Misregulation of ELK1, AP1, and E12 Transcription Factor Networks Is Associated with Melanoma Progression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. RNA-seq Data Grouping and Sample Selection
2.2. RNA-seq Analysis
2.3. ChIP-seq and Super-Enhancer Analysis
2.4. Co-Expression Network Analysis
2.5. Single-Cell RNA-seq (scRNA-seq) Analysis
3. Results
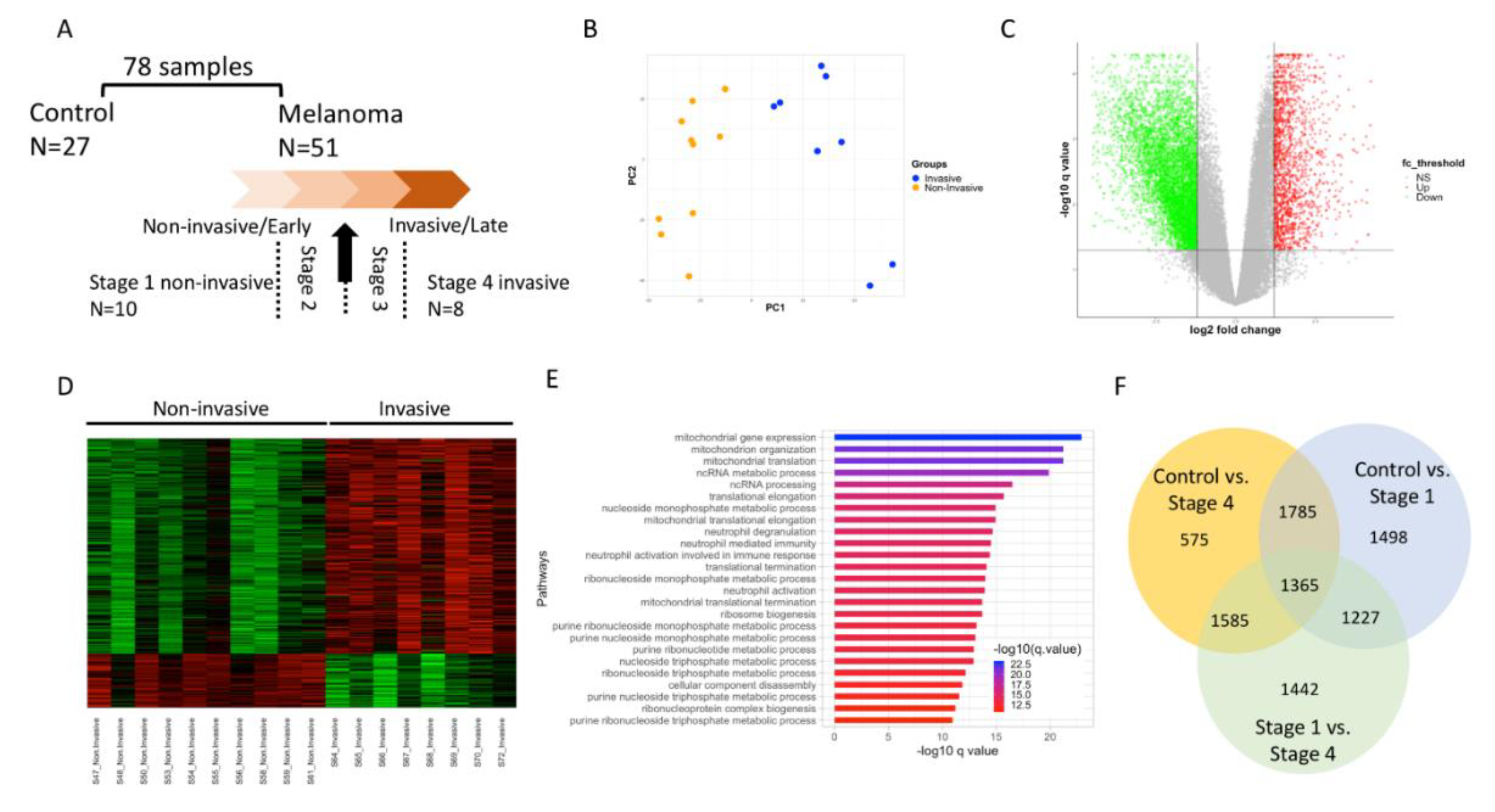
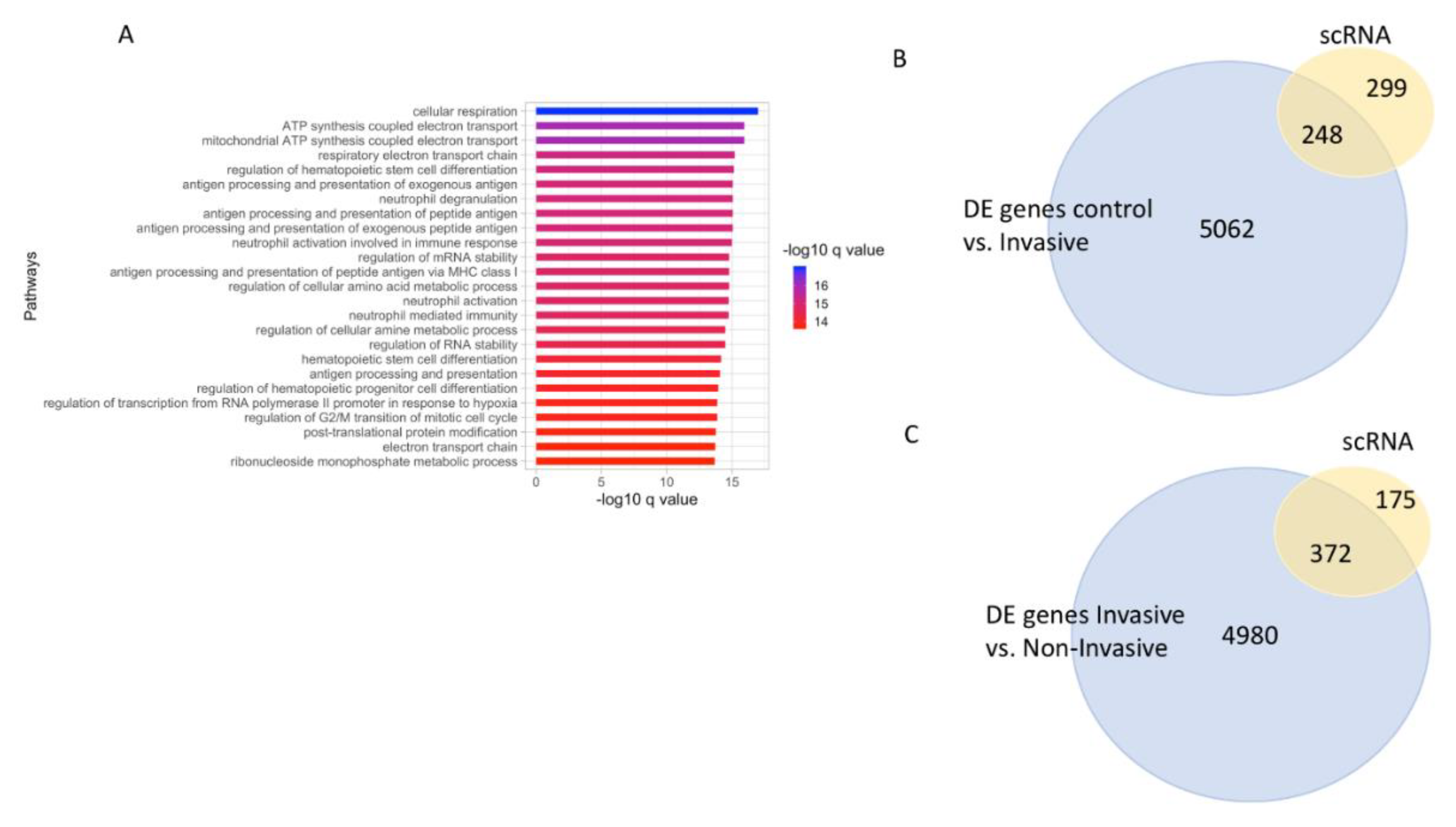
3.1. Sample Processing and Differential Expression Analysis to Understand Melanoma Cancer Progression
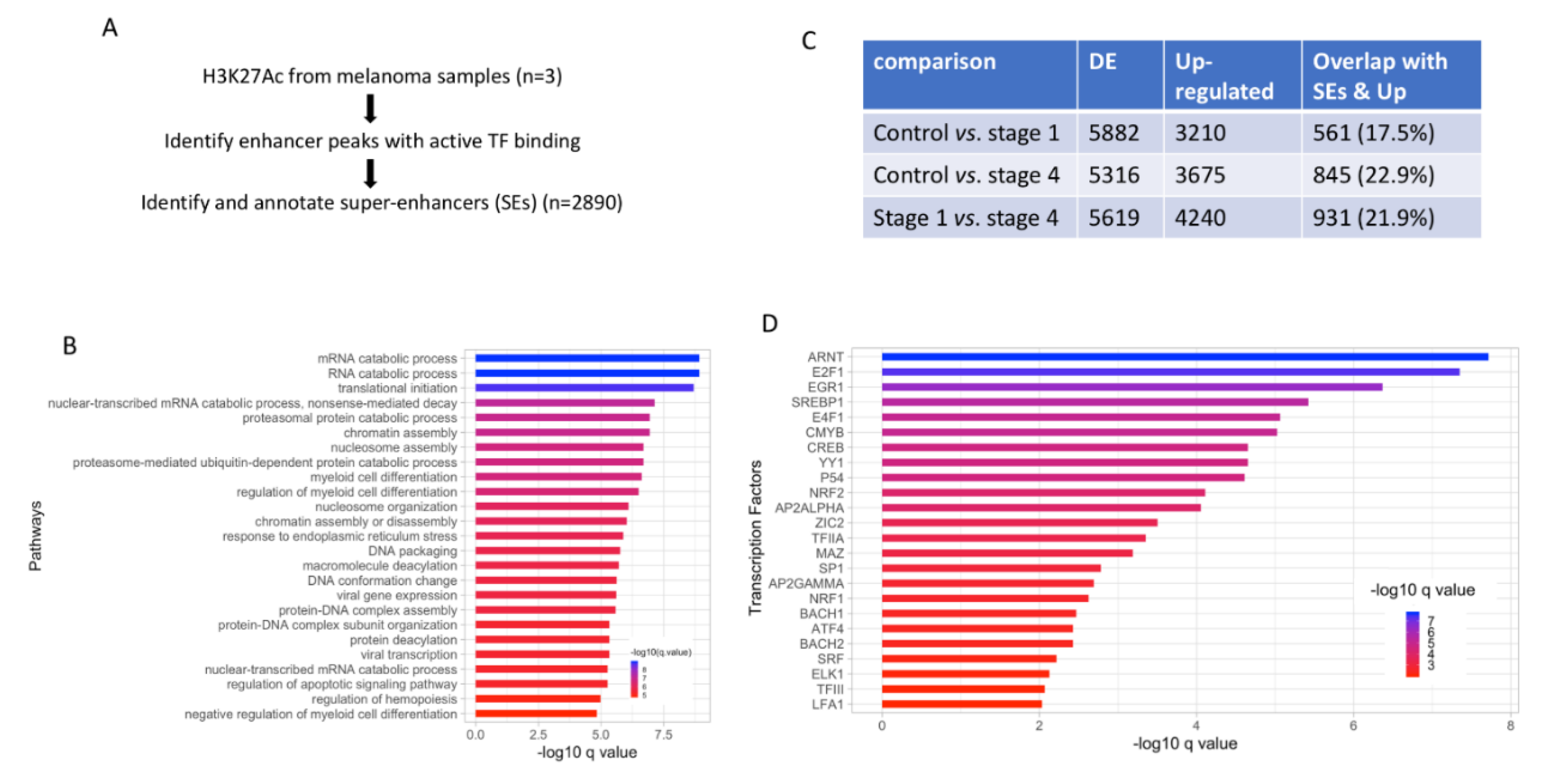
3.2. Potential Link between Upregulated DE Genes and Super-Enhancers
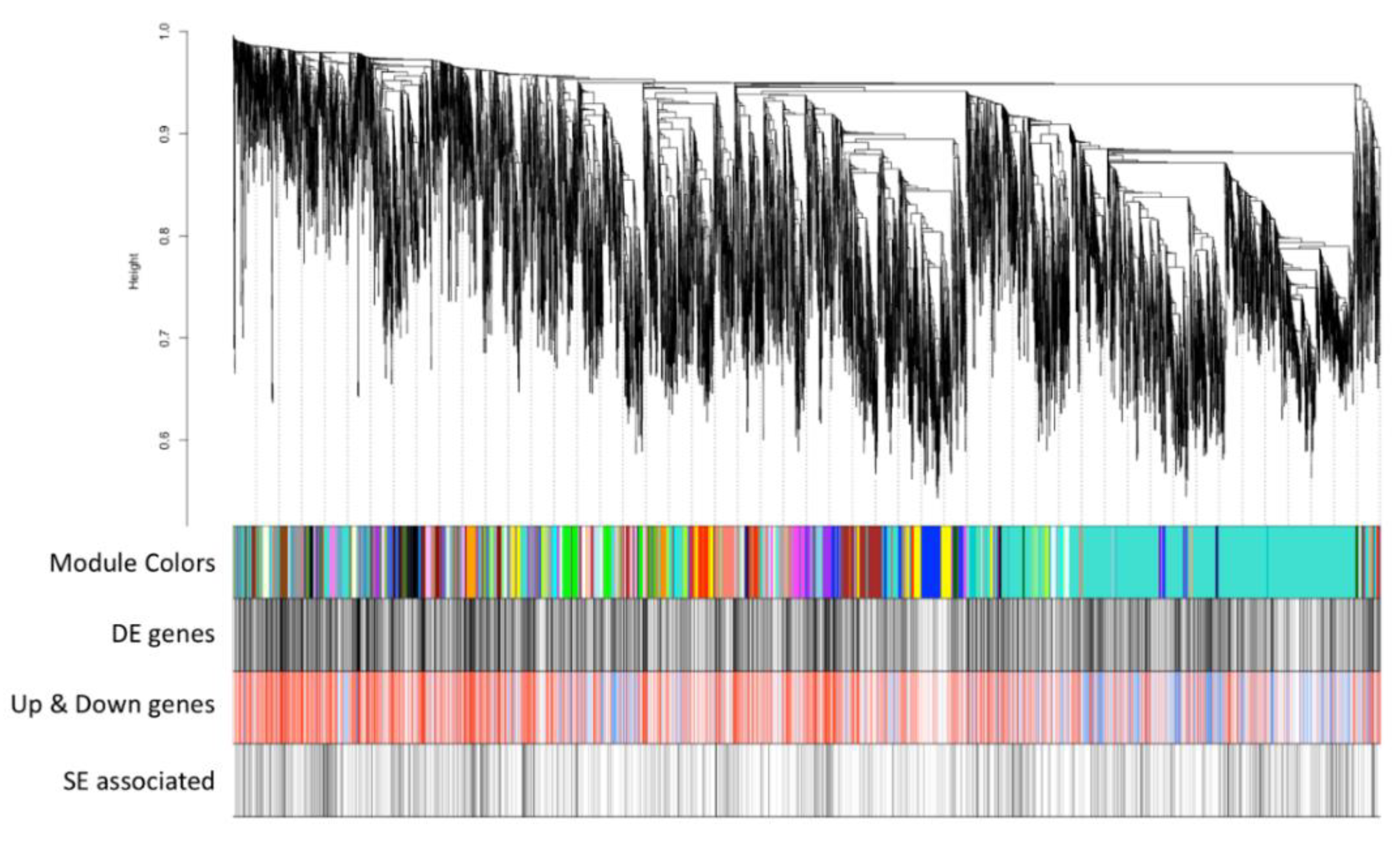
3.3. Co-Expression Network Analysis to Understand Cancer Progression
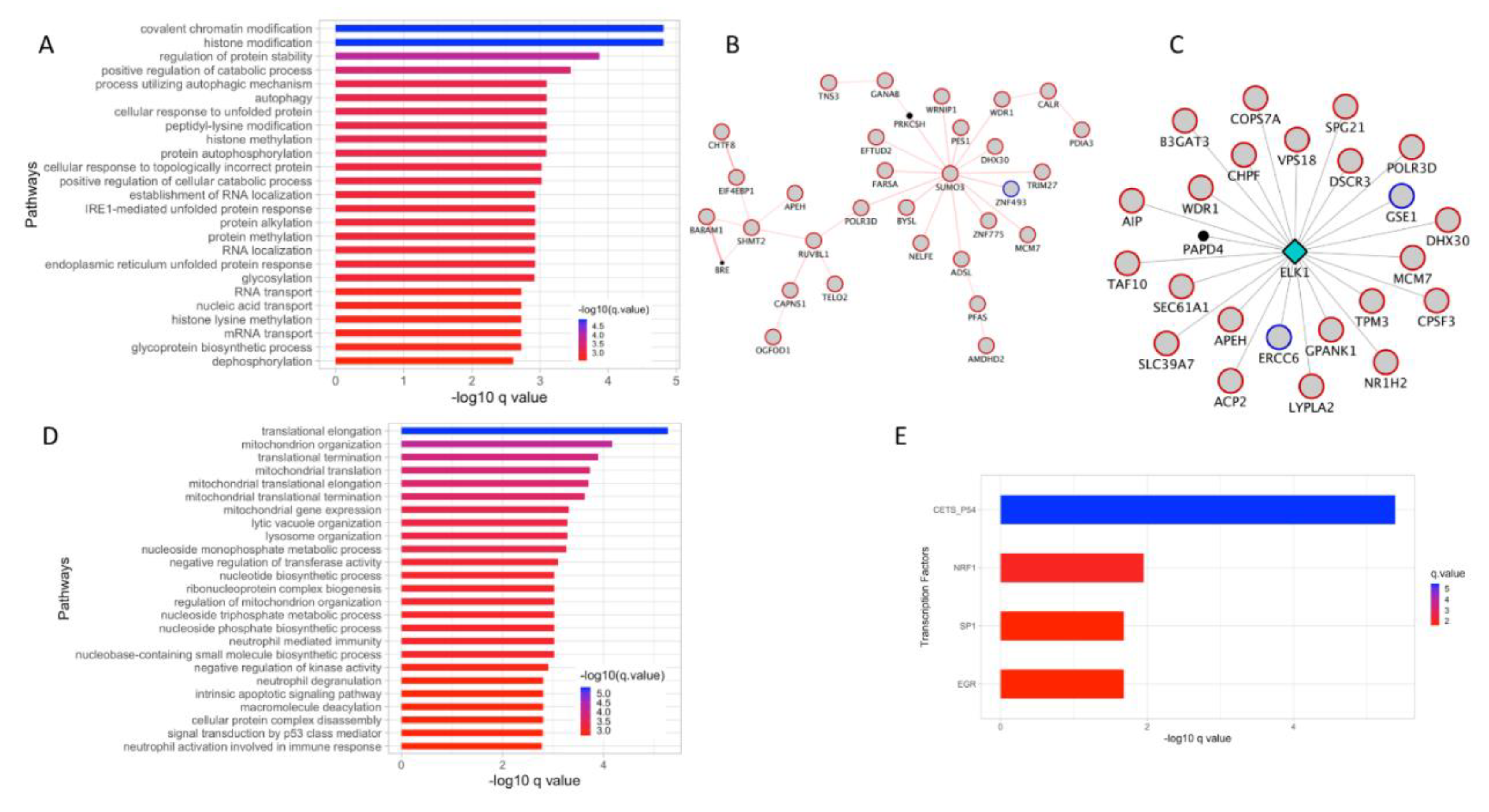
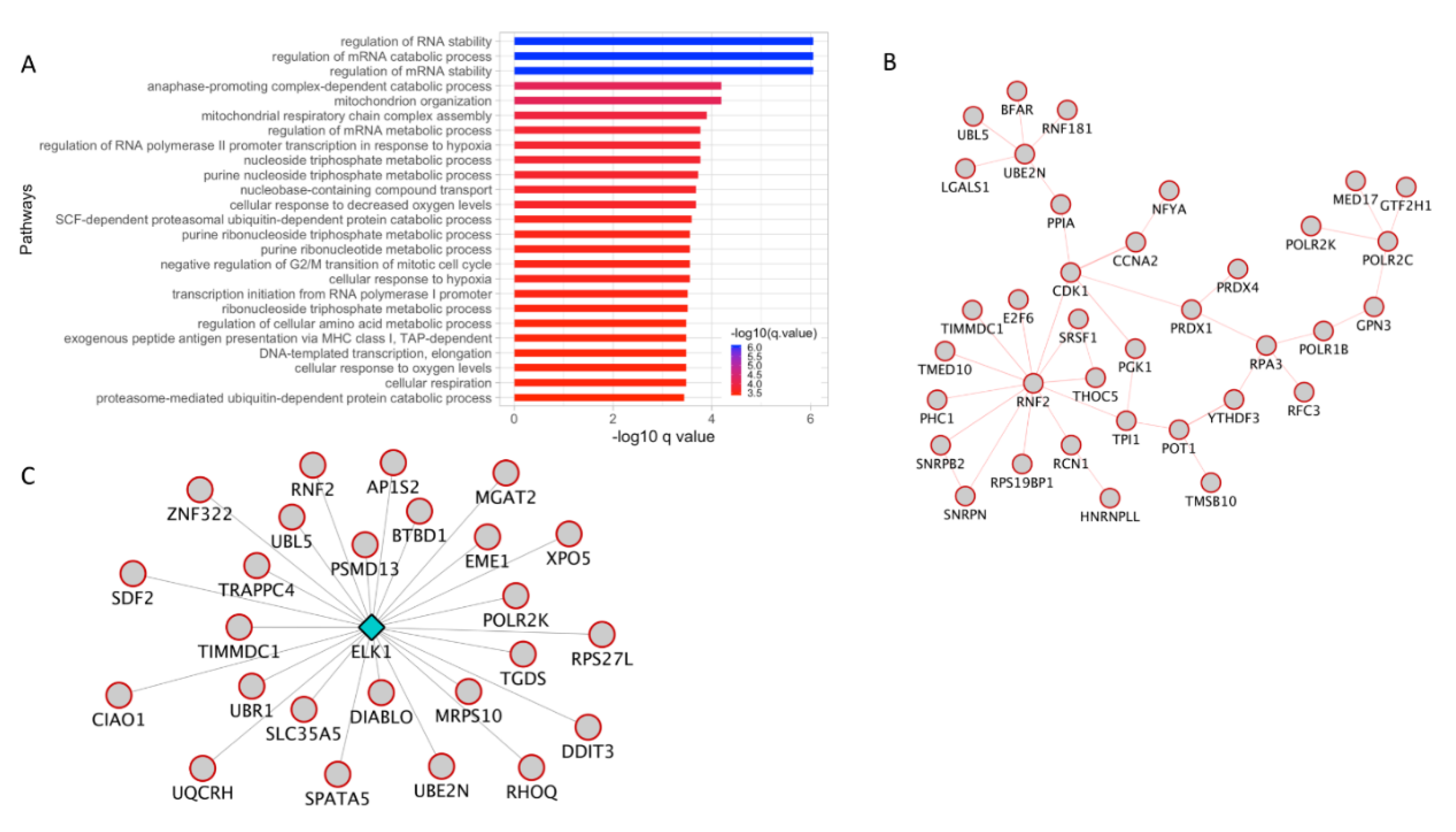
3.4. Sumoylation and ELK1 TF Targets Interactome is Highly Expressed in Invasive Melanoma
3.5. Super-Enhancer (SE) Associated Genes from the Turquoise Module Implicate Four TFs in Melanoma Progression
3.6. Protein Ubiquitination Process and ELK1 TF Targets Are Upregulated in Invasive Melanoma
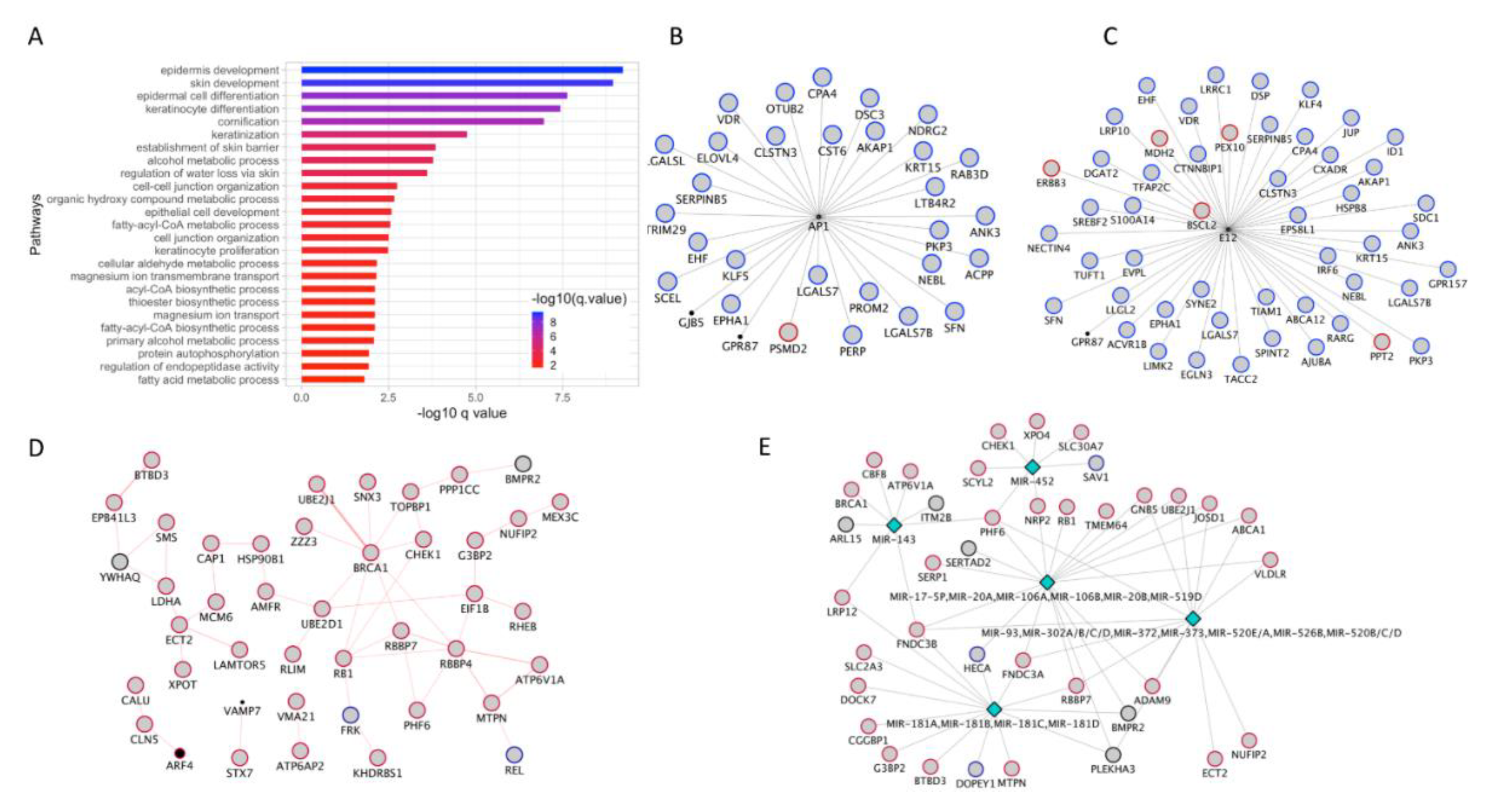
3.7. Co-Expression Module Associated with Skin/Keratinocyte Associated Processes
3.8. Identification of BRCA1 Hub and Involvement of MicroRNA
3.9. Single Cell (sc) RNA-seq Analysis of Melanoma Overlaps with Genes Upregulated with Stage 4 Melanoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AJCC | American Joint Committee on Cancer |
| ChIP | chromatin immunoprecipitation |
| DE | differential expression |
