]
- Böhly, N.; Kistner, M.; Bastians, H. Mild replication stress causes aneuploidy by deregulating microtubule dynamics in mitosis. Cell Cycle 2019, 18, 1–14. [Google Scholar] [CrossRef]
- Wilhelm, T.; Olziersky, A.-M.; Harry, D.; De Sousa, F.; Vassal, H.; Eskat, A.; Meraldi, P. Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat. Commun. 2019, 10, 3585. [Google Scholar] [CrossRef] [Green Version]
- Bakhoum, S.F.; Kabeche, L.; Murnane, J.P.; Zaki, B.I.; Compton, D.A. DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov. 2014, 4, 1281–1289. [Google Scholar] [CrossRef] [Green Version]
- Rutledge, S.D.; Douglas, T.A.; Nicholson, J.M.; Vila-Casadesús, M.; Kantzler, C.L.; Wangsa, D.; Barroso-Vilares, M.; Kale, S.D.; Logarinho, E.; Cimini, D. Selective advantage of trisomic human cells cultured in non-standard conditions. Sci. Rep. 2016, 6, 22828. [Google Scholar] [CrossRef] [Green Version]
- Duesberg, P.; Stindl, R.; Hehlmann, R. Origin of multidrug resistance in cells with and without multidrug resistance genes: Chromosome reassortments catalyzed by aneuploidy. Proc. Natl. Acad. Sci. USA 2001, 98, 11283–11288. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.X.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.M.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsova, A.Y.; Seget, K.; Moeller, G.K.; de Pagter, M.S.; de Roos, J.A.D.M.; Dürrbaum, M.; Kuffer, C.; Müller, S.; Zaman, G.J.R.; Kloosterman, W.P.; et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 2015, 14, 2810–2820. [Google Scholar] [CrossRef]
- Vitale, I.; Galluzzi, L.; Vivet, S.; Nanty, L.; Dessen, P.; Senovilla, L.; Olaussen, K.A.; Lazar, V.; Prudhomme, M.; Golsteyn, R.M.; et al. Inhibition of Chk1 kills tetraploid tumor cells through a p53-dependent pathway. PLoS ONE 2007, 2, e1337. [Google Scholar] [CrossRef] [PubMed]
- Marxer, M.; Foucar, C.E.; Man, W.Y.; Chen, Y.; Ma, H.T.; Poon, R.Y.C. Tetraploidization increases sensitivity to Aurora B kinase inhibition. Cell Cycle 2012, 11, 2567–2577. [Google Scholar] [CrossRef] [PubMed]
- Rello-Varona, S.; Vitale, I.; Kepp, O.; Senovilla, L.; Jemaá, M.; Métivier, D.; Castedo, M.; Kroemer, G. Preferential killing of tetraploid tumor cells by targeting the mitotic kinesin Eg5. Cell Cycle 2009, 8, 1030–1035. [Google Scholar] [CrossRef]
- Jemaà, M.; Manic, G.; Lledo, G.; Lissa, D.; Reynes, C.; Morin, N.; Chibon, F.; Sistigu, A.; Castedo, M.; Vitale, I.; et al. Whole-genome duplication increases tumor cell sensitivity to MPS1 inhibition. Oncotarget 2016, 7, 885–901. [Google Scholar] [CrossRef]
- Coward, J.; Harding, A. Size does matter: Why polyploid tumor cells are critical drug targets in the war on cancer. Front. Oncol. 2014, 4, 123. [Google Scholar] [CrossRef]
- Kleivi, K.; Teixeira, M.R.; Eknaes, M.; Diep, C.B.; Jakobsen, K.S.; Hamelin, R.; Lothe, R.A. Genome signatures of colon carcinoma cell lines. Cancer Genet. Cytogenet. 2004, 155, 119–131. [Google Scholar] [CrossRef]
- Knutsen, T.; Padilla-Nash, H.M.; Wangsa, D.; Barenboim-Stapleton, L.; Camps, J.; McNeil, N.; Difilippantonio, M.J.; Ried, T. Definitive molecular cytogenetic characterization of 15 colorectal cancer cell lines. Genes Chromosom. Cancer 2010, 49, 204–223. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Bodmer, W.F. Analysis of P53 mutations and their expression in 56 colorectal cancer cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknæs, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Castedo, M.; Coquelle, A.; Vivet, S.; Vitale, I.; Kauffmann, A.; Dessen, P.; Pequignot, M.O.; Casares, N.; Valent, A.; Mouhamad, S.; et al. Apoptosis regulation in tetraploid cancer cells. EMBO J. 2006, 25, 2584–2595. [Google Scholar] [CrossRef] [Green Version]
