Probiotic-Treated Super-Charged NK Cells Efficiently Clear Poorly Differentiated Pancreatic Tumors in Hu-BLT Mice

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
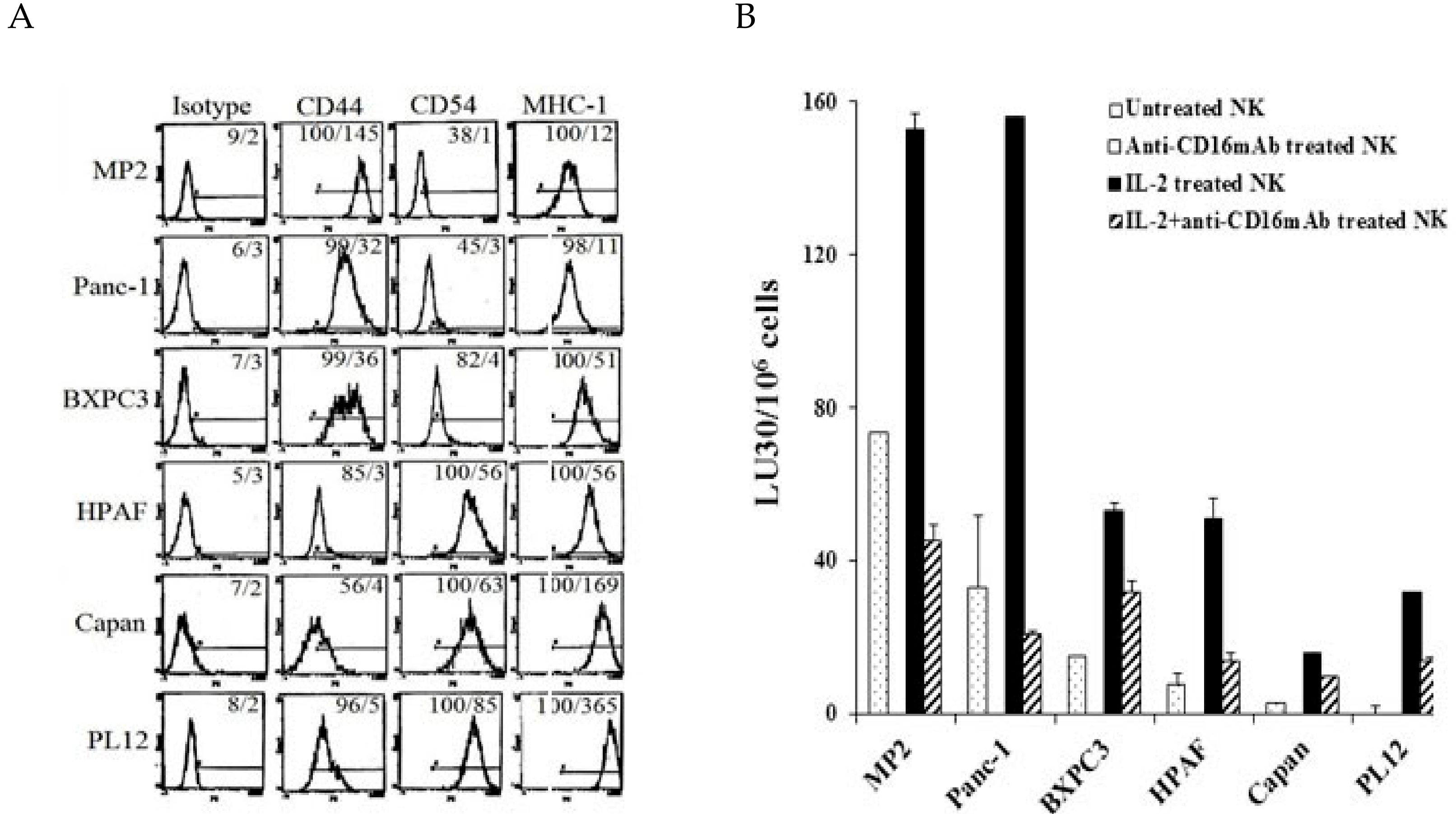
2.1. Correlation between NK Cell Cytotoxicity and the Stage of Differentiation in Pancreatic Tumors
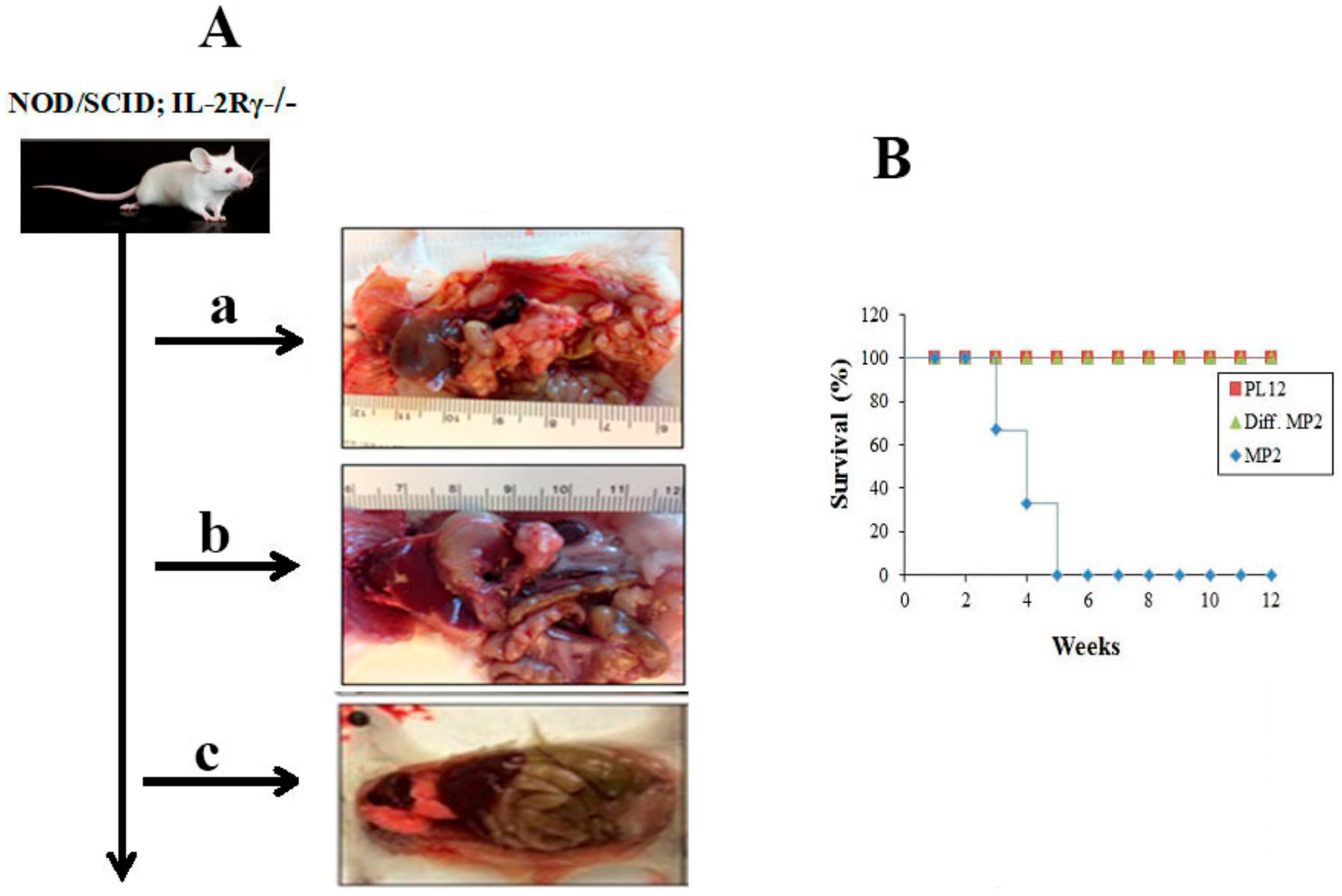
2.2. Curtailed Pancreatic Tumor Growth and Long-Term Survival of Mice after Implantation of NK-Differentiated MP2 and Patient-Derived Differentiated PL12 Tumors
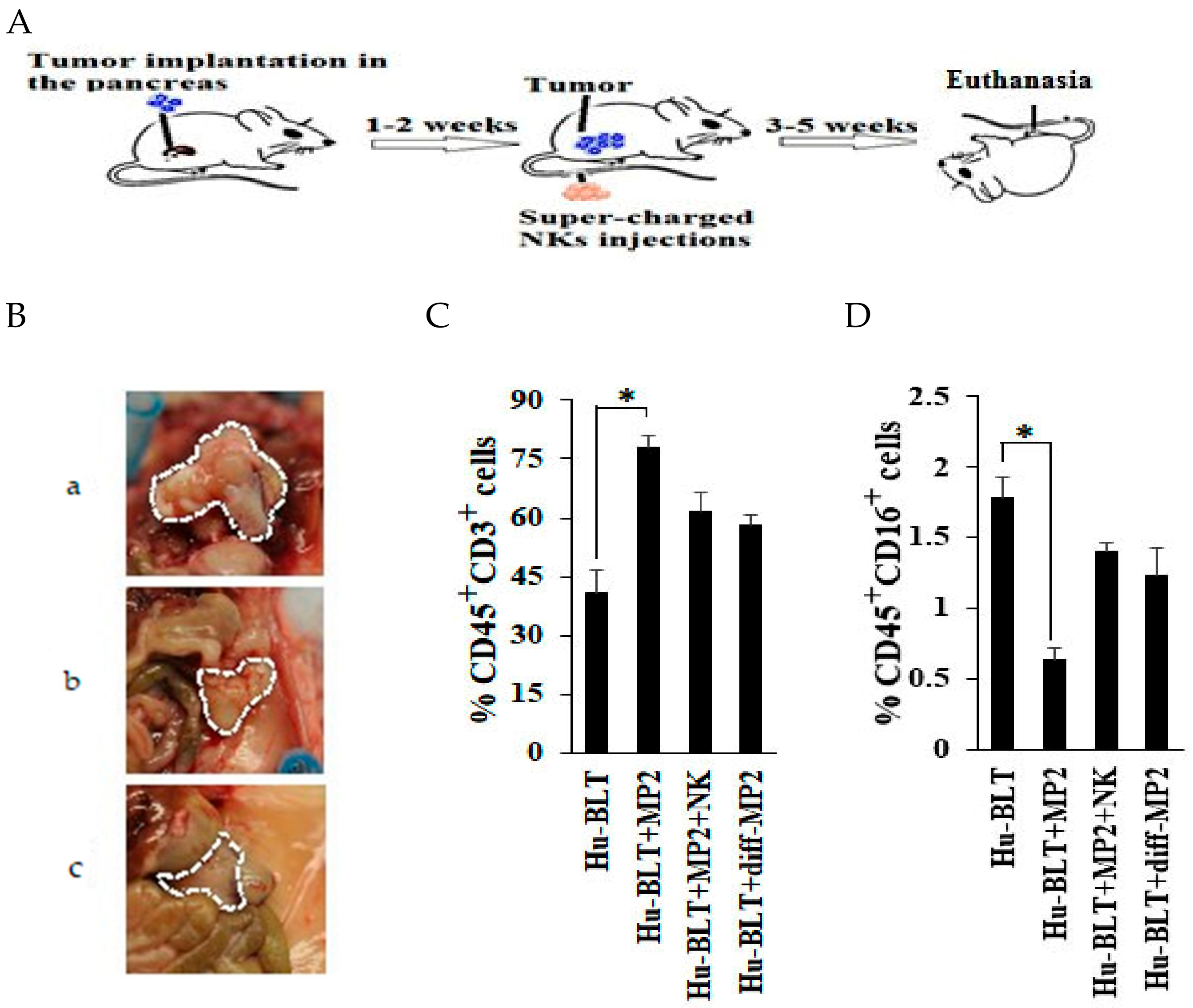
2.3. NK-Differentiated MP2 Tumors Did Not Grow Visible Tumors in the Pancreas of Hu-BLT Mice
2.4. Single Injection of NK Cells Inhibited Tumor Growth in Mice Implanted with MP2 Tumors
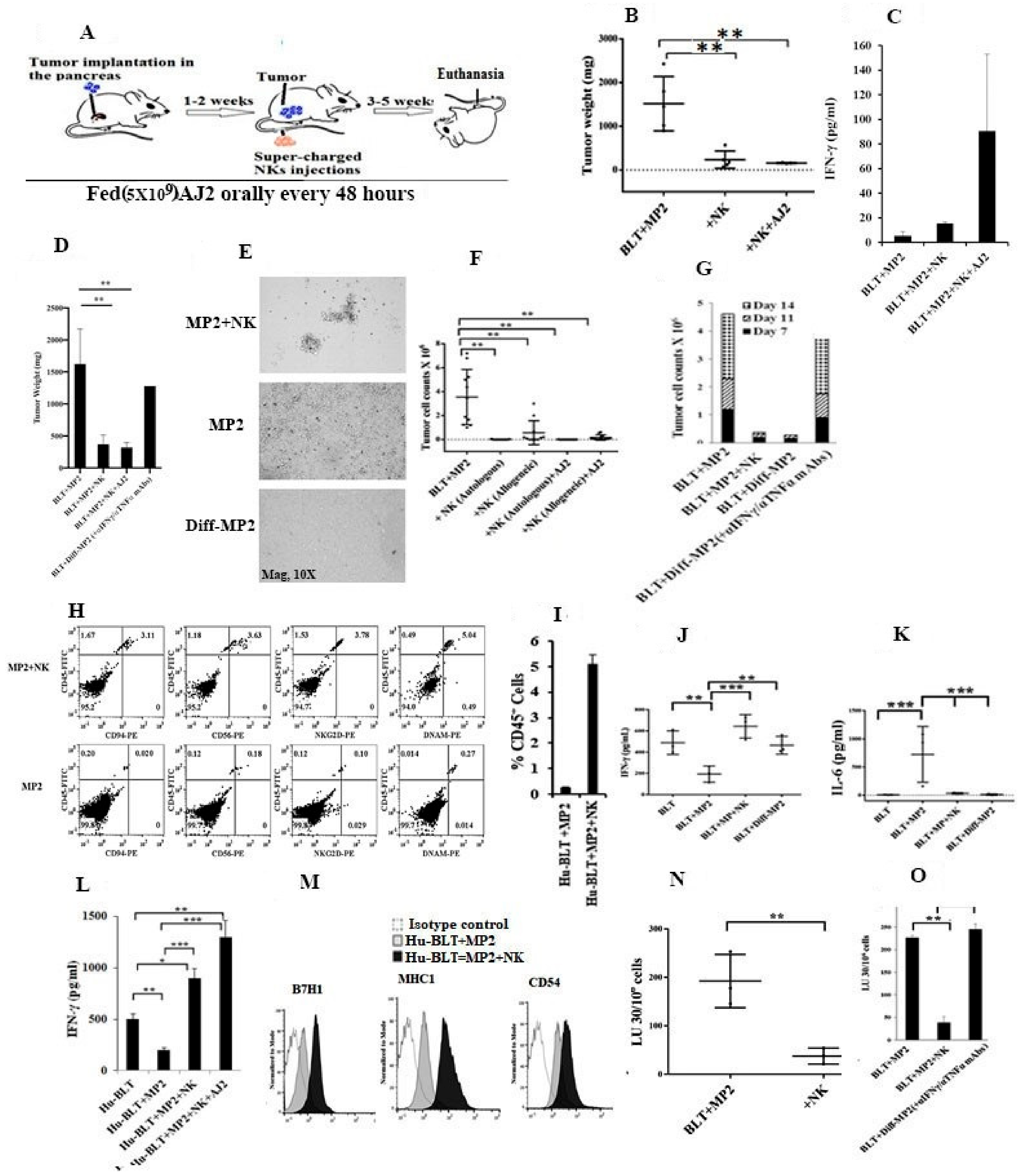
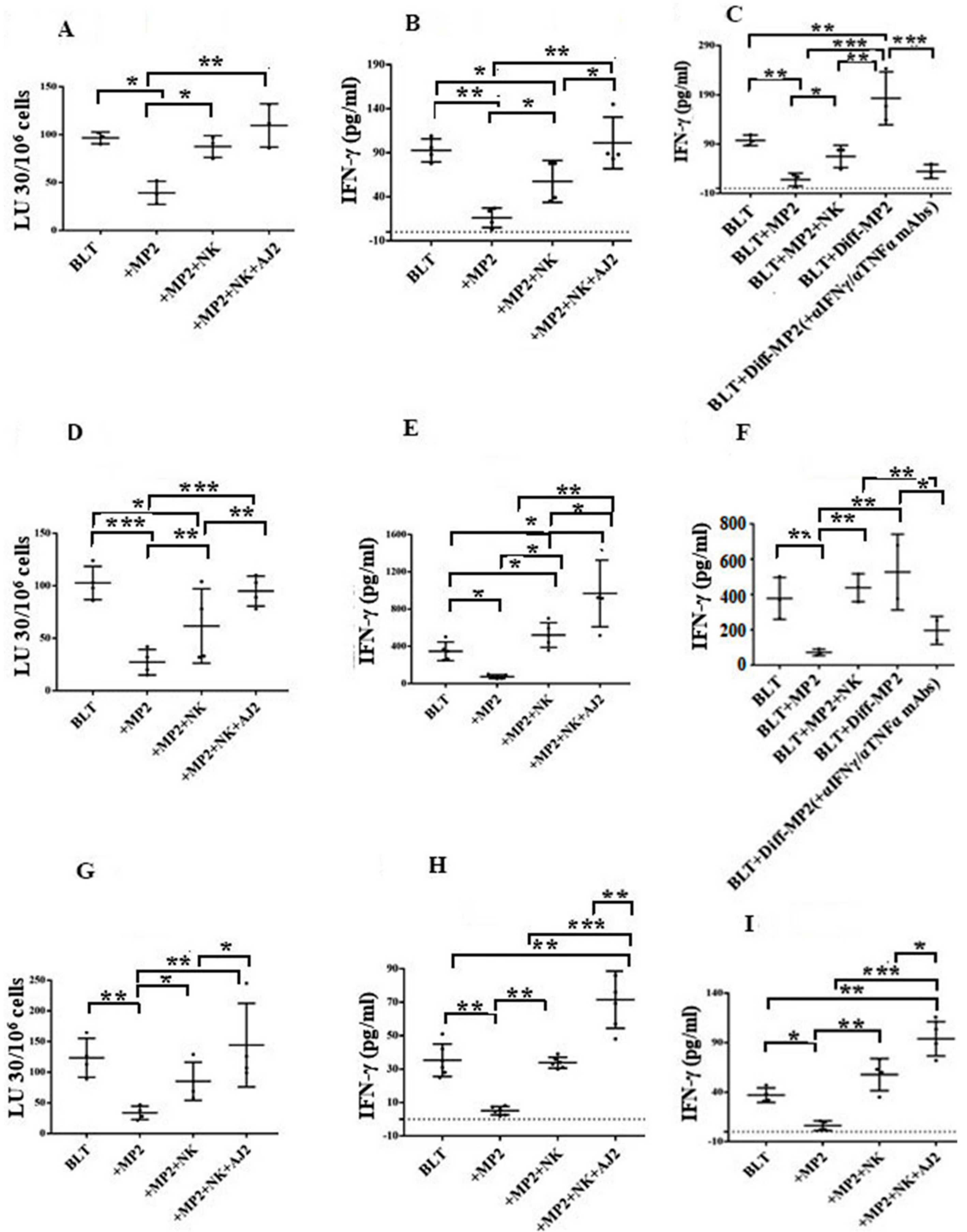
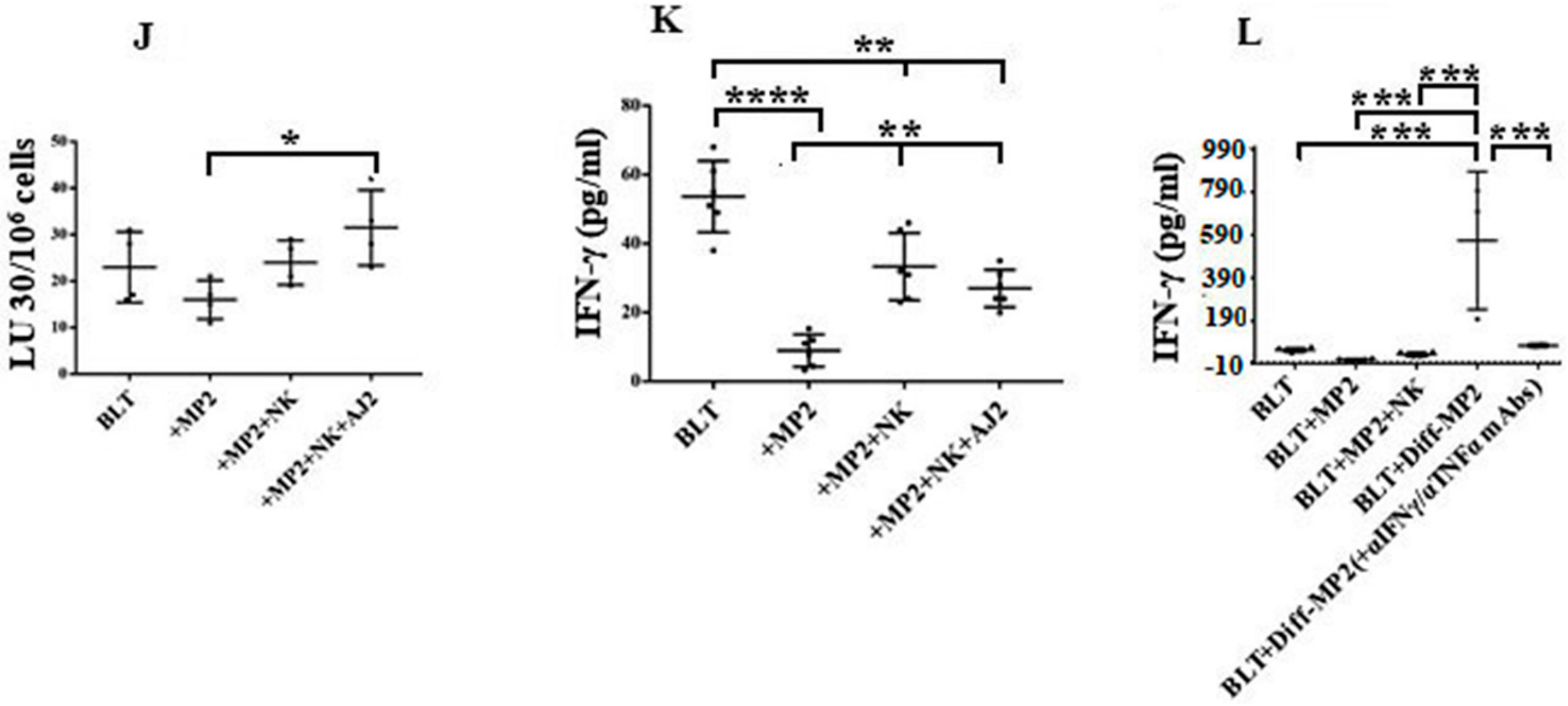
2.5. Suppression of NK Cell Cytotoxicity and Decreased Secretion of IFN-γ in Tumor-Bearing Mice within All Tissue Compartments; Restoration by Super-Charged NK Cells
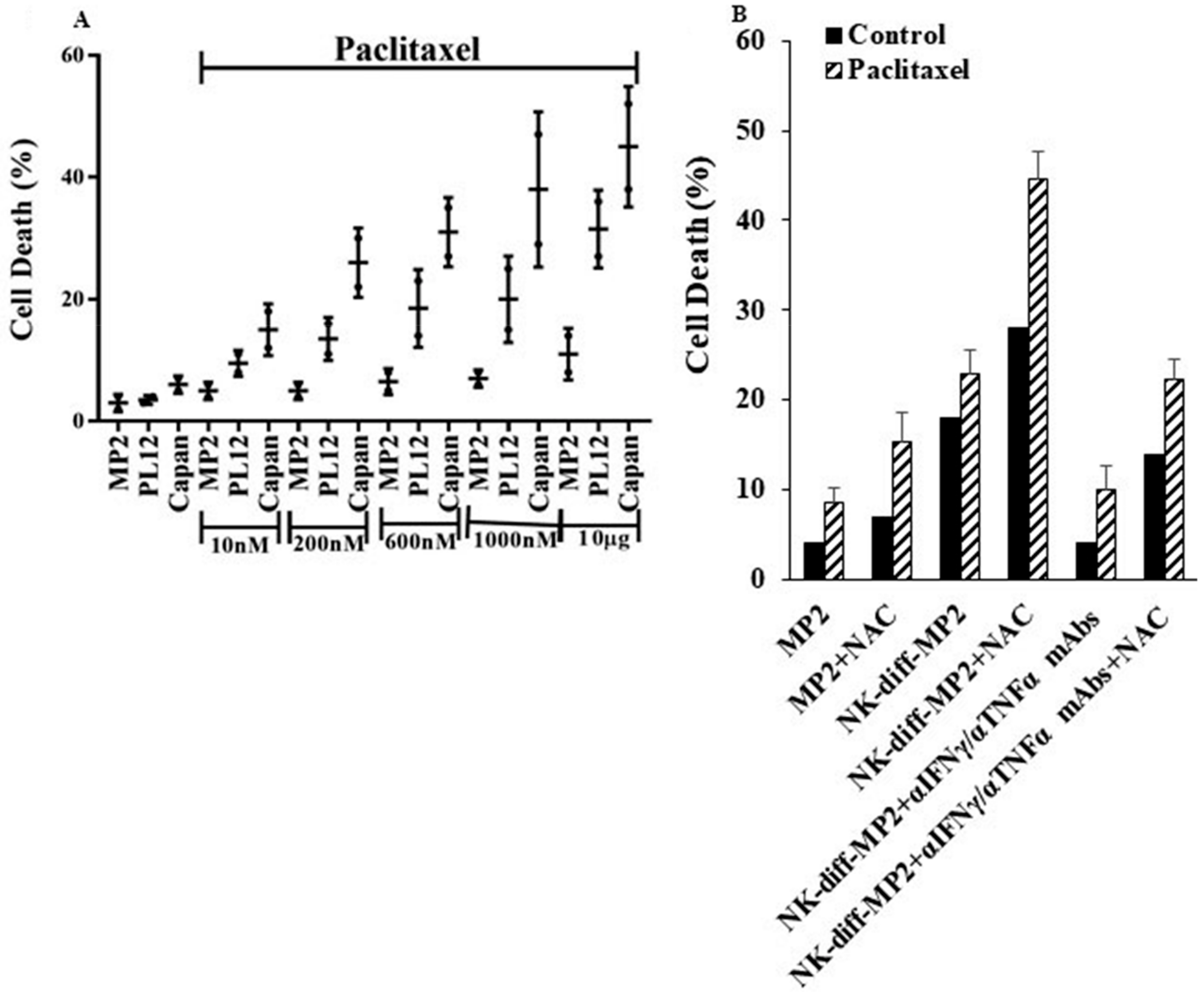
2.6. Paclitaxel Induce Cell Death in NK-Differentiated MP2 Tumors Treated with/without N-acetyl Cysteine (NAC)
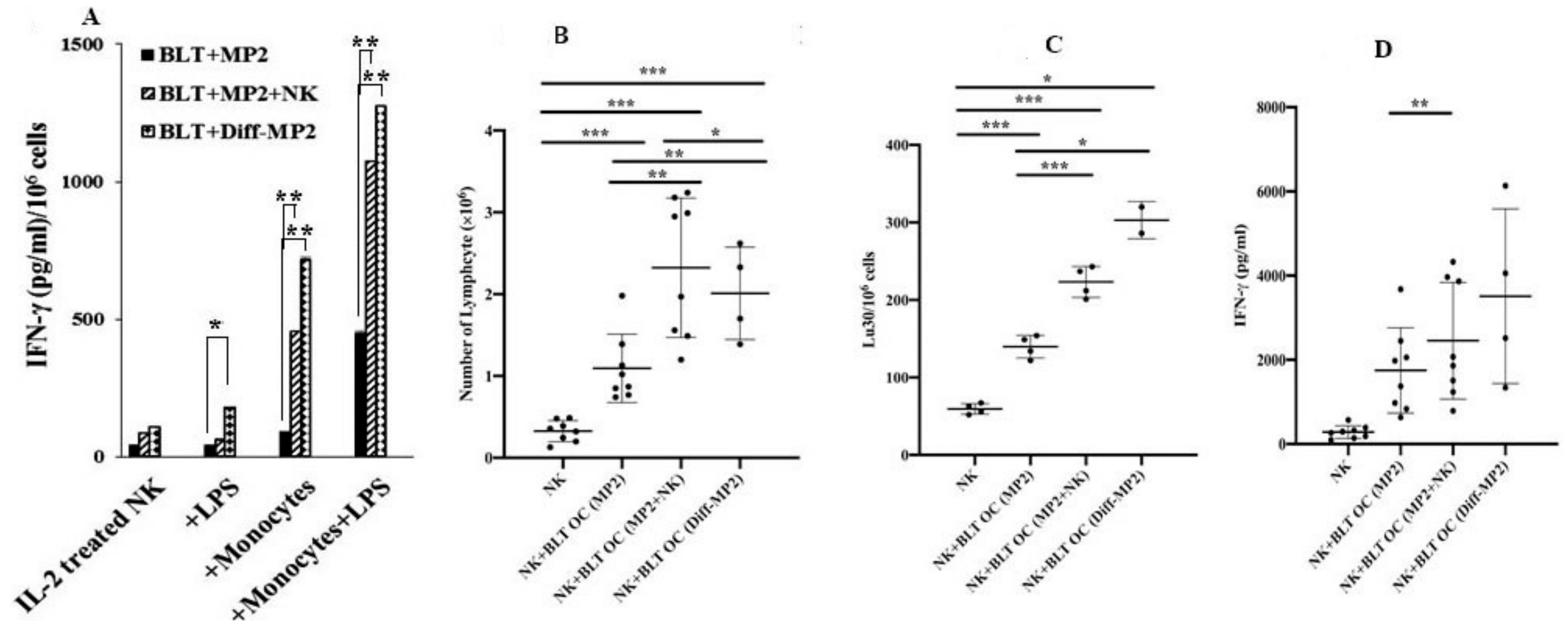
2.7. Monocytes or Osteoclasts from NK Injected Tumor Bearing Mice or NK-Differentiated Tumor Bearing Mice Had Higher Capacity to Activate NK Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Reagents, and Antibodies
4.2. Ethics Approval and Consent to Participate
4.3. Purification of Human NK Cells and Monocytes
4.4. Analysis of Human Pancreatic Cancer Cell Growth in Immune-Deficient (NSG) and Humanized-BLT Mice
4.5. Cell Dissociation and Cell Culture of Tissues from hu-BLT and NSG Mice
4.6. Isolations of NK Cells, T Cells and Monocytes from hu-BLT Mice
4.7. Generation of Osteoclasts and Expansion of Human and hu-BLT NK Cells
4.8. In-Vitro MP2 and OSCSCs Cancer Stem Cell Differentiation
4.9. Enzyme-Linked Immunosorbent Assays (ELISAs) and Multiplex Cytokine Assay
4.10. Surface Staining and Cell Death Assays
4.11. 51Cr Release Cytotoxicity Assay
4.12. Statistical Analysis
5. Conclusions
Availability of Data and Material
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Hu-BLT mouse | Humanized-bone marrow/liver/thymus mouse |
| NK cells | Natural killer cells |
| NSG mouse | NOD scid gamma mouse |