| GEO | gene expression omnibus |
| MsigDB | molecular signature database |
| PPI | protein-protein interaction |
| RNA | ribonucleic acid |
| sc | single cell |
| Seq | sequence |
| SE | super-enhancer |
| TF | Transcription factor |
| WGCNA | weighted gene co-expression network analysis |
References
- Cramer, S.F. Malignant melanoma in situ: Another perspective. Hum. Pathol. 1991, 22, 626–627. [Google Scholar] [CrossRef]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tyminska, A. Skin melanocytes: Biology and development. Postepy Dermatol. Alergol. 2013, 30, 30–41. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, S.A.; Finlay, G.J.; Baguley, B.C.; Askarian-Amiri, M.E. Signaling Pathways in Melanogenesis. Int. J. Mol. Sci. 2016, 17, 1144. [Google Scholar]
- De Angelis, R.; Sant, M.; Coleman, M.P.; Francisci, S.; Baili, P.; Pierannunzio, D.; Trama, A.; Visser, O.; Brenner, H.; Ardanaz, E.; et al. Cancer survival in Europe 1999–2007 by country and age: Results of EUROCARE—5-a population-based study. Lancet Oncol. 2014, 15, 23–34. [Google Scholar] [CrossRef]
- Schadendorf, D.; Hauschild, A. Melanoma in 2013: Melanoma—The run of success continues. Nat. Rev. Clin. Oncol. 2014, 11, 75–76. [Google Scholar] [CrossRef]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- FitzGerald, M.G.; Harkin, D.P.; Silva-Arrieta, S.; MacDonald, D.J.; Lucchina, L.C.; Unsal, H.; O’Neill, E.; Koh, J.; Finkelstein, D.M.; Isselbacher, K.J.; et al. Prevalence of germ-line mutations in p16, p19ARF, and CDK4 in familial melanoma: Analysis of a clinic-based population. Proc. Natl. Acad. Sci. USA 1996, 93, 8541–8545. [Google Scholar] [CrossRef] [Green Version]