- Chumduri, C.; Gillissen, B.; Richter, A.; Richter, A.; Milojkovic, A.; Overkamp, T.; Müller, A.; Pott, C.; Daniel, P.T. Apoptosis resistance, mitotic catastrophe, and loss of ploidy control in Burkitt lymphoma. J. Mol. Med. 2015, 93, 559–572. [Google Scholar] [CrossRef]
- Vecchione, L.; Gambino, V.; Raaijmakers, J.; Schlicker, A.; Fumagalli, A.; Russo, M.; Villanueva, A.; Beerling, E.; Bartolini, A.; Mollevi, D.G.; et al. A Vulnerability of a subset of colon cancers with potential clinical utility. Cell 2016, 165, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Glynne-Jones, R.; Wyrwicz, L.; Tiret, E.; Brown, G.; Rödel, C.; Cervantes, A.; Arnold, D. Rectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv22–iv40. [Google Scholar] [CrossRef]
- Jun, S.; Jung, Y.-S.; Suh, H.N.; Wang, W.; Kim, M.J.; Oh, Y.S.; Lien, E.M.; Shen, X.; Matsumoto, Y.; McCrea, P.D.; et al. LIG4 mediates Wnt signalling-induced radioresistance. Nat. Commun. 2016, 7, 10994. [Google Scholar] [CrossRef] [Green Version]
- Kendziorra, E.; Ahlborn, K.; Spitzner, M.; Rave-Fränk, M.; Emons, G.; Gaedcke, J.; Kramer, F.; Wolff, H.A.; Becker, H.; Beissbarth, T.; et al. Silencing of the Wnt transcription factor TCF4 sensitizes colorectal cancer cells to (chemo-) radiotherapy. Carcinogenesis 2011, 32, 1824–1831. [Google Scholar] [CrossRef]
- Baptistella, A.R.; Landemberger, M.C.; Dias, M.V.S.; Giudice, F.S.; Rodrigues, B.R.; da Silva, P.P.C.E.; Cassinela, E.K.; Lacerda, T.C.; Marchi, F.A.; Leme, A.F.P.; et al. Rab5C enhances resistance to ionizing radiation in rectal cancer. J. Mol. Med. 2019, 97, 855–869. [Google Scholar] [CrossRef]
- Lluis, F.; Pedone, E.; Pepe, S.; Cosma, M.P. Periodic Activation of Wnt/β-Catenin Signaling Enhances Somatic Cell Reprogramming Mediated by Cell Fusion. Cell Stem Cell 2008, 3, 493–507. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Touil, Y.; Igoudjil, W.; Corvaisier, M.; Dessein, A.-F.; Vandomme, J.; Monté, D.; Stechly, L.; Skrypek, N.; Langlois, C.; Grard, G.; et al. Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clin. Cancer Res. 2014, 20, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Otto, S.P. The evolutionary consequences of polyploidy. Cell 2007, 131, 452–462. [Google Scholar] [CrossRef] [Green Version]
- López, S.; Lim, E.L.; Horswell, S.; Haase, K.; Huebner, A.; Dietzen, M.; Mourikis, T.P.; Watkins, T.B.K.; Rowan, A.; Dewhurst, S.M.; et al. Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat. Genet. 2020, 52, 283–293. [Google Scholar] [CrossRef]
- Thompson, D.A.; Desai, M.M.; Murray, A.W. Ploidy controls the success of mutators and nature of mutations during budding yeast evolution. Curr. Biol. 2006, 16, 1581–1590. [Google Scholar] [CrossRef] [Green Version]
- Euhus, D.M.; Hudd, C.; Laregina, M.C.; Johnson, F.E. Tumor measurement in the nude mouse. J. Surg. Oncol. 1986, 31, 229–234. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galofré, C.; Gönül Geyik, Ö.; Asensio, E.; Wangsa, D.; Hirsch, D.; Parra, C.; Saez, J.; Mollà, M.; Yüce, Z.; Castells, A.; et al. Tetraploidy-Associated Genetic Heterogeneity Confers Chemo-Radiotherapy Resistance to Colorectal Cancer Cells. Cancers 2020, 12, 1118. https://doi.org/10.3390/cancers12051118
Galofré C, Gönül Geyik Ö, Asensio E, Wangsa D, Hirsch D, Parra C, Saez J, Mollà M, Yüce Z, Castells A, et al. Tetraploidy-Associated Genetic Heterogeneity Confers Chemo-Radiotherapy Resistance to Colorectal Cancer Cells. Cancers. 2020; 12(5):1118. https://doi.org/10.3390/cancers12051118
Chicago/Turabian StyleGalofré, Claudia, Öykü Gönül Geyik, Elena Asensio, Darawalee Wangsa, Daniela Hirsch, Carolina Parra, Jordi Saez, Meritxell Mollà, Zeynep Yüce, Antoni Castells, and et al. 2020. "Tetraploidy-Associated Genetic Heterogeneity Confers Chemo-Radiotherapy Resistance to Colorectal Cancer Cells" Cancers 12, no. 5: 1118. https://doi.org/10.3390/cancers12051118