| PD-1 | Programmed death ligand 1 |
| MP2 | MiaPaCa-2 pancreatic cancer stem cells |
| OSCSCs | Oral squamous cancer stem cells |
| MHC-Class I | Major histocompatibility complex molecule class I |
| IFN-γ | Interferon-gamma |
| TNF-α | Tumor necrosis factor-α |
| IL-6 | Interleukin-6 |
| CSCs | Cancer stem cells |
| FBS | Fetal bovine serum |
| rhIL-2 | Recombinant human IL-2 |
| M-CSF | Macrophage colony-stimulating factor |
| RANKL | Receptor activator of nuclear factor kappa-B ligand |
| ELISA | Enzyme-Linked Immunosorbent Assays |
| BM | Bone marrow |
| GVHD | Graft vs. Host Disease |
| PBMCs | Peripheral blood mononuclear cells |
| NAC | N-acetyl cysteine |
| MDSCs | Myeloid-derived suppressor cells |
References
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III Study Comparing Gemcitabine Plus Cetuximab Versus Gemcitabine in Patients With Advanced Pancreatic Adenocarcinoma: Southwest Oncology Group-Directed Intergroup Trial S0205. J. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Lee, C.J.; Simeone, D.M. Identification of human pancreatic cancer stem cells. Methods Mol. Biol. 2009, 568, 161–173. [Google Scholar] [PubMed]
- Bao, B.; Azmi, A.S.; Aboukameel, A.; Ahmad, A.; Bolling-Fischer, A.; Sethi, S.; Ali, S.; Li, Y.; Kong, D.; Banerjee, S.; et al. Pancreatic cancer stem-like cells display aggressive behavior mediated via activation of FoxQ1. J. Biol. Chem. 2014, 289, 14520–14533. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Qin, R.; Wei, C.; Wang, M.; Shi, C.; Tian, R.; Peng, C. Pancreatic Cancer Cells Resistant to Chemoradiotherapy Rich in "Stem-Cell-Like" Tumor Cells. Dig. Dis. Sci. 2011, 56, 741–750. [Google Scholar] [CrossRef]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H.; et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, C.M.; Nieto, A.; Cortes, J.L.; Montes, R.M.; Catalina, P.; Cobo, F.; Barroso-Del-Jesus, A.; Concha, A. The low rate of HLA class I molecules on the human embryonic stem cell line HS293 is associated with the APM components’ expression level. Cell Biol. Int. 2007, 31, 1072–1078. [Google Scholar] [CrossRef]