- Zuo, L.; Weger, J.; Yang, Q.; Goldstein, A.M.; Tucker, M.A.; Walker, G.J.; Hayward, N.; Dracopoli, N.C. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat. Genet. 1996, 12, 97–99. [Google Scholar] [CrossRef]
- Lin, W.M.; Baker, A.C.; Beroukhim, R.; Winckler, W.; Feng, W.; Marmion, J.M.; Laine, E.; Greulich, H.; Tseng, H.; Gates, C.; et al. Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer Res. 2008, 68, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [Green Version]
- Kunz, M.; Loffler-Wirth, H.; Dannemann, M.; Willscher, E.; Doose, G.; Kelso, J.; Kottek, T.; Nickel, B.; Hopp, L.; Landsberg, J.; et al. RNA-seq analysis identifies different transcriptomic types and developmental trajectories of primary melanomas. Oncogene 2018, 37, 6136–6151. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.X.; Li, Y.; Chen, G.Z. Network-based co-expression analysis for exploring the potential diagnostic biomarkers of metastatic melanoma. PLoS ONE 2018, 13, e0190447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badal, B.; Solovyov, A.; Di Cecilia, S.; Chan, J.M.; Chang, L.W.; Iqbal, R.; Aydin, I.T.; Rajan, G.S.; Chen, C.; Abbate, F.; et al. Transcriptional dissection of melanoma identifies a high-risk subtype underlying TP53 family genes and epigenome deregulation. JCI Insight 2017, 2, 92102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [Green Version]
- Sobin, L.H.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumours, 7th ed.; Wiley-Blackwell: Chichester, UK; Hoboken, NJ, USA, 2010; 309p. [Google Scholar]