- Suarez-Alvarez, B.; Rodriguez, R.M.; Calvanese, V.; Blanco-Gelaz, M.A.; Suhr, S.T.; Ortega, F.; Otero, J.; Cibelli, J.B.; Moore, H.; Fraga, M.F.; et al. Epigenetic Mechanisms Regulate MHC and Antigen Processing Molecules in Human Embryonic and Induced Pluripotent Stem Cells. PLoS ONE 2010, 5, e10192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryschich, E.; Nötzel, T.; Hinz, U.; Autschbach, F.; Ferguson, J.; Simon, I.; Weitz, J.; Fröhlich, B.; Klar, E.; Büchler, M.W.; et al. Control of T-Cell-mediated immune response by HLA class I in human pancreatic carcinoma. Clin. Cancer Res. 2005, 11, 498–504. [Google Scholar] [PubMed]
- Pandha, H.; Rigg, A.; John, J.; Lemoine, N. Loss of expression of antigen-presenting molecules in human pancreatic cancer and pancreatic cancer cell lines. Clin. Exp. Immunol. 2007, 148, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Jewett, A.; Man, Y.G.; Tseng, H.C. Dual functions of natural killer cells in selection and differentiation of stem cells; role in regulation of inflammation and regeneration of tissues. J. Cancer 2013, 4, 12–24. [Google Scholar] [CrossRef]
- Tseng, H.-C.; Arasteh, A.; Paranjpe, A.; Teruel, A.; Yang, W.; Behel, A.; Alva, J.A.; Walter, G.; Head, C.; Ishikawa, T.O.; et al. Increased Lysis of Stem Cells but Not Their Differentiated Cells by Natural Killer Cells; De-Differentiation or Reprogramming Activates NK Cells. PLoS ONE 2010, 5, e11590. [Google Scholar] [CrossRef] [Green Version]
- Jewett, A.; Arasteh, A.; Tseng, H.C.; Behel, A.; Arasteh, H.; Yang, W.; Cacalano, N.A.; Paranjpe, A. Strategies to rescue mesenchymal stem cells (MSCs) and dental pulp stem cells (DPSCs) from NK cell mediated cytotoxicity. PLoS ONE 2010, 5, e9874. [Google Scholar] [CrossRef]
- Jewett, A.; Cacalano, N.A.; Head, C.; Teruel, A. Coengagement of CD16 and CD94 receptors mediates secretion of chemokines and induces apoptotic death of naive natural killer cells. Clin. Cancer Res. 2006, 12, 1994–2003. [Google Scholar] [CrossRef] [Green Version]
- Warabi, M.; Kitagawa, M.; Hirokawa, K. Loss of MHC class II expression is associated with a decrease of tumor-infiltrating T cells and an increase of metastatic potential of colorectal cancer: Immunohistological and histopathological analyses as compared with normal colonic mucosa and adenomas. Pathol. Res. Pract. 2000, 196, 807–815. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Luo, Y. Association Between Activation of the Programmed Cell Death-1 (PD-1)/Programmed Death-Ligand 1 (PD-L1) Pathway and Pain in Patients with Cancer. Med. Sci. Monit. 2019, 25, 1275–1282. [Google Scholar] [CrossRef]
- Shu, X.; Liu, H.; Pan, Y.; Sun, L.; Yu, L.; Sun, L.; Yang, Z.; Ran, Y. Distinct biological characterization of the CD44 and CD90 phenotypes of cancer stem cells in gastric cancer cell lines. Mol. Cell. Biochem. 2019, 459, 35–47. [Google Scholar] [CrossRef]
- Ruiz-Cabello, F.; Klein, E.; Garrido, F. MHC antigens on human tumors. Immunol. Lett. 1991, 29, 181–189. [Google Scholar] [CrossRef]
- Tseng, H.C.; Bui, V.; Man, Y.G.; Cacalano, N.; Jewett, A. Induction of Split Anergy Conditions Natural Killer Cells to Promote Differentiation of Stem Cells through Cell-Cell Contact and Secreted Factors. Front. Immunol. 2014, 5, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, K.; Topchyan, P.; Kozlowska, A.K.; Ohanian, N.; Chiang, J.; Maung, P.O.; Park, S.H.; Ko, M.W.; Fang, C.; Nishimura, I.; et al. Super-charged NK cells inhibit growth and progression of stem-like/poorly differentiated oral tumors in vivo in humanized BLT mice; effect on tumor differentiation and response to chemotherapeutic drugs. Oncoimmunology 2018, 7, e1426518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selmani, Z.; Naji, A.; Zidi, I.; Favier, B.; Gaiffe, E.; Obert, L.; Borg, C.; Saas, P.; Tiberghien, P.; Rouas-Freiss, N.; et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4(+)CD25(high)FOXP3(+) regulatory T cells. Stem Cells 2008, 26, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, G.M.; Capobianco, A.; Abdelrazik, H.; Becchetti, F.; Mingari, M.C.; Moretta, L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: Role of indoleamine 2, 3-dioxygenase and prostaglandin E2. Blood 2008, 111, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Bui, V.T.; Tseng, H.C.; Kozlowska, A.; Maung, P.O.; Kaur, K.; Topchyan, P.; Jewett, A. Augmented IFN-gamma and TNF-alpha Induced by Probiotic Bacteria in NK Cells Mediate Differentiation of Stem-Like Tumors Leading to Inhibition of Tumor Growth and Reduction in Inflammatory Cytokine Release; Regulation by IL-10. Front. Immunol. 2015, 6, 576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westin, J.R.; Chu, F.; Zhang, M.; Fayad, L.E.; Kwak, L.W.; Fowler, N.; Romaguera, J.; Hagemeister, F.; Fanale, M.; Samaniego, F.; et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: A single group, open-label, phase 2 trial. Lancet Oncol. 