- Edge, S.B. American Joint Committee on Cancer. AJCC Cancer Staging Manual, 7th ed.; Springer: New York, NY, USA, 2010; 648p. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 14 February 2020).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Zheng, R.; Wan, C.; Mei, S.; Qin, Q.; Wu, Q.; Sun, H.; Chen, C.H.; Brown, M.; Zhang, X.; Meyer, C.A.; et al. Cistrome Data Browser: Expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019, 47, D729–D735. [Google Scholar] [CrossRef]
- Gelato, K.A.; Schockel, L.; Klingbeil, O.; Ruckert, T.; Lesche, R.; Toedling, J.; Kalfon, E.; Heroult, M.; Lejeune, P.; Monning, U.; et al. Super-enhancers define a proliferative PGC-1alpha-expressing melanoma subgroup sensitive to BET inhibition. Oncogene 2018, 37, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Mai, T.L.; Hu, G.M.; Chen, C.M. Visualizing and Clustering Protein Similarity Networks: Sequences, Structures, and Functions. J. Proteome Res. 2016, 15, 2123–2131. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Franz, M.; Rodriguez, H.; Lopes, C.; Zuberi, K.; Montojo, J.; Bader, G.D.; Morris, Q. GeneMANIA update 2018. Nucleic Acids Res. 2018, 46, W60–W64. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo Yau, H.; Ettayebi, I.; de Carvalho, D.D. The Cancer Epigenome: Exploiting Its Vulnerabilities for Immunotherapy. Trends Cell Biol. 2019, 29, 31–43. [Google Scholar] [CrossRef]