2014, 15, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, J.; Atomi, Y.; Nagawa, H.; Kuroda, A.; Mutoh, T.; Minami, M.; Juji, T. Functional-analysis of TCR-gamma-delta+ T cells in tumor-infiltrating lymphocytes (TIL) of human pancreatic-cancer. Clin. Exp. Immunol. 1993, 93, 442–447. [Google Scholar] [CrossRef]
- Degrate, L.; Nobili, C.; Franciosi, C.; Caprotti, R.; Brivio, F.; Romano, F.; Leone, B.E.; Trezzi, R.; Uggeri, F. Interleukin-2 immunotherapy action on innate immunity cells in peripheral blood and tumoral tissue of pancreatic adenocarcinoma patients. Langenbecks Arch. Surg. 2009, 394, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Pages, M.N.; Verspaget, H.W.; Peña, A.S.; Lamers, C.B. Natural killer cell activity in patients with adenocarcinoma in the upper gastrointestinal tract. J. Clin. Lab. Immunol. 1991, 35, 27–32. [Google Scholar] [PubMed]
- Duan, X.; Deng, L.; Chen, X.; Lu, Y.; Zhang, Q.; Zhang, K.; Hu, Y.; Zeng, J.; Sun, W. Clinical significance of the immunostimulatory MHC class I chain-related molecule A and NKG2D receptor on NK cells in pancreatic cancer. Med. Oncol. 2011, 28, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-P.; Zhu, Y.; Zhang, J.J.; Xu, Z.K.; Qian, Z.Y.; Dai, C.C.; Jiang, K.R.; Wu, J.L.; Gao, W.T.; Li, Q.; et al. Comprehensive analysis of the percentage of surface receptors and cytotoxic granules positive natural killer cells in patients with pancreatic cancer, gastric cancer, and colorectal cancer. J. Transl. Med. 2013, 11, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, K.; Cook, J.; Park, S.H.; Topchyan, P.; Kozlowska, A.; Ohanian, N.; Fang, C.; Nishimura, I.; Jewett, A. Novel Strategy to Expand Super-Charged NK Cells with Significant Potential to Lyse and Differentiate Cancer Stem Cells: Differences in NK Expansion and Function between Healthy and Cancer Patients. Front. Immunol. 2017, 8, 297. [Google Scholar] [CrossRef] [Green Version]
- Kaur, K.; Chang, H.H.; Cook, J.; Eibl, G.; Jewett, A. Suppression of Gingival NK Cells in Precancerous and Cancerous Stages of Pancreatic Cancer in KC and BLT-Humanized Mice. Front. Immunol. 2017, 8, 1606. [Google Scholar] [CrossRef] [Green Version]
- Kaur, K.; Chang, H.H.; Topchyan, P.; Cook, J.M.; Barkhordarian, A.; Eibl, G.; Jewett, A. Deficiencies in Natural Killer Cell Numbers, Expansion, and Function at the Pre-Neoplastic Stage of Pancreatic Cancer by KRAS Mutation in the Pancreas of Obese Mice. Front. Immunol. 2018, 9, 1229. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [Green Version]
- Venstrom, J.M.; Pittari, G.; Gooley, T.A.; Chewning, J.; Spellman, S.R.; Haagenson, M.D.; Gallagher, M.M.; Malkki, M.; Petersdorf, E.W.; Dupont, B.; et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N. Engl. J. Med. 2012, 367, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Iliopoulou, E.G.; Kountourakis, P.; Karamouzis, M.V.; Doufexis, D.; Ardavanis, A.; Baxevanis, C.N.; Rigatos, G.; Papamichail, M.; Perez, S.A. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol. Immunother. 2010, 59, 1781–1789. [Google Scholar] [CrossRef]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [Green Version]
- Re, F.; Staudacher, C.; Zamai, L.; Vecchio, V.; Bregni, M. Killer cell Ig-like receptors ligand-mismatched, alloreactive natural killer cells lyse primary solid tumors. Cancer 2006, 107, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlowska, A.K.; Topchyan, P.; Kaur, K.; Tseng, H.C.; Teruel, A.; Hiraga, T.; Jewett, A. Differentiation by NK cells is a prerequisite for effective targeting of cancer stem cells/poorly differentiated tumors by chemopreventive and chemotherapeutic drugs. J. Cancer 2017, 8, 537–554. [Google Scholar] [CrossRef] [PubMed]
- Sipos, B.; Möser, S.; Kalthoff, H.; Török, V.; Löhr, M.; Klöppel, G. A comprehensive characterization of pancreatic ductal carcinoma cell lines: Towards the establishment of an in vitro research platform. Virchows Arch. 2003, 442, 444–452. [Google Scholar] [CrossRef]
- Kozlowska, A.K.; Tseng, H.C.; Kaur, K.; Topchyan, P.; Inagaki, A.; Bui, V.T.; Kasahara, N.; Cacalano, N.; Jewett, A. Resistance to cytotoxicity and sustained release of interleukin-6 and interleukin-8 in the presence of decreased interferon-gamma after differentiation of glioblastoma by human natural killer cells. Cancer Immunol. Immunother. 2016, 65, 1085–1097. [Google Scholar] [CrossRef] [Green Version]
- Kaur, K.; Nanut, M.P.; Ko, M.W.; Safaie, T.; Kos, J.; Jewett, A. Natural killer cells target and differentiate cancer stem-like cells/undifferentiated tumors: Strategies to optimize their growth and expansion for effective cancer immunotherapy. Curr. Opin. Immunol. 2018, 51, 170–180. [Google Scholar] [CrossRef]