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.J.; Dicker, A.J.; Dahler, A.L.; Saunders, N.A. E2F as a regulator of keratinocyte proliferation: Implications for skin tumor development. J. Investig. Dermatol. 1997, 109, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef]
- Van der Stoop, P.; Boutsma, E.A.; Hulsman, D.; Noback, S.; Heimerikx, M.; Kerkhoven, R.M.; Voncken, J.W.; Wessels, L.F.; van Lohuizen, M. Ubiquitin E3 ligase Ring1b/Rnf2 of polycomb repressive complex 1 contributes to stable maintenance of mouse embryonic stem cells. PLoS ONE 2008, 3, e2235. [Google Scholar] [CrossRef] [Green Version]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef]
- Tutt, A.; Gabriel, A.; Bertwistle, D.; Connor, F.; Paterson, H.; Peacock, J.; Ross, G.; Ashworth, A. Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr. Biol. 1999, 9, 1107–1110. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [Green Version]
- Cabrini, G.; Bezzerri, V.; Mancini, I.; Nicolis, E.; Dechecchi, M.C.; Tamanini, A.; Lampronti, I.; Piccagli, L.; Bianchi, N.; Borgatti, M.; et al. Targeting transcription factor activity as a strategy to inhibit pro-inflammatory genes involved in cystic fibrosis: Decoy oligonucleotides and low-molecular weight compounds. Curr. Med. Chem. 2010, 17, 4392–4404. [Google Scholar] [CrossRef] [PubMed]
- Aljada, A.; Ghanim, H.; Mohanty, P.; Kapur, N.; Dandona, P. Insulin inhibits the pro-inflammatory transcription factor early growth response gene-1 (Egr)-1 expression in mononuclear cells (MNC) and reduces plasma tissue factor (TF) and plasminogen activator inhibitor-1 (PAI-1) concentrations. J. Clin. Endocrinol. Metab. 2002, 87, 1419–1422. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, G.M.; Pitarresi, J.R.; Balakrishnan, S.; Ostrowski, M.C. The ETS family of oncogenic transcription factors in solid tumours. Nat. Rev. Cancer 2017, 17, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Pallai, R.; Bhaskar, A.; Sodi, V.; Rice, L.M. Ets1 and Elk1 transcription factors regulate cancerous inhibitor of protein phosphatase 2A expression in cervical and endometrial carcinoma cells. Transcription 2012, 3, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Engel, I.; Murre, C. Ectopic expression of E47 or E12 promotes the death of E2A-deficient lymphomas. Proc. Natl. Acad. Sci. USA 1999, 96, 996–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhu, S.; Ignatova, A.; Park, S.T.; Sun, X.H. Regulation of the expression of cyclin-dependent kinase inhibitor p21 by E2A and Id proteins. Mol. Cell. Biol. 1997, 17, 5888–5896. [Google Scholar] [CrossRef] [Green Version]
- Seberg, H.E.; van Otterloo, E.; Cornell, R.A. Beyond MITF: Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment Cell Melanoma Res. 2017, 30, 454–466. [Google Scholar] [CrossRef] [Green Version]
- Ye, N.; Ding, Y.; Wild, C.; Shen, Q.; Zhou, J. Small molecule inhibitors targeting activator protein 1 (AP-1). J. Med. Chem. 2014, 57, 6930–6948. [Google Scholar] [CrossRef]
- Fan, M.; Goodwin, M.E.; Birrer, M.J.; Chambers, T.C. The c-Jun NH(2)-terminal protein kinase/AP-1 pathway is required for efficient apoptosis induced by vinblastine. Cancer Res. 2001, 61, 4450–4458. [Google Scholar]
- Weiss, C.; Bohmann, D. Deregulated repression of c-Jun provides a potential link to its role in tumorigenesis. Cell Cycle 2004, 3, 111–113. [Google Scholar] [CrossRef]