- Jewett, A.; Kos, J.; Fong, Y.; Ko, M.W.; Safaei, T.; Perišić Nanut, M.; Kaur, K. NK cells shape pancreatic and oral tumor microenvironments; role in inhibition of tumor growth and metastasis. Semin. Cancer Biol. 2018, 53, 178–188. [Google Scholar] [CrossRef]
- Tseng, H.C.; Cacalano, N.; Jewett, A. Split anergized Natural Killer cells halt inflammation by inducing stem cell differentiation, resistance to NK cell cytotoxicity and prevention of cytokine and chemokine secretion. Oncotarget 2015, 6, 8947–8959. [Google Scholar] [CrossRef] [Green Version]
- Paranjpe, A.; Cacalano, N.A.; Hume, W.R.; Jewett, A. N-acetylcysteine protects dental pulp stromal cells from HEMA-induced apoptosis by inducing differentiation of the cells. Free Radic. Biol. Med. 2007, 43, 1394–1408. [Google Scholar] [CrossRef] [Green Version]
- Jewett, A.; Tseng, H.C. Potential rescue, survival and differentiation of cancer stem cells and primary non-transformed stem cells by monocyte-induced split anergy in natural killer cells. Cancer Immunol. Immunother. 2012, 61, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, A.; Cacalano, N.A.; Hume, W.R.; Jewett, A. N-acetyl cysteine mediates protection from 2-hydroxyethyl methacrylate induced apoptosis via nuclear factor kappa B-dependent and independent pathways: Potential involvement of JNK. Toxicol. Sci. 2009, 108, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Yu, Y.; Zhou, L.; Ma, J.; Tang, K.; Xu, P.; Ji, T.; Liang, X.; Lv, J.; Dong, W.; et al. Circulating Tumor Microparticles Promote Lung Metastasis by Reprogramming Inflammatory and Mechanical Niches via a Macrophage-Dependent Pathway. Cancer Immunol. Res. 2018, 6, 1046–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, S.; Hong, P.; Arumugam, B.; Pokomo, L.; Boyer, J.; Koizumi, N.; Kittipongdaja, P.; Chen, A.; Bristol, G.; Galic, Z.; et al. A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood 2010, 115, 1534–1544. [Google Scholar] [CrossRef] [Green Version]
- Vatakis, D.N.; Koya, R.C.; Nixon, C.C.; Wei, L.; Kim, S.G.; Avancena, P.; Bristol, G.; Baltimore, D.; Kohn, D.B.; Ribas, A.; et al. Antitumor activity from antigen-specific CD8 T cells generated in vivo from genetically engineered human hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2011, 108, E1408–E1416. [Google Scholar] [CrossRef] [Green Version]
- Jewett, A.; Cavalcanti, M.; Bonavida, B. Pivotal role of endogenous TNF-alpha in the induction of functional inactivation and apoptosis in NK cells. J. Immunol. 1997, 159, 4815–4822. [Google Scholar]
- Jewett, A.; Bonavida, B. Interferon-alpha activates cytotoxic function but inhibits interleukin-2-mediated proliferation and tumor necrosis factor-alpha secretion by immature human natural killer cells. J. Clin. Immunol. 1995, 15, 35–44. [Google Scholar] [CrossRef]
- Jewett, A.; Bonavida, B. Target-induced inactivation and cell death by apoptosis in a subset of human NK cells. J. Immunol. 1996, 156, 907–915. [Google Scholar]
- Jewett, A.; Wang, M.Y.; Teruel, A.; Poupak, Z.; Bostanian, Z.; Park, N.H. Cytokine dependent inverse regulation of CD54 (ICAM1) and major histocompatibility complex class I antigens by nuclear factor kappaB in HEp2 tumor cell line: Effect on the function of natural killer cells. Hum. Immunol. 2003, 64, 505–520. [Google Scholar] [CrossRef]
- Kozlowska, A.K.; Kaur, K.; Topchyan, P.; Jewett, A. Novel strategies to target cancer stem cells by NK cells; studies in humanized mice. Front. Biosci. (Landmark Ed.) 2017, 22, 370–384. [Google Scholar] [PubMed] [Green Version]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, K.; Kozlowska, A.K.; Topchyan, P.; Ko, M.-W.; Ohanian, N.; Chiang, J.; Cook, J.; Maung, P.O.; Park, S.-H.; Cacalano, N.; et al. Probiotic-Treated Super-Charged NK Cells Efficiently Clear Poorly Differentiated Pancreatic Tumors in Hu-BLT Mice. Cancers 2020, 12, 63. https://doi.org/10.3390/cancers12010063
Kaur K, Kozlowska AK, Topchyan P, Ko M-W, Ohanian N, Chiang J, Cook J, Maung PO, Park S-H, Cacalano N, et al. Probiotic-Treated Super-Charged NK Cells Efficiently Clear Poorly Differentiated Pancreatic Tumors in Hu-BLT Mice. Cancers. 2020; 12(1):63. https://doi.org/10.3390/cancers12010063
Chicago/Turabian StyleKaur, Kawaljit, Anna Karolina Kozlowska, Paytsar Topchyan, Meng-Wei Ko, Nick Ohanian, Jessica Chiang, Jessica Cook, Phyu Ou Maung, So-Hyun Park, Nicholas Cacalano, and et al. 2020. "Probiotic-Treated Super-Charged NK Cells Efficiently Clear Poorly Differentiated Pancreatic Tumors in Hu-BLT Mice" Cancers 12, no. 1: 63. https://doi.org/10.3390/cancers12010063