- Kim, H.; Lee, J.E.; Cho, E.J.; Liu, J.O.; Youn, H.D. Menin, a tumor suppressor, represses JunD-mediated transcriptional activity by association with an mSin3A-histone deacetylase complex. Cancer Res. 2003, 63, 6135–6139. [Google Scholar] [PubMed]
- Agarwal, S.K.; Guru, S.C.; Heppner, C.; Erdos, M.R.; Collins, R.M.; Park, S.Y.; Saggar, S.; Chandrasekharappa, S.C.; Collins, F.S.; Spiegel, A.M.; et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 1999, 96, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Pfarr, C.M.; Mechta, F.; Spyrou, G.; Lallemand, D.; Carillo, S.; Yaniv, M. Mouse JunD negatively regulates fibroblast growth and antagonizes transformation by ras. Cell 1994, 76, 747–760. [Google Scholar] [CrossRef]
- Han, Z.J.; Feng, Y.H.; Gu, B.H.; Li, Y.M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabellino, A.; Andreani, C.; Scaglioni, P.P. The Role of PIAS SUMO E3-Ligases in Cancer. Cancer Res. 2017, 77, 1542–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, J.D.; Kahle, K.T.; Sun, T.; Meerbrey, K.L.; Schlabach, M.R.; Schmitt, E.M.; Skinner, S.O.; Xu, Q.; Li, M.Z.; Hartman, Z.C.; et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 2012, 335, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; de Lichy, M.; Bille, K.; Dessen, P.; d’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef]
- Pinder, J.B.; Attwood, K.M.; Dellaire, G. Reading, writing, and repair: The role of ubiquitin and the ubiquitin-like proteins in DNA damage signaling and repair. Front. Genet. 2013, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.X.; Ma, M.; Sang, M.X.; Wang, X.X.; Song, H.; Liu, Z.K.; Zhu, S.C. Radiosensitization of esophageal carcinoma cells by knockdown of RNF2 expression. Int. J. Oncol. 2016, 48, 1985–1996. [Google Scholar] [CrossRef]
- Liu-Smith, F.; Dellinger, R.; Meyskens, F.L., Jr. Updates of reactive oxygen species in melanoma etiology and progression. Arch. Biochem. Biophys. 2014, 563, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Fried, L.; Arbiser, J.L. The reactive oxygen-driven tumor: Relevance to melanoma. Pigment Cell Melanoma Res. 2008, 21, 117–122. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, K.; Baird, M.; Fischer, R.; Chaitankar, V.; Seifuddin, F.; Chen, Y.-C.; Tunc, I.; Waterman, C.M.; Pirooznia, M. Misregulation of ELK1, AP1, and E12 Transcription Factor Networks Is Associated with Melanoma Progression. Cancers 2020, 12, 458. https://doi.org/10.3390/cancers12020458
Singh K, Baird M, Fischer R, Chaitankar V, Seifuddin F, Chen Y-C, Tunc I, Waterman CM, Pirooznia M. Misregulation of ELK1, AP1, and E12 Transcription Factor Networks Is Associated with Melanoma Progression. Cancers. 2020; 12(2):458. https://doi.org/10.3390/cancers12020458
Chicago/Turabian StyleSingh, Komudi, Michelle Baird, Robert Fischer, Vijender Chaitankar, Fayaz Seifuddin, Yun-Ching Chen, Ilker Tunc, Clare M. Waterman, and Mehdi Pirooznia. 2020. "Misregulation of ELK1, AP1, and E12 Transcription Factor Networks Is Associated with Melanoma Progression" Cancers 12, no. 2: 458. https://doi.org/10.3390/cancers